

Abstract
Essential tremor (ET) is the most common pathological tremor in humans. The traditional view of ET, as a mono-symptomatic condition, is being replaced by an appreciation of the spectrum of clinical features, with both motor and non-motor elements. These features are not distributed homogeneously across patients. In addition, post-mortem studies are now demonstrating distinct structural changes in ET. There is growing evidence that ET may be a family of diseases rather than a single entity. Further, this aging-associated, progressive disorder is associated with neuronal loss and postmortem changes that occur in traditional neurodegenerative disorders.
Keywords: Essential tremor, neurodegenerative, clinical, pathology, syndrome
Introduction to Essential Tremor and Questions Posed
Essential tremor (ET), the commonest pathological tremor in humans, is among the more ubiquitous neurological diseases, with a prevalence (age ≥40 years) of 4.0%.1, 2 Its most recognizable clinical feature is an 8–12 Hz kinetic tremor of the arms (i.e., tremor during voluntary movement)(Figure 1), which often is later accompanied by head and voice tremors. The condition is global, affecting human beings in a variety of settings, ranging from the remote Okapa sub-district of Papua New Guinea to the urban Washington Heights-Inwood community in northern Manhattan, New York.1, 2 The traditional view of ET as a mono-symptomatic condition characterized only by kinetic or postural tremor has been supplanted in recent years. First, the tremor phenomenology is broad and many patients have other motor manifestations3 and non-motor features, including cognitive and psychiatric.3, 4 Furthermore, this expanded constellation of clinical features is not distributed homogeneously across patients. For example, some patients develop head tremor while others do not. Some dement while others do not; some develop Parkinson’s disease (PD) while others do not.5 Recent studies of the pathology of ET have demonstrated several distinctive structural changes, including neuronal loss.6–10 With evidence of clinical and pathological heterogeneity emerging, several important questions arise. First, is ET a single disease entity or a family of diseases? Second, is ET neurodegenerative?
Figure 1.

Kinetic tremor is apparent in an ET patient’s drawing of an Archimedes spiral.
Essential Tremor or the Essential Tremors?
Neurology is replete with historical examples in which clinical-pathological investigations have resulted in the partitioning of disease entities and the refinement of classification. For example, many patients with upper motor neurons signs were clinically lumped together, yet in the 1800s, Charcot and others, observed that among such patients who had sclerosis on postmortem examination, one sub-group had a pattern that could be characterized as ‘amyotrophic lateral sclerosis’ (ALS) while a second sub-group had a pattern that could be characterized as ‘multiple sclerosis’ (MS). We now know that ALS and MS are different diseases with distinct mechanisms, clinical features, and treatments. Likewise, in the mid-twentieth century, advances in neuropathology and careful clinical-pathological studies re-defined the concept of parkinsonism as a family of disease entities rather than a single disease. Idiopathic Parkinson’s disease (PD) could be distinguished from other entities such as progressive supranuclear palsy and corticobasal ganglionic degeneration. It is conceivable that we are now arriving at such a primary juncture in our understanding of ET. Evidence of clinical heterogeneity and pathological heterogeneity is emerging, raising the question as to whether it is possible to re-formulate ET as a cluster of separable clinical-pathological entities, that is, a family of diseases – “the essential tremors”.
