

Abstract
Background/Aims
Glycogen storage disease type Ib (GSD-Ib) patients deficient in a glucose-6-phosphate transporter (G6PT) manifest disturbed glucose homeostasis, myeloid dysfunctions, and hepatocellular adenoma (HCA). This study was conducted to evaluate whether maintaining normoglycemia in GSD-Ib could prevent HCA.
Methods
We infused neonatal GSD-Ib mice with adeno-associated virus (AAV) carrying G6PT and examined their metabolic and myeloid phenotypes for the 72-week study.
Results
The AAV vector delivered the G6PT transgene to the liver and bone marrow. Long-term metabolic correction was achieved alongside a transient myeloid correction. Hepatic G6PT activity was 50% of wild-type levels at 2 weeks post-infusion but declined rapidly thereafter to reach 3% of wild-type levels by age 6 to 72 weeks. Despite this, the infused mice maintained normoglycemia throughout the study, exhibited near normal growth and normalized serum metabolite profiles. However, all five AAV-treated GSD-Ib mice that lived over 50 weeks accumulated excessive hepatic glycogen and fat. Two mice developed steatohepatitis and multiple HCAs with one undergoing malignant transformation.
Conclusion
Normoglycemia alone cannot prevent hepatic steatosis and glycogen accumulation or the development of HCAs in GSD-Ib, providing one explanation why GSD-Ib patients maintaining normoglycemia under intense dietary therapy continue at risk for this long-term complication.
Keywords: glycogen storage disease type Ib, glucose-6-phosphate transporter, hepatocellular adenoma, malignant transformation, adeno-associated virus, gene therapy
1. Introduction
Glycogen storage disease type Ib (GSD-Ib, (MIM232220) is an autosomal recessive disorder caused by deficiencies in a glucose 6-phosphate transporter (G6PT) (1, 2). The primary function of G6PT is to translocate glucose-6-phosphate (G6P) from the cytoplasm into the lumen of the endoplasmic reticulum (ER) where it is hydrolyzed to glucose and phosphate either by the liver/kidney/intestine-restricted glucose-6-phosphatase-α (G6Pase-α) (1, 2) or the ubiquitously expressed G6Pase-β (3, 4). The concerted action of G6PT and G6Pase-α is required to maintain blood glucose homeostasis between meals and a deficiency of either protein results in an abnormal metabolic phenotype characterized by fasting hypoglycemia, hepatomegaly, nephromegaly, hyperlipidemia, hyperuricemia, lactic acidemia, and growth retardation (1, 2). The concerted action of G6PT and G6Pase-β is vital for normal neutrophil functions (5–7) and a deficiency of either protein results in an abnormal myeloid phenotype characterized by neutropenia, neutrophil dysfunctions and an abnormal increase in serum levels of granulocyte colony stimulating factor (G-CSF) (2, 6–9). The current treatment for GSD-Ib consists of a dietary therapy, including continuous nasogastric infusion of glucose (10) or frequent oral administration of uncooked cornstarch (11) to correct the loss of glucose homeostasis, augmented with G-CSF therapy (12, 13) to restore myeloid functions. The combined dietary and G-CSF therapies alleviate the metabolic and myeloid abnormalities of GSD-Ib patients significantly and greatly improve their prognosis. However, a significant percentage of adult patients under intensive dietary therapy continue to suffer from long-term complications, including hepatocellular adenoma (HCA) with risk for malignant transformation (14–17).
To study the biology and pathogenesis of GSD-Ib and to develop novel therapies for this disorder, we generated G6PT-deficient mice that manifest all known metabolic and myeloid dysfunctions characteristics of human GSD-Ib (9). If left untreated, the mice rarely survive weaning, mimicking the lethality of the untreated human GSD-Ib. To correct for the loss of glucose homeostasis, we also developed a glucose therapy for GSD-Ib mice that involves glucose injections twice daily starting immediately after birth (9). Unfortunately, the glucose therapy cannot sustain the life much beyond weaning and less than 15% of the mice live beyond age 2 months. Therefore we need additional tools to study the long-term complication of this disorder. One approach is to use gene therapy to reintroduce the G6PT gene.
G6PT is a hydrophobic protein that anchors in the ER membranes by 10 transmembrane domains (18). Using GSD-Ib mice, we have investigated the feasibility of viral vector-mediated G6PT gene transfer in vivo. We have shown that a recombinant adenovirus-mediated G6PT gene transfer can deliver the transgene to the liver and bone marrow and correct the metabolic and myeloid abnormalities of this disorder (19), suggesting there is a potential in gene therapy. However, recombinant adenovirus-mediated gene transfer is short-lived because of the rapid loss of vector-mediated gene expression (19, 20). Therefore in this study we evaluate the feasibility of an adeno-associated virus serotype 8 (AAV8) vector, that has been shown to direct efficient hepatic gene transfer (21, 22), to mediate gene replacement therapy for GSD-Ib. We also examine whether the treated mice that survive to adulthood will develop HCA. Our study shows that an AAV8 vector delivers the G6PT transgene to the liver and bone marrow, and corrects the metabolic and myeloid abnormalities in GSD-Ib mice. The infused animals no longer require glucose therapy and maintain a near normal metabolic phenotype including normoglycemia for the length of the 72-week study. However, HCAs developed in the infused animals after 51 weeks with one undergoing malignant transformation.
2. Materials and Methods
2.1. Infusion of GSD-Ib mice with AAV8-hG6PT
AAV8-hG6PT was packaged by co-transfection of 293 cells with UF11-hG6PT, an AAV vector containing human G6PT driven by the chicken β-actin promoter/CMV enhancer that directs high levels of transgene expression in the liver (23), and pDG8 (24) at University of Florida Powell Gene Therapy Center (Gainesville, FL). The virus preparations were quantified by dot-blot for the physical titer (25).