Heterogeneity (Clinical, Therapeutic Response, Pathological, and Etiological)
Although clinical phenotypic variability by itself is not an argument for separate diseases, its presence further opens that possibility. Clinically, the view of ET as a single neurological sign no longer seems tenable. First, the tremor phenomenology itself is multifaceted. Although kinetic and postural tremors are the core features upon which diagnostic criteria are generally based (Table 1), intention tremor,11 and tremor at rest12 may also occur in subsets of patients. The relative severity of different tremor types (kinetic > postural 13), the favored sites of anatomical involvement (arm more often than head, and head more often than jaw 14), and the typical direction of spread over time (from arms to head rather than the converse)15 are distinctive, adding a degree of subtlety and complexity to the recognition and diagnosis of a disorder that is often viewed as relatively ordinary and featureless. Indeed, perhaps due to a lack of familiarity with these features, mis-diagnosis is exceedingly common; 30–50% of patients diagnosed with ET do not have ET,16 which may make this one of the most commonly mis-diagnosed neurological disorders. Moreover, kinetic tremor of the arms, though not the head, may be a side-effect of many medications, which further contributes to this diagnostic difficulty (Table 2). There are motor features aside from tremor. In a large number of studies,11, 17 postural instability and mild to moderate gait ataxia, beyond that observed in normal aging, have been demonstrated in subgroups of ET patients. In addition to these gait problems, eye motion abnormalities (impaired smooth pursuit initiation and pathological suppression of the vestibulo-ocular reflex time constant) were recently described in a study of 17 ET cases.18 Third, there is a growing appreciation of the existence of a variety of non-motor features, including cognitive, psychiatric, and sensory. Cognitive features, especially mild problems with executive function and memory, were first reported in 200119 and soon after by numerous other investigators.20 Furthermore, a population-based study in Madrid21 first demonstrated in 2006 that the odds of prevalent dementia were nearly twice as high among the subset of ET cases with older onset disease when compared with age-matched controls. The same group later demonstrated 5 that the risk of developing incident dementia was nearly twice as high in the ET cases than controls. Also, a number of psychiatric correlates have been observed. The presence of specific personality traits22, 23 has been demonstrated. In one such study, 22 the observed personality profile was not related to functional disability or tremor severity, suggesting that it could be a primary disease feature rather than a response to disabling tremor.22 Anxiety,24 depressive symptoms,25 and social phobia 26 have been shown to occur in ET patients more often than in controls. Traditionally, these have been viewed as psychiatric responses to disabling tremor. Yet in one recent prospective study,4 depressive symptoms preceded the onset of the tremor by several years (i.e., the presence of baseline self-reported depression was associated with an increased risk of developing incident ET during follow-up). Sensory abnormalities, including olfactory deficits in some studies27 and hearing loss in others,28 have been reported in ET cases compared with age-matched controls, further drawing attention to the domain of non-motor manifestations. Although a single disease may be have a broad array of clinical manifestations (e.g., Huntington’s disease), such clinical variety can also be an indication that one is dealing with a group of diseases (e.g., parkinsonisms such as PD, progressive supranuclear palsy, and corticobasal ganglionic degeneration).
Table 1.
Two sets of clinical diagnostic criteria for definite essential tremor. The Movement Disorder Society has proposed consensus criteria for ET.52, 53 The Washington Heights–Inwood Genetic Study of Essential Tremor criteria54 specify the minimal severity of tremor that is required, and are also widely used in genetic and epidemiological studies.1, 55–58
| Consensus statement of the Movement Disorder Society on tremor52, 53 |
| Inclusion criteria |
|
| Exclusion criteria |
|
| Washington Heights Inwood Genetic Study of Essential Tremor criteria54 |
|
0 to +3 tremor ratings: 0, no visible tremor; +1, tremor is of low amplitude, barely perceivable, or intermittent; +2, tremor is of moderate amplitude (1–2 cm) and usually present, and is clearly oscillatory; +3, tremor is of large amplitude (>2 cm), violent, and jerky, resulting in difficulty completing the task because of spilling or inability to hold a pen to paper
Table 2.
Medications that may produce kinetic tremor
| Amiodarone |
| Bronchodilators |
| Cyclosporin |
| Lithium |
| Methylphenidate |
| Phenylpropanolamine |
| Procainamide |
| Pseudoephedrine |
| Selective serotonin reuptake inhibitors |
| Steroids |
| Theophylline |
| Thyroxine |
| Tricyclic antidepressants |
| Valproic acid |
It is important to emphasize that these clinical features are heterogeneously distributed across ET patients. Although some patterns are now becoming apparent (e.g., patients with intention tremor have more gait difficulties and eye motion abnormalities,18 patients with rest tremor generally have longstanding disease with severe kinetic tremor,12 dementia is associated with older age of onset of ET,5 older age of onset and unilateral onset are associated with more rapid progression29, 30), a clear separation of distinct clinical subtypes has yet to emerge.
The mainstays of therapy for ET are propranolol and primidone, although several promising new agents have been introduced in recent years (Table 3); surgical treatment (deep brain stimulation) is also highly effective.3 One re-occurring feature of pharmacotherapeutic trails in ET is that the response to a particular medication is usually patchy, with approximately one-half of the patients evidencing some degree of tremor reduction and others none at all;31 this phenomenon is frequently observed by practitioners as well. Among other possibilities (e.g., differences in disease duration across treated patients), this heterogeneity of response could be a marker of different underlying disease mechanisms in subsets of patients, although this remains to be demonstrated.
Table 3.
Medications used to treat ET
| Medication | Dosage | Potential side effects |
|---|---|---|
| Acetazolamide | Up to 500 mg/d | Weakness, fatigue |
| Alprazolam | 0.125–3 mg/d | Sedation, fatigue, potential for abuse |
| Atenolol | 50–150 mg/d | Lightheadedness, nausea, cough, dry mouth, sleepiness |
| Botulinum toxin A (hand tremor) | 50–100 U | Hand/finger weakness, reduced grip strength, pain at injection site, stiffness, cramping, haematoma, paraesthesias |
| Botulinum toxin A (head tremor) | 40–400 U | Neck weakness, post-injection pain |
| Botulinum toxin A (voice tremor) | 0.6–15 U | Breathiness, weak voice, swallowing difficulty |
| Clonazepam | 0.5–6 mg/d | Drowsiness |
| Clozapine | Up to 200 mg/d | Sedation, potential agranulocytosis (0.8% at one year) |
| Flunarizine | 10 mg/d | Sedation, weight gain, depression, dystonia, parkinsonism |
| Gabapentin | 1,200–1,800 mg/d | Lethargy, fatigue, decreased libido, dizziness, nervousness, shortness of breath |
| Levetiracetam | Up to 3,000 mg/d | Depressed mood, drowsiness, fatigue |
| Methazolamide | Up to 300 mg/d | Somnolence, nausea, paresthesias, headache |
| Nimodipine | 120 mg/d | Headache, heartburn |
| Nadolol | 120–240 mg/d | None |
| Olanzapine | 20 mg/d | Drowsiness, sedation, weight gain, diabetes |
| 1-octanol | 1 mg/kg/d | Lethargy, asthenia, headache |
| Phenobarbital | Up to 150 mg/d | Drowsiness, fatigue ataxia |
| Pregabalin | Up to 600 mg/d | Dizziness, somnolence |
| Primidone | Up to 750 mg/d | Sedation, drowsiness, fatigue, nausea, giddiness, vomiting, ataxia, malaise, dizziness, unsteadiness, confusion, vertigo, acute toxic reaction |
| Propranolol | 60–800 mg/d | Reduced arterial pressure, reduced pulse rate, tachycardia, bradycardia, impotency, drowsiness, exertional dyspnoea, confusion, headache, dizziness |
| Long-acting propranolol | 80–320 mg/d | Skin eruption, transient dizziness |
| Sodium oxybate | Up to 7.5 g/d | Dizziness, headache, sedation, ataxia |
| Sotalol | 75–200 mg/d | Decreased alertness |
| Topiramate | Up to 400 mg/d | Appetite suppression, weight loss, paraesthesias, anorexia, concentration difficulties |
| Zonisamide | Up to 200 mg/d | Ataxia, dizziness, somnolence, agitation, anorexia |
Disease mechanisms in ET have been elusive. Despite its high prevalence, until recently, few ET brains were examined and little information was available about the pathology. An intensive effort was launched in 2003 to bank ET brains.6 In contrast with previous studies, these brains were systematically examined to quantify cerebellar and other brainstem pathologies and were compared to control brains. These analyses, based on 33 ET brains, indicated that the structural pathological changes appeared to be of two types.6, 7, 9, 32 Most commonly (75%), brains were characterized by clear cerebellar degenerative changes, including a 40% reduction in number of Purkinje cells, a six-fold increase numbers of torpedoes (i.e., swellings of the Purkinje cell axon that likely represent a cellular response to injury), and Purkinje cell heterotopia and dendrite swellings. These brains did not have Lewy bodies. The remaining brains were characterized by Lewy bodies confined mainly to the locus ceruleus with relative to total sparing of other brainstem structures (see Table 2 in6). The prevalence of Lewy bodies was significantly greater than that observed in similarly-aged controls, indicating that they were not likely to be incidental.6 Furthermore, this particular pattern of Lewy body involvement in ET has not been described in series of “atypical” Lewy body brains (i.e., brains that do not follow the Braak staging scheme).33 The ET brains with Lewy bodies did not have excessive torpedoes or Purkinje cell loss.6 These two pathological patterns were labeled “cerebellar ET” and “Lewy body variant of ET” [LBVET]).6, 34 Other recent series of ET brains have also confirmed a heterogeneous pathology involving Purkinje cell loss in the cerebellum in some cases and changes, including cell loss, in the locus ceruleus in others.8 These recent postmortem studies have helped identify degenerative structural alterations in the cerebellum and its connecting pathways in ET. How changes in the locus ceruleus could produce ET is less clear, although neurons of the locus ceruleus synapse with cerebellar Purkinje cell dendrites.35 These projections are important for the normal development and maintenance of Purkinje cells.36 Impaired activity in the locus ceruleus could result in a diminution of stimulatory output from that locus to the Purkinje cells.