All animal studies were conducted under an animal protocol approved by the NICHD Animal Care and Use Committee. Glucose therapy, consisting of intra-peritoneal or subcutaneous injection of 25 to 200 μl of 15% glucose solution, was administrated to the GSD-Ib mice immediately upon birth and every 12 h thereafter, as described previously (9). Mice that survived weaning were given unrestricted access to Mouse Chow (Zeigler Bros., Inc., Gardners, PA). GSD-Ib is an autosomal recessive disorder (1, 2). Since the phenotypes of wild-type and heterozygous mice are identical, we use the designation “control” or “unaffected” to refer to both wild-type and heterozygous mice.
For gene therapy, neonatal GSD-Ib mice were infused via the temporal vein with AAV8-hG6PT at 6 × 1013 viral particles (vg)/kg. The glucose therapy was terminated immediately in AAV8-hG6PT-infused mice and the weaning mice were given Mouse Chow ad libitum.
2.2 RT-PCR analysis
Bone marrow cells from femoral and tibiae bones were harvested by flushing with 3 ml of Iscove’s modified Dulbecco’s medium containing 2% fetal bovine serum. Total RNA was isolated from liver or bone marrow cells using the TRIzol Reagent (Invitrogen Life Technologies, Carlsbad, CA). The introduced hG6PT transcripts were quantified by RT-PCR using a hG6PT-specific primer pair that detects the transduced hG6PT but not the endogenous mouse G6PT transcripts (Fig. 1).
Fig. 1.

RT-PCR analysis of hG6PT mRNA expression in the transduced liver and bone marrow. The hG6PT transcript was examined in the liver (A) and bone marrow (B) of neonatally AAV8-hG6PT-infused GSD-Ib mice (−/−/AAV) during postnatal development using an hG6PT-specific primer pair that detects the transduced human transcript but not the endogenous mouse G6PT transcripts.
2.3. G6P uptake assay
Microsomal preparations and G6P uptake measurement were performed as described previously (9). In G6P uptake assays, microsomes were incubated in a reaction mixture (100 μl) containing 50 mM sodium cacodylate buffer, pH 6.5, 250 mM sucrose, and 0.2 mM [U-14C]G6P (50 μCi/μmol, American Radiolabeled Chemicals, St Louis, MO). The reaction was stopped by filtering immediately through a nitrocellulose membrane (BA85, Schleicher & Schuell, Keene, NH). Microsomes permeabilized with 0.2% deoxycholate, to abolish G6P uptake, were used as negative controls.
2.4. Hematological and phenotype analyses
Blood samples were collected from the tail vein using EDTA-containing CAPIJECT tubes (TerumoMedical Co., Elkton, MD) for the differential leukocyte counts. Manual 200-cell leukocyte differential counts of peripheral blood cells were performed on Hema 3 (Fisher Scientific, Pittsburgh, PA.) stained smears. Serum glucose, total cholesterol, and uric acid were analyzed using kits obtained from Thermo Electron (Louisville, CO). Triglycerides were measured with a kit from Sigma Diagnostics (St Louis, MO) and lactate was measured by a kit from Trinity Biotech (St. Louis, MO). G-CSF was quantified using Quantikine ELISA kits (R&D Systems Inc., Minneapolis, MN).
For hematoxylin and eosin (H&E) and Masson’s trichrome staining, tissues were preserved in 10% neutral buffered formalin, embedded in paraffin, and sectioned at 4–6 microns thickness. The stained sections were visualized using the Axioskop2 plus microscope and the AxioVision 4.5 software (Carl Zeiss, Thornwood, NY). The composite pictures of the anaplastic carcinoma in the 52-week-old GSD-Ib mice with HCA were stitched together using the Photomerge feature of Adobe Photoshop CS3 (Adobe System Incorporated, San Jose, CA).
2.5. Statistical analysis
The unpaired t test was performed using the GraphPad Prism Program, version 4 (GraphPad Software, San Diego, CA). Values were considered statistically significant at p < 0.05.
3. Results
3.1. Neonatal AAV8-hG6PT infusion directs G6PT expression in the liver and bone marrow
GSD-Ib mice under dietary glucose therapy continue to suffer from frequent hypoglycemic seizures with less than 15% living beyond 2 months (9). For gene therapy, neonatal GSD-Ib mice were infused with 6 × 1013 vg/kg of AAV8-hG6PT via the temporal vein while simultaneously, the glucose therapy was terminated. There is no premature death of the infused animals before the age of 3 months.
GSD-Ib is characterized by disturbed glucose homeostasis and myeloid dysfunctions (1, 2). We therefore examined the expression of the hG6PT transgene in the liver and the bone marrow. Hepatic G6P uptake activity in the mice receiving gene therapy was restored to 50% of the levels of their unaffected littermates at 2 weeks post-infusion (Table 1). However, hepatic G6P uptake activity decreased rapidly to only 3% of the control activity at age 6 to 7 weeks but then stabilized for the duration of the 72-week study (Table 1).
Table 1.
Hepatic G6P uptake activity in neonatal AAV8-hG6PT-infused GSD-Ib mice
| Mice | Age weeks | G6P uptake Activity nmol/mg/3 mg |
|---|---|---|
| +/+ (n = 3) | 2 | 0.284 ± 0.04 |
| −/−/AAV8-hG6PT (n = 3) | 2 | 0.142 ± 0.02 (49.5) |
| −/− (n = 5) | 2–10 | 0.003 ± 0.001 |
| +/+ (n = 10) | 6–72 | 0.505 ± 0.026 |
| −/−/AAV8-hG6PT (n = 5) | 6–7 | 0.018 ± 0.006* (3.0) |
| −/−/AAV8-hG6PT (n = 5) | 51–72 | 0.018 ± 0.003* (3.0) |
GSD-Ib (−/−) mice were infused neonatally with 6 × 1013 vg/kg of AAV8-hG6PT as described under Materials and Methods. Age-matched wild-type & heterozygous mice (+/+) mice were used as positive controls. Untreated GSD-Ib mice (−/−) were used as negative controls. Data are presented as mean ± SEM. Numbers in parentheses represent percent of control activity.
p < 0.05 for comparisons between infused and untreated GSD-Ib mice.