On an etiological level, ET is often considered to be genetic.37 There are many examples of families in which the proband and multiple relatives have ET and in which the pattern of inheritance is consistent with an autosomal dominant model. In 1997, linkage was demonstrated to a region on chromosome 2p 38 and, in that same year, to chromosome 3q in other families.39 A third study demonstrated linkage to a region on chromosome 6p in several families.40 Aside from these three studies, other studies that have failed to demonstrate linkage to these three regions, indicating that there will likely be more than three genes responsible for this disease.37, 41 It is important to note, however, that the genetic studies have not progressed further and the specific ET genes have been not yet been identified.37–40 In the absence of a specific genotype for ET, there are as yet no specific genotype-phenotype correlations, although is appears that young onset cases are generally familial (i.e., likely to have a genetic susceptibility).42 Aside from genetic factors, there is also a growing understanding that environmental factors are likely to contribute to the etiology of ET as well, indicating that there is further heterogeneity on an etiological level. Several lines of evidence support the role of these factors. First, while commonly stated that 50% or more of ET cases have a genetic basis, the precise derivation of this estimate is unclear and its validity is also doubtful.43 Indeed, some estimates are as low as 17%.43 In twin studies 44, 45 concordance in monozygotic twins was only 60% in one study and 63% in another. Second, the well-known existence of intra-familial differences in age of onset, location of tremor, and severity of tremor 46 also suggests that environmental (or perhaps other genetic) factors may be serving as modifiers of underlying susceptibility genotypes. In terms of environmental factors, a growing number of case-control studies 47 has implicated several specific toxins, namely β-carboline alkaloids (e.g., harmine and harmane, a group of highly tremorogenic dietary chemicals) and lead; and further studies of these putative environmental toxins are needed.
Organizing the Heterogeneity
Information presented in the previous section may be organized by etiologies, pathologies and the clinical features (Figure 2). Given the sheer prevalence and ubiquity of this condition, the historical tendency to split disease entities as new knowledge arises, the appreciation of a broader variety of clinical features in several separate domains (e.g., tremor, other motor, cognitive, psychiatric), the observation that these are not uniformly present in ET patients, the evidence that multiple genes will likely be responsible for this disease, and preliminary evidence of distinct pathological patterns, it is likely that ET will turn out to be a family of diseases rather than a single disease entity. It is likely that this family of diseases, united by the presence of kinetic tremor, would be separable on the basis of etiological, clinical, therapeutic response and pathological features, although further work is needed to explore these relationships.
Figure 2.