The transgene expression in the liver was also examined by RT-PCR using an hG6PT-specific probe that detects the transduced hG6PT but not the endogenous mouse G6PT transcripts (Fig. 1). The results paralleled those seen in hepatic G6PT activity. Initially high levels of hG6PT mRNA were delivered to the liver at 2 weeks post-infusion, but expression declined rapidly during postnatal development, then stabilized for the duration of the 72-week study (Fig. 1A). RT-PCR analysis also showed that, at 2 weeks post-infusion, the hG6PT transgene was delivered to the bone marrow (Fig. 1B). However, hG6PT expression in the bone marrow became undetectable by 6 weeks post-infusion (Fig. 1B), suggesting that the expression of the introduced transgene in the bone marrow is transient.
3.2. Neonatal AAV8-hG6PT-infusion transiently corrects myeloid abnormalities of GSD-Ib
The GSD-Ib mice are severely neutropenic at age 1–3 weeks (9) and neutrophil counts in 2-week-old GSD-Ib mice only average 18.3% of the counts in their unaffected littermates (Fig. 2A). AAV8-hG6PT infusion improved neutropenia manifested by GSD-Ib mice and neutrophil counts in 2-week-old infused animals increased to 53.5% of the counts in the control littermates (Fig. 2A). The neutropenic condition in GSD-Ib mice improves with age (9). Neutrophil counts in 5–7 week-old untreated and AAV8-hG6PT-infused GSD-Ib mice averaged 38% and 47%, respectively, of the counts in the control mice (Fig. 2A). However, the number is not statistically different, suggesting that AAV8-hG6PT-mediated correction of neutropenia in GSD-Ib is transient.
Fig. 2.

Analysis of myeloid functions in AAV8-hG6PT-infused GSD-Ib mice. Neutrophil counts and serum G-CSF levels were examined in AAV8-hG6PT-infused GSD-Ib mice (−/−/AAV) at 2 and z weeks post-infusion. Age-matched unaffected (+/+) and GSD-Ib (−/−) mice were used as controls. (A) Blood neutrophil counts. (B) Serum G-CSF levels. Results are mean ± SEM from a minimal of 7 animals in each group. *p < 0.05; **p < 0.005.
Serum G-CSF values in 2-week-old GSD-Ib mice were 19.2-fold higher than those in the unaffected littermates (Fig. 2B). AAV8-hG6PT infusion normalized plasma cytokine levels and the levels of G-CSF in 2-week-old infused GSD-Ib− mice were only 2.4-fold higher than those in the control animals (Fig. 2B). The abnormal increase in serum G-CSF improved with age (9) and G-CSF values in 5–7 week-old untreated and AAV8-hG6PT-infused GSD-Ib mice were 3.1 and 2.5-fold, respectively, higher than those in the unaffected littermates (Fig. 2B), further confirmation that AAV8-hG6PT-mediated correction of myeloid abnormalities is transient.
3.3. Neonatal AAV8-hG6PT-infusion corrects metabolic abnormalities of GSD-Ib
GSD-Ib mice manifest hypoglycemia with a mean serum glucose level of 58 mg/dl (9). Consequently, GSD-Ib mice suffer from frequent hypoglycemic seizures and in the absence of any form of therapy die within the first two weeks of life (9). None of the AAV8-hG6PT-infused GSD-Ib mice suffered frequent hypoglycemic episodes in the absence of glucose therapy. Serum glucose levels in AAV8-hG6PT-infused GSD-Ib mice were normalized to 95.4 ± 6.3 mg/dl (n = 27) throughout the 72-week of the study (Fig. 3A). AAV8-hG6PT infusion also normalized serum cholesterol, triglyceride, uric acid, and lactic acid profiles (Fig. 3A). Of the 15 infused mice, three died prematurely at 15, 29, and 45 weeks post-infusion, suggesting that restoration of 3% of hepatic G6PT activity is borderline for the survival of the treated GSD-Ib mice.
Fig. 3.

Analyses of metabolic functions in AAV8-hG6PT-infused GSD-Ib mice. Metabolic functions were analyzed in neonatally AAV8-hG6PT-infused GSD-Ib mice (−/−/AAV) during postnatal development. Age-matched unaffected (+/+) littermates were used as controls. (A) Serum glucose, cholesterol, triglyceride, uric acid, and lactic acid levels. N = 26 for each group. (B) The body weight of AAV8-hG6PT-infused GSD-Ib (●) and their unaffected (○) littermates during postnatal development. Data are presented as mean ± SEM. (C) The weights of the liver relative to total body weight. N = 10 for each group. *p < 0.05; **p < 0.005.
GSD-Ib mice under dietary glucose therapy are growth retarded and at 2-week-old their average body weight is approximately 61% that of their control littermates (9). AAV8-hG6PT infusion markedly improved growth and the body weights of the infused animals to a level comparable to their unaffected littermates and remained so for the duration of the 72-week study (Fig. 3B).
Hepatomegaly is another clinical presentation in GSD-Ib. The average liver weight of 6–10 week-old GSD-Ib mice was elevated to 10.5% of body weight, compared to the control value of 4.0% (9). AAV8-hG6PT infusion moderately reduced hepatomegaly in GSD-Ib mice and the liver weight in 6- to 72-week-old infused GSD-Ib mice averaged 7.9 % of body weight (Fig. 3C).
3.4. AAV8-hG6PT-infused GSD-Ib mice developed HCA
The liver of human (1, 2) and mouse (9) GSD-Ib is characterized by glycogen and fat storage, leading to hepatomegaly. Moreover, HCA with risk for malignant transformation remains a long-term presentation in GSD-Ib patients maintaining normoglycemia (14–17).
In the liver of the control littermates, no abnormal liver histology is observed, although mild steatosis is observed in older mice, most noticeable in the liver of the 72-week-old control littermate (Fig. 4A). By 51 weeks the GSD-Ib mice treated with the AAV-hG6PT exhibited an excessive accumulation of glycogen in their hepatocytes with prominent lipid vacuoles in the liver parenchyma, evident of marked steatosis (Fig. 4A). While neutrophils were readily detectable in the liver of all 5 GSD-Ib mice that lived to age 51weeks and beyond, multifocal mild to moderate neutrophil infiltration was observed primarily in the 52- and 72-week-old GSD-Ib mice (Fig. 4B) which also carried multiple HCAs.
Fig. 4.
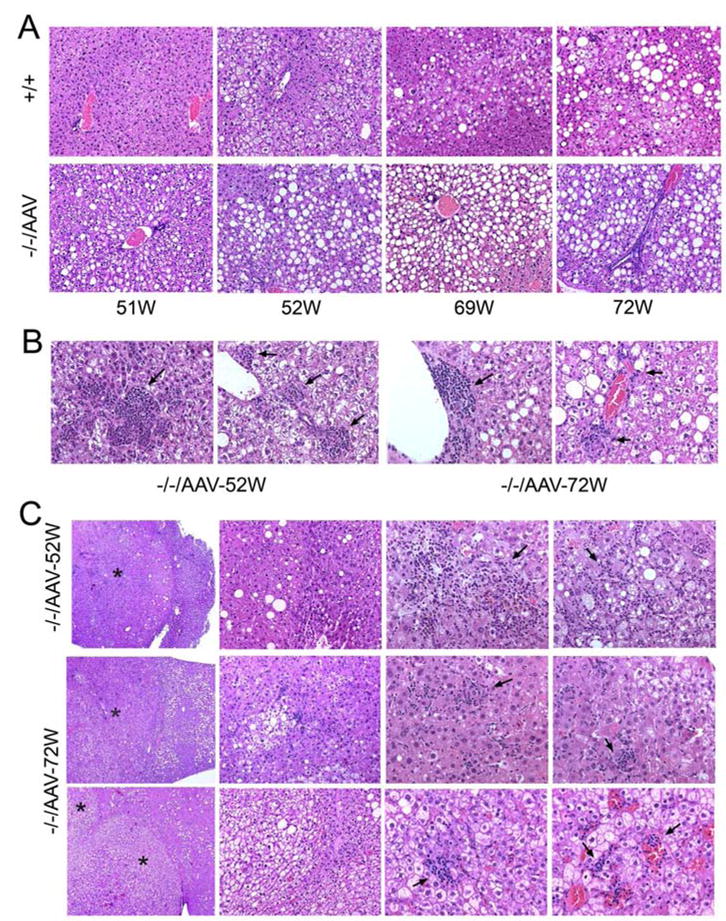
Histological analysis of the livers from the AAV8-hG6PT-infused GSD-Ib mice. (A) H&E stained liver sections from unaffected (+/+) and AAV8-hG6PT-infused GSD-Ib mice (−/−/AAV) at age 51 to 72 weeks. Representative sections are shown. Note of the glycogen and fat storage in the AAV8-hG6PT-infused GSD-Ib mice. Magnifications of ×200. (B) H&E stained liver sections showing neutrophil infiltration in the non-lesion parenchyma of the 52- and 72-week-old AAV8-hG6PT-infused GSD-Ib mice. Representative sections are shown. Arrows denote infiltrating neutrophils. Magnifications of ×400. (C) H&E stained liver sections showing HCAs in the 52- and 72-week-old AAV8-hG6PT-infused GSD-Ib mice. Representative sections showed fatty changes, glycogen storage and infiltrating neutrophils (denoted by arrows) at magnifications from left to right, ×50, ×200, ×400 and ×400. The HCAs are denoted by asterisks.
The HCA seen in the 52- and 72-week-old GSD-Ib mice are well circumscribed, non-encapsulated, with mild to moderate neutrophil infiltration, and mild steatosis (Fig. 4C). While all HCA nodules contained glycogenated hypertrophied hepatocytes, the double HCA nodules seen in the 72-week-old GSD-Ib mouse were excessively glycogenated (Fig. 4C, bottom panels). The lipid vacuoles were identified primarily at the boundary to the normal liver parenchyma (Fig. 4C). Small arteries derived from sinusoidal beds along with infiltrated neutrophils were also identified in the HCA nodules (Fig. 4C). Immunohistochemical staining using an antibody to cytokeratin AE1/AE3 failed to detect Mallory hyaline in the HCA nodules (data not shown).
Masson’s trichrome staining of the liver of control mice showed a few areas of interstitial fibrosis which was also observed in the 51-, 63-, and 69-week-old infused animals without HCA (data not shown). In contrast, Masson’s trichrome staining revealed focal fibrosis in the liver of the two GSD-Ib mice that developed HCA. In the non-lesion hepatic parenchyma, marked focal fibrosis/necrosis was observed in the 52-week-old GSD-Ib mouse carrying HCAs (Fig. 5A). Likewise, multifocal mild interstitial fibrosis was observed in the non-tumorous tissue near the capsular surface of the 72-week-old GSD-Ib mouse with HCAs (Fig. 5A). The HCA nodules exhibited variable degree of intra-tumor fibrosis. In the 52-week-old GSD-Ib mouse, Masson’s trichrome staining showed marked focal fibrosis in the HCA nodules (Fig. 5B). In the 72-week-old GSD-Ib mouse, moderate fibrosis was observed in one HCA nodule, while the other HCA nodules exhibited only very mild fibrosis (Fig. 5B).