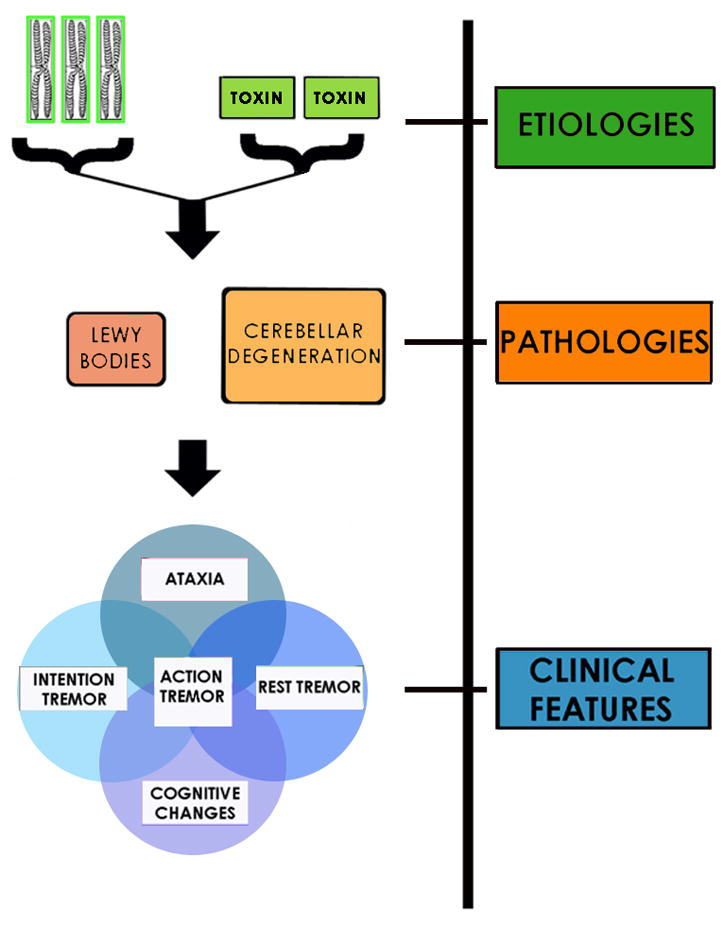
The heterogeneity in ET may be organized by (1) disease etiology, (2) tissue-level changes that occur after the disease process is initiated and as it develops, and (3) clinical features that are the end product of these underlying pathological processes. Here is one such possible schema.
Is ET Neurodegenerative?
A question implicitly raised in the above discussion is whether this disease (or these diseases) are neurodegenerative. The idea that ET could be neurodegenerative is not new. In 1948, Critchley and Greenfield wrote: “Although anatomical proof is as yet lacking, there are at least a number of clinical points to make question whether “essential tremor” may not, at times any rate, represent an incomplete or a premature variant of one of the cerebellar atrophies.”48 Although not further elaborated by those authors, these clinical points include its insidious onset, association with advanced aging (i.e., both incidence and prevalence increase with aging), gradual yet progressive nature, and the presence of “cerebellar” features (e.g., intention tremor and ataxia) on examination.
Neurodegenerative diseases traditionally have been defined as diseases that begin insidiously, pursue a gradually progressive course that may continue for many years, and are characterized by the selective involvement of anatomically and physiologically related systems of neurons due to intrinsic processes rather than an identifiable outside influence (e.g., vascular, auto-immune). Cell loss is also considered by many to be a prominent feature of these diseases.49 Furthermore, their occurrence often increases markedly with advancing age. What is the evidence that ET is neurodegenerative? The clinical points noted above are important. ET has an insidious onset46, 50 and then follows a gradual yet progressive clinical course.29, 30 There is a marked and continued rise in disease occurrence in advanced ages.1, 2 This clinical constellation is somewhat compelling; however, none of these features in isolation is specific to neurodegenerative diseases. On a tissue-based level, the evidence is more compelling. Selective involvement of an anatomically and physiologically related system of neurons, Purkinje cells, has been reported in ET cases both in our recent series (Figure 3)6, 7 and in the other large recent series.8 When quantified using different methods, Purkinje cell loss is significant. There is an approximate 40% loss of these cells compared to age-matched control brains,6, 7 which persists even when one adjusts for age and other confounding pathologies (e.g., mild Alzheimer’s type changes). Additional evidence that the Purkinje cells are diseased is that there are significantly more torpedoes in the ET brain, where their numbers are six-times higher than expected for age, and a preponderance of displaced (i.e., heterotopic) Purkinje cells as well as Purkinje cells with dendritic swellings.6 In contrast to these cases, the ET cases with normal cerebella have Lewy bodies,6, 9 which are lesions that have long been considered important in the pathogenesis of another neurodegenerative tremor disorder, namely, PD. While none of these pathological changes in the cerebellum are disease-specific (e.g., other forms of cerebellar degeneration may be characterized by Purkinje cell loss and torpedo formation), this just indicates that the changes seen in the cerebellum in ET occur more broadly in the cerebellar degenerations. Aside from this structural-pathological evidence suggesting a neurodegenerative process, other evidence suggests that ET is neurodegenerative. While many of these features in isolation are not specific to neurodegenerative diseases, the constellation of findings, all present in the same disease, is more compelling (Table 4). For example, there is a longstanding clinical association between ET and PD; indeed, having ET increases the risk of developing incident PD four to five-fold.51 Furthermore, having older onset ET increases the risk of developing Alzheimer’s disease nearly two-fold.5 This association between ET and subsequent development of these neurodegenerative diseases suggests that ET may share pathogenic mechanisms with these disorders.