Fig. 5.

Multifocal fibrosis in the livers of AAV8-hG6PT-infused GSD-Ib mice carrying HCA. (A) Masson’s trichrome staining of non-tumorous liver sections in the 52- and 72-week-old AAV8-hG6PT-infused GSD-Ib mice (−/−/AAV) at magnifications from left to right, ×50, ×400, and ×400. (B) Masson’s trichrome staining of HCA-containing nodules in the 52- and 72-week-old AAV8-hG6PT-infused GSD-Ib mice at magnifications from left to right, ×50, ×400, and ×400. Representative sections are shown. Interstatitial fibrosis or necrosis is shown by the blue colored staining of the collagen fibers.
H&E stained non-nodular tissue of the two HCA-containing mice also included multifocal area of necrosis occasionally admixed with degenerate neutrophils which was most marked in the liver of the 52-week-old infused mouse (Fig. 6A).
Fig. 6.

Hepatic necrosis and malignant transformation in the AAV8-hG6PT-infused GSD-Ib mice. (A) H&E stained liver sections in the 52- and 72-week-old AAV8-hG6PT-infused GSD-Ib mice (−/−/AAV). Representative sections showed multiple necrotic foci at magnifications from left to right, ×50, ×200, and ×200. (B) H&E stained liver sections showing a poorly differentiated anaplatic carcinoma in the 52-week-old AAV8-hG6PT-infused GSD-Ib mice with HCA. Top panels, the composite pictures of the carcinoma stitched together using the Photomerge feature of Adobe Photoshop CS3 as described under Materials and Methods at a magnification of ×17; the 2 rows of lower panels, representative sections at a magnification of ×400.
3.5. Malignant transformation occurred in one HCA-carrying GSD-Ib mice
One poorly differentiated anaplastic carcinoma developed in a 52-week-old GSD-Ib mouse carrying multiple HCA. The carcinoma was an infiltrative, non-encapsulated mass of moderately anisocytotic and anisokaryotic polygonal cells surrounded by large areas of necrosis admixed with hemorrhage (Fig. 6B).
4. Discussion
In the last two and half decades, intensive dietary therapy (10, 11) has enabled GSD-I patients to maintain normoglycemia and avoid the early metabolic manifestations of the disease. However, 50–70% of patients remain at risk for the longer-term complication of HCA that may undergo malignant transformation (14–17). To investigate the relationship between hepatic G6PT activity, serum glucose levels, and the incidence of HCA, we had previously generated G6PT-deficient GSD-Ib mice that mimic the phenotype of the human disorder (9). However, we were unable to examine the long-term complications of GSD-Ib because the current standard for care, a dietary glucose therapy, failed to prevent premature death of GSD-Ib animals from hypoglycemic seizures (9). In this study, we explored an AAV vector-mediated G6PT gene transfer, demonstrated its efficacy in preventing premature deaths, maintaining normoglycemia, and then examined whether GSD-Ib mice maintaining normoglycemia would still develop HCA. We show that neonatal infusion of GSD-Ib mice with AAV8-hG6PT delivered the G6PT transgene to the liver and bone marrow. While high levels of hG6PT transgene expression occurred for 2 weeks post-infusion, transgene expression declined rapidly in both tissues over the next 4 weeks. In the bone marrow of the infused GSD-Ib mice, G6PT expression became non-detectable at 6 weeks post-infusion, consistent with the transient correction of neutropenia and abnormal elevation in serum G-CSF levels. In the liver of the infused animals, low levels of G6PT activity at approximately 3% of wild-type levels persisted for the duration of the 72-week study. This level of hepatic G6PT activity enabled the GSD-Ib mice to maintain normoglycemia, achieve near normal growth, normalized serum profiles, and moderately reduced hepatomegaly. However, after 51 weeks, the AAV-hG6PT-infused GSD-Ib mice manifested excessive hepatic glycogen accumulation and steatosis resulting in the development of multiple HCAs. Our study suggests that restoration of 3% normal hepatic G6PT activity while maintain normoglycemia cannot prevent hepatic steatosis and excessive glycogen storage leading to the clinical manifestation of HCAs, explaining why human GSD-I patients maintaining normoglycemia continue to develop HCAs.
The rapid decline of hepatic transgene expression may reflect the extrachromosomal forms of the infused AAV vectors (26) and the neonatal age of the mice receiving gene transfer. A recent study showed that recombinant AAV8 transduces the neonatal mouse liver with high efficiency but there is a rapid reduction in vector genome numbers and stable transgene expression has been observed only in a small percentage of hepatocytes (27). Indeed, persistent long-term hepatic transgene expression mediated by recombinant AAV8 has been observed when administered to mature mice (28, 29) and rats (30) where there is a much lower rate of hepatic cell division.
Neutrophils are terminally differentiated short-lived leukocytes that are replenished from the multi-potent hematopoietic stem cells in the bone marrow (31, 32). Therefore, it was not surprising that the AAV8-hG6PT-mediated gene transfer to the bone marrow only transiently corrected myeloid abnormalities in GSD-Ib mice. Recently, sustained transgene expression has been achieved in hematopoietic stem cells of transplant recipient mice using self-complementary AAV vectors in conjunction with a hematopoietic cell-specific promoter/enhancer (33). These are worthy of further consideration and may be more attractive in future gene therapy studies for GSD-Ib.