Figure 3.
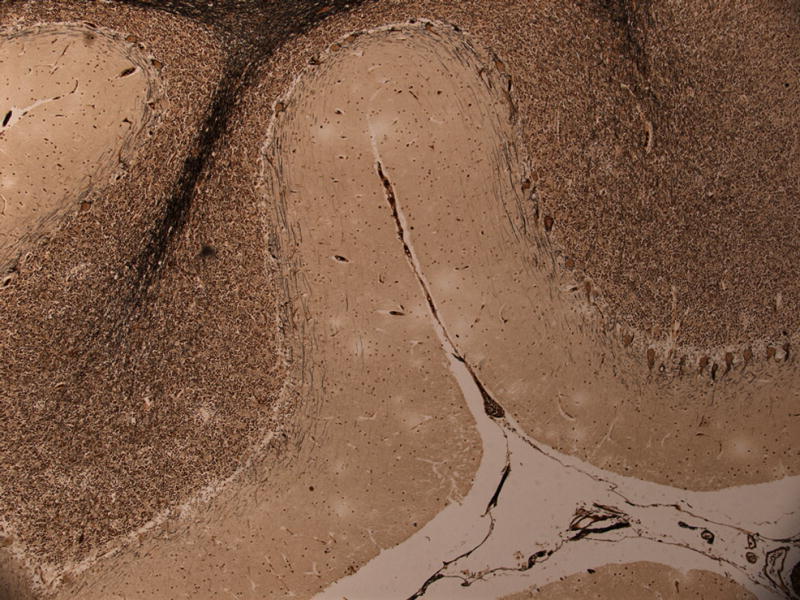
When quantified using different methods, Purkinje cell loss in ET is significant compared to age-matched controls. Bielschowsky-stained section (50× magnification) of the cerebellar cortex in a 79 year old ET case showing normal Purkinje cellularity (right) vs. a region with segmental loss of Purkinje cells (left).
Table 4.
Evidence suggesting that ET is neurodegenerative
| Evidence | Comment/Rationale | PD* | AD* | Cautionary Caveat |
|---|---|---|---|---|
| Purkinje cell loss or Lewy bodies on postmortem studies | Selective involvement of anatomically and physiologically related systems of neurons is considered an important feature of neurodegenerative disorders. Lewy bodies are a feature of PD, a neurodegenerative disease. | Yes | Yes1 | Additional postmortem studies are needed to confirm the results of currently-emerging large postmortem series.6, 8 |
| Clinical resemblance to other cerebellar atrophies and Purkinje cell loss | This suggests shared disease mechanisms. | No | No | |
| Marked rise in occurrence with advanced age | PD and AD are associated with advanced age | Yes | Yes | |
| Clinically progressive in most patients | PD and AD are relentlessly clinically-progressive | Yes | Yes | Diseases that are not neurodegenerative may sometimes be clinically-progressive. |
| Somatotopic spread of tremor over time | Tremor often begins in one body region, and as the disease worsens, spreads to others. | Yes | Yes2 | |
| Olfactory Deficit | Olfactory deficits are reported in PD and AD | Yes | Yes | Not specific to neurodegenerative disorders. Not a consistent finding across all studies of ET. |
| Loss of body mass index59 | Neurodegnerative diseases are often associated with loss of body mass index | Yes | Yes | Not specific to neurodegenerative disorders. Not a consistent finding across all studies of ET. |
| Increased risk of mortality60 | Neurodegenerative disorders are associated with increased risk of mortality | Yes | Yes | Not specific to neurodegenerative disorders. Not a consistent finding across all studies of ET. |
| Increased risk of AD | This association with ET suggests that ET may share pathogenic features with this neurodegenerative disease. | Yes | ||
| Increased risk of PD | This association with ET suggests that ET may share pathogenic features with this neurodegenerative disease. | Yes |
This feature or a similar feature is a characteristic of PD or AD.
AD = Alzheimer’s disease, PD = Parkinson’s disease, ET = essential tremor
Neuronal loss in the hippocampus and neocortex are features of AD.
As AD progresses, different cortical regions may become involved.