The AAV8-hG6PT-infused GSD-Ib mice harboring 3% normal hepatic G6PT activity maintained normoglycemia but all manifested hepatomegaly evident by excessive glycogen accumulation and steatosis. In the 2 GSD-Ib mice that developed HCAs, their livers exhibited marked steatosis, neutrophil infiltration, fibrosis, and hepatocellular injury reminiscent of non-alcoholic steatohepatitis (34). Since persistent transgene expression occurred in only a small percentage of hepatocytes in neonatal AAV8-mediated gene transfer (27), the majority of hepatocytes still lacked a functional G6PT and continued to accumulate glycogen and fat vacuoles. Glycogen is known to induce inflammation (35, 36) and steatosis leads to lipotoxicity which causes apoptosis, necrosis, oxidative stress and inflammation (34). Taken together, the excessive glycogen and lipid accumulation in the liver could be the cause of the long-term consequence of HCAs developed in 2 of the 5 infused GSD-Ib mice.
HCA has been classified into 4 subtypes (37, 38): 1) mutations in hepatocyte nuclear factor 1α (HNF-1α), characterized by steatosis, no inflammatory infiltrates, and lack of cytological abnormalities; 2) mutations in β-catenin, characterized by frequent cytological abnormalities and a high risk of malignant transformation; 3) HCA with no HNF-1α or β-catenin mutation but with inflammatory infiltrates; and 4) HCA with no HNF-1α or β-catenin mutation and no inflammatory infiltrates. The HCAs seen in GSD-I patients tend to be small, multiple, and non-encapsulated (14–17) with histological features including fatty change, polymorphonuclear infiltrates, fibrosis and Mallory hyaline (39, 40). In 2 GSD-I patients, the HCAs have been characterized with one harboring the β-catenin mutation and the other with no mutation and no inflammatory infiltrates (37), suggesting heterogeneity in GSD-I patients. The HCAs developed in GSD-Ib mice are multiple and non-encapsulated with histological changes of steatosis, neutrophil infiltration, and fibrosis but no Mallory hyaline. Future studies will be directed toward elucidating the nature of the HCAs identified in the GSD-Ib mice.
Despite the numbers of AAV8-hG6PT-treated GSD-Ib mice that lived to age 51 weeks or older are small, the 40% incidence differs little from the incidence of 50–70% seen in human GSD-I patients (14–17). AAV vectors have the best safety profiles among the viruses widely used in gene therapy because they can infect a wide range of cell types, and have low immunogenicity, few toxic effects, and no known association with any human disease (41, 42). However, Donsante et al (43) have recently showed that neonatal mice treated with an AAV vector expressing the human β-glucuronidase gene driven by a β-actin promoter/CMV enhancer showed an increased incidence of hepatocellular carcinoma, suggesting tumorigenicity of the AAV vectors. In human GSD-I patients carrying HCA, approximately 10% undergo malignant transformation (16). In the 2 HCA-containing GSD-Ib mice, 1 underwent malignant transformation. While this might appear to indicate a higher malignant potential in the mouse model, there is insufficient statistical support for such a conclusion at present. The G6PT gene in AAV8-hG6PT is under the control of the chicken β-actin promoter/CMV enhancer (23). Whether the AAV vectors used in this study contributed to malignant transformation observed in the infused GSD-Ib mice remained to be determined.
In summary, using a murine model of GSD-Ib, we have shown that an AAV vector-mediated gene transfer delivered the G6PT transgene to multiple tissues including the liver and bone marrow, correcting metabolic and myeloid dysfunctions in GSD-Ib. However, the levels of G6PT expression while maintaining normoglycemia failed to prevent excessive hepatic glycogen and lipid accumulation leading to the development of HCAs. The results are in agreement with the observation that human GSD-I patients maintaining normoglycemia suffer this long-term complication.
Acknowledgments
This research was supported by the Intramural Research Programs of the NICHD, NIH. The authors gratefully acknowledge the University of Florida Powell Gene Therapy Center for producing the AAV vectors used in this study.
Abbreviations
- GSD-Ib
glycogen storage disease type Ib
- G6P
glucose-6-phosphate
- G6PT
glucose-6-phosphate transporter
- HCA
hepatocellular adenoma
- AAV
adeno-associated virus
- G6P
glucose-6-phosphate
- G6Pase
glucose-6-phosphatase
- G-CSF
granulocyte colony stimulating factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chou JY, Matern D, Mansfield BC, Chen Y-T. Type I glycogen storage diseases: disorders of the glucose-6-phosphatase complex. Curr Mol Med. 2002;2:121–143. doi: 10.2174/1566524024605798. [DOI] [PubMed] [Google Scholar]
- 2.Chou JY, Mansfield BC. Glucose-6-phosphate transporter: the key to glycogen storage disease type Ib. In: Broer S, Wagner CA, editors. Membrane Transporter Diseases. New York: Springer; 2003. pp. 191–205. [Google Scholar]
- 3.Shieh J-J, Pan C-J, Mansfield BC, Chou JY. Glucose-6-phosphate hydrolase, widely expressed outside the liver, can explain age-dependent resolution of hypoglycemia in glycogen storage disease type Ia. J Biol Chem. 2003;278:47098–47103. doi: 10.1074/jbc.M309472200. [DOI] [PubMed] [Google Scholar]
- 4.Ghosh A, Shieh J-J, Pan C-J, Chou JY. Histidine-167 is the phosphate acceptor in glucose-6-phosphatase-β forming a phosphohistidine-enzyme intermediate during catalysis. J Biol Chem. 2004;279:12479–12483. doi: 10.1074/jbc.M313271200. [DOI] [PubMed] [Google Scholar]
- 5.Kim SY, Nguyen ATD, Gao J-L, Murphy PM, Mansfield BC, Chou JY. Bone-Marrow Derived Cells Require a Functional Glucose-6-Phosphate Transporter for Normal Myeloid Functions. J Biol Chem. 2006;281:28794–28801. doi: 10.1074/jbc.M604964200. [DOI] [PubMed] [Google Scholar]
- 6.Cheung YY, Kim SY, Yiu WH, Pan CJ, Jun HS, Ruef RA, et al. Impaired neutrophil activity and increased susceptibility to bacterial infection in mice lacking glucose-6-phosphatase-beta. J Clin Invest. 2007;117:784–793. doi: 10.1172/JCI30443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim SY, Jun HS, Mansfield BC, Chou JY. Neutrophil stress and apoptosis underlie myeloid dysfunction in glycogen storage disease type Ib. Blood. 2008;111:5704–5711. doi: 10.1182/blood-2007-12-129114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gitzelmann R, Bosshard NU. Defective neutrophil and monocyte functions in glycogen storage disease type 1b: a literature review. Eur J Pediatr. 1993;152:S33–S38. doi: 10.1007/BF02072085. [DOI] [PubMed] [Google Scholar]
- 9.Chen L-Y, Shieh J-J, Lin B, Pan C-J, Gao J-L, Murphy PM, et al. Impaired glucose homeostasis, neutrophil rafficking and function in mice lacking the glucose-6-phosphate transporter. Hum Mol Genet. 2003;12:2547–2558. doi: 10.1093/hmg/ddg263. [DOI] [PubMed] [Google Scholar]
- 10.Greene HL, Slonim AE, O’Neill JA, Jr, Burr IM. Continuous nocturnal intragastric feeding for management of type 1 glycogen-storage disease. N Engl J Med. 1776;294:423–425. doi: 10.1056/NEJM197602192940805. [DOI] [PubMed] [Google Scholar]
- 11.Chen YT, Cornblath M, Sidbury JB. Cornstarch therapy in type I glycogen storage disease. N Engl J Med. 1984;310:171–175. doi: 10.1056/NEJM198401193100306. [DOI] [PubMed] [Google Scholar]
- 12.Schroten H, Roesler J, Breidenbach T, Wendel U, Elsner J, Schweitzer S, et al. Granulocyte and granulocyte-macrophage colony-stimulating factors for treatment of neutropenia in glycogen storage disease type Ib. J Pediatr. 1991;119:748–754. doi: 10.1016/s0022-3476(05)80290-2. [DOI] [PubMed] [Google Scholar]
- 13.Roe TF, Coates TD, Thomas DW, Miller JH, Gilsanz V. Brief report: treatment of chronic inflammatory bowel disease in glycogen storage disease type Ib with colony-stimulating factors. N Engl J Med. 1992;326:1666–1669. doi: 10.1056/NEJM199206183262504. [DOI] [PubMed] [Google Scholar]
- 14.Bianchi L. Glycogen storage disease I and hepatocellular tumours. Eur J Pediatr. 1993;52 (Suppl 1):S63–S70. doi: 10.1007/BF02072092. [DOI] [PubMed] [Google Scholar]
- 15.Labrune P, Trioche P, Duvaltier I, Chevalier P, Odievre M. Hepatocellular adenomas in glycogen storage disease type I and III: a series of 43 patients and review of the literature. J Pediatr Gastroenterol Nutr. 1997;24:276–279. doi: 10.1097/00005176-199703000-00008. [DOI] [PubMed] [Google Scholar]
- 16.Lee PJ. Glycogen storage disease type I: pathophysiology of liver adenomas. Eur J Pediatr. 2002;161 (Suppl 1):S46–S49. doi: 10.1007/s00431-002-1002-0. [DOI] [PubMed] [Google Scholar]
- 17.Rake JP, Visser G, Labrune P, Leonard JV, Ullrich K, Smit GP. Glycogen storage disease type I: diagnosis, management, clinical course and outcome. Results of the European Study on Glycogen Storage Disease Type I (ESGSD I) Eur J Pediatr. 2002;161 (Suppl 1):S20–S34. doi: 10.1007/s00431-002-0999-4. [DOI] [PubMed] [Google Scholar]
- 18.Pan C-J, Lin B, Chou JY. Transmembrane topology of human Glucose-6-phosphate transporter. J Biol Chem. 1999;274:13865–13869. doi: 10.1074/jbc.274.20.13865. [DOI] [PubMed] [Google Scholar]
- 19.Yiu WH, Pan C-J, Allamarvdasht A, Kim SY, Chou JY. Glucose-6-phosphate transporter gene therapy corrects metabolic and myeloid abnormalities in glycogen storage disease type Ib mice. Gene Ther. 2007;14:219–226. doi: 10.1038/sj.gt.3302869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson JM. Adenovirus-mediated gene transfer to liver. Adv Drug Deliv Rev. 2001;46:205–209. doi: 10.1016/s0169-409x(00)00125-3. [DOI] [PubMed] [Google Scholar]
- 21.Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci USA. 2002;99:11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas CE, Storm TA, Huang Z, Kay MA. Rapid uncoating of vector genomes is the key to efficient liver transduction with pseudotyped adeno-associated virus vectors. J Virol. 2004;78:3110–3122. doi: 10.1128/JVI.78.6.3110-3122.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu L, Daly T, Gao C, Flotte TR, Song S, Byrne BJ, et al. CMV-beta-actin promoter directs higher expression from an adeno-associated viral vector in the liver than the cytomegalovirus or elongation factor 1 alpha promoter and results in therapeutic levels of human factor X in mice. Hum Gene Ther. 2001;12:563–573. doi: 10.1089/104303401300042500. [DOI] [PubMed] [Google Scholar]