Conclusion
There is some evidence to suggest that ET is a family of diseases rather than a single entity (Table 5). These disorders, perhaps better termed “the essential tremors”, are aging-associated, progressive, and associated with cell loss and other types of changes (Lewy body formation) that traditionally occur in neurodegenerative disorders. Future study is needed to continue to shape our evolving notion of the entity that we currently refer to as “essential tremor”.
Table 5.
Key Points
|
Acknowledgments
Funding Source: R01 NS42859 from the National Institutes of Health (Bethesda, MD); the Parkinson’s Disease Foundation (New York, NY); the Arlene Bronstein Essential Tremor Research Fund (Columbia University); and the Claire O’Neil Essential Tremor Research Fund (Columbia University).
I would like to thank Blair Ford M.D., Steven Frucht M.D., and Vinita Sehgal M.D. for reading this manuscript and providing critical commentary.
Footnotes
Disclosure: The author reports no conflicts of interest.
References
- 1.Dogu O, Sevim S, Camdeviren H, et al. Prevalence of essential tremor: door-to-door neurologic exams in Mersin Province, Turkey. Neurology. 2003 Dec 23;61(12):1804–1806. doi: 10.1212/01.wnl.0000099075.19951.8c. [DOI] [PubMed] [Google Scholar]
- 2.Louis ED, Ottman R, Hauser WA. How common is the most common adult movement disorder? estimates of the prevalence of essential tremor throughout the world. Mov Disord. 1998 Jan;13(1):5–10. doi: 10.1002/mds.870130105. [DOI] [PubMed] [Google Scholar]
- 3.Benito-Leon J, Louis ED. Essential tremor: emerging views of a common disorder. Nat Clin Pract Neurol. 2006 Dec;2(12):666–678. doi: 10.1038/ncpneuro0347. quiz 662p following 691. [DOI] [PubMed] [Google Scholar]
- 4.Louis ED, Benito-Leon J, Bermejo-Pareja F. Self-reported depression and anti-depressant medication use in essential tremor: cross-sectional and prospective analyses in a population-based study. Eur J Neurol. 2007 Oct;14(10):1138–1146. doi: 10.1111/j.1468-1331.2007.01923.x. [DOI] [PubMed] [Google Scholar]
- 5.Bermejo-Pareja F, Louis ED, Benito-Leon J. Risk of incident dementia in essential tremor: A population-based study. Mov Disord. 2007 May 21; doi: 10.1002/mds.21553. [DOI] [PubMed] [Google Scholar]
- 6.Louis ED, Faust PL, Vonsattel JP, et al. Neuropathological changes in essential tremor: 33 cases compared with 21 controls. Brain. 2007 Dec;130(Pt 12):3297–3307. doi: 10.1093/brain/awm266. [DOI] [PubMed] [Google Scholar]
- 7.Axelrad JE, Louis ED, Honig LS, et al. Reduced purkinje cell number in essential tremor: a postmortem study. Arch Neurol. 2008 Jan;65(1):101–107. doi: 10.1001/archneurol.2007.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shill HA, Adler CH, Sabbagh MN, et al. Pathologic findings in prospectively ascertained essential tremor subjects. Neurology. 2008 Apr 15;70(16 Pt 2):1452–1455. doi: 10.1212/01.wnl.0000310425.76205.02. [DOI] [PubMed] [Google Scholar]
- 9.Louis ED, Vonsattel JP. The emerging neuropathology of essential tremor. Mov Disord. 2007 Nov 12;23(2):174–182. doi: 10.1002/mds.21731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ross GWDD, Cerosimo M, et al. Pathological investigation of essential tremor. Neurology. 2004;62(Suppl 5):A537–A538. [Google Scholar]
- 11.Deuschl G, Wenzelburger R, Loffler K, Raethjen J, Stolze H. Essential tremor and cerebellar dysfunction clinical and kinematic analysis of intention tremor. Brain. 2000 Aug;123 (Pt 8):1568–1580. doi: 10.1093/brain/123.8.1568. [DOI] [PubMed] [Google Scholar]
- 12.Cohen O, Pullman S, Jurewicz E, Watner D, Louis ED. Rest tremor in patients with essential tremor: prevalence, clinical correlates, and electrophysiologic characteristics. Arch Neurol. 2003 Mar;60(3):405–410. doi: 10.1001/archneur.60.3.405. [DOI] [PubMed] [Google Scholar]
- 13.Brennan KC, Jurewicz