- 24.Grimm D, Kern A, Rittner K, Kleinschmidt JA. Novel tools for production and purification of recombinant adenoassociated virus vectors. Hum Gene Ther. 1998;10:2745–2760. doi: 10.1089/hum.1998.9.18-2745. [DOI] [PubMed] [Google Scholar]
- 25.Hauswirth WW, Lewin AS, Zolotukhin S, Muzyczka N. Production and purification of recombinant adeno-associated virus. Methods Enzymol. 2000;316:743–761. doi: 10.1016/s0076-6879(00)16760-6. [DOI] [PubMed] [Google Scholar]
- 26.Nakai H, Yant SR, Storm TA, Fuess S, Meuse L, Kay MA. Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J Virol. 2001;75:6969–6976. doi: 10.1128/JVI.75.15.6969-6976.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cunningham SC, Dane AP, Spinoulas A, Alexander IE. Gene delivery to the juvenile mouse liver using AAV2/8 vectors. Mol Ther. 2008;16:1081–1088. doi: 10.1038/mt.2008.72. [DOI] [PubMed] [Google Scholar]
- 28.Conlon TJ, Cossette T, Erger K, Choi YK, Clarke T, Scott-Jorgensen M, et al. Efficient hepatic delivery and expression from a recombinant adeno-associated virus 8 pseudotyped α1-antitrypsin vector. Mol Ther. 2005;12:867–875. doi: 10.1016/j.ymthe.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 29.Nakai H, Fuess S, Storm TA, Muramatsu S, Nara Y, Kay M. Unrestricted hepatocyte transduction with adeno-associated virus serotype 8 vectors in mice. J Virol. 2005;79:214–224. doi: 10.1128/JVI.79.1.214-224.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seppen J, Bakker C, de Jong B, Kunne C, van den Oever K, Vandenberghe K, et al. Adeno-associated virus vector serotypes mediate sustained correction of bilirubin UDP glucuronosyltransferase deficiency in rats. Mol Ther. 2006;13:1085–1092. doi: 10.1016/j.ymthe.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 31.Cumano A, Godin I. Ontogeny of the hematopoietic system. Annu Rev Immunol. 2007;25:745–785. doi: 10.1146/annurev.immunol.25.022106.141538. [DOI] [PubMed] [Google Scholar]
- 32.Iwasaki H, Akashi K. Myeloid lineage commitment from the hematopoietic stem cell. Immunity. 2007;26:726–740. doi: 10.1016/j.immuni.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 33.Maina N, Han Z, Li X, Hu Z, Zhong L, Bischof D, et al. Recombinant self-complementary adeno-associated virus serotype vector-mediated hematopoietic stem cell transduction and lineage-restricted, long-term transgene expression in a murine serial bone marrow transplantation model. Hum Gene Ther. 2008;19:376–383. doi: 10.1089/hum.2007.143. [DOI] [PubMed] [Google Scholar]
- 34.Marra F, Gastaldelli A, Svegliati Baroni G, Tell G, Tiribelli C. Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends Mol Med. 2008;14:72–81. doi: 10.1016/j.molmed.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 35.Yamashita T, Ishibashi Y, Nagaoka I, Kasuya K, Masuda K, Warabi H, et al. Studies of glycogen-induced inflammation of mice. Dynamics of inflammatory responses and influence of antiinflammatory drugs and protease inhibitors. Inflammation. 1982;6:87–101. doi: 10.1007/BF00910722. [DOI] [PubMed] [Google Scholar]
- 36.Mulligan MS, Lentsch AB, Miyasaka M, Ward PA. Cytokine and adhesion molecule requirements for neutrophil recruitment during glycogen-induced peritonitis. Inflamm Res. 1998;47:251–255. doi: 10.1007/s000110050326. [DOI] [PubMed] [Google Scholar]
- 37.Zucman-Rossi J, Jeannot E, Nhieu JT, Scoazec JY, Guettier C, Rebouissou S, et al. Genotype-phenotype correlation in hepatocellular adenoma: new classification and relationship with HCC. Hepatology. 2006;43:515–524. doi: 10.1002/hep.21068. [DOI] [PubMed] [Google Scholar]
- 38.Bioulac-Sage P, Rebouissou S, Thomas C, Blanc JF, Saric J, Sa Cunha A, et al. Hepatocellular adenoma subtype classification using molecular markers and immunohistochemistry. Hepatology. 2007;46:740–748. doi: 10.1002/hep.21743. [DOI] [PubMed] [Google Scholar]
- 39.Poe R, Snover DC. Adenomas in glycogen storage disease type 1. Two cases with unusual histologic features. Am J Surg Pathol. 1988;12:477–483. doi: 10.1097/00000478-198806000-00008. [DOI] [PubMed] [Google Scholar]
- 40.Volmar KE, Burchette JL, Creager AJ. Hepatic adenomatosis in glycogen storage disease type Ia: report of a case with unusual histology. Arch Pathol Lab Med. 2003;127:e402–e405. doi: 10.5858/2003-127-e402-HAIGSD. [DOI] [PubMed] [Google Scholar]
- 41.Carter PJ, Samulski RJ. Adeno-associated viral vectors as gene delivery vehicles. Int J Mol Med. 2000;6:17–27. doi: 10.3892/ijmm.6.1.17. [DOI] [PubMed] [Google Scholar]
- 42.Flotte TR. Gene therapy progress and prospects: recombinant adeno-associated virus (rAAV) vectors. Gene Ther. 2004;11:805–810. doi: 10.1038/sj.gt.3302233. [DOI] [PubMed] [Google Scholar]
- 43.Donsante A, Miller DG, Li Y, Vogler C, Brunt EM, Russell DW, et al. AAV vector integration sites in mouse hepatocellular carcinoma. Science. 2007;317:477. doi: 10.1126/science.1142658. [DOI] [PubMed] [Google Scholar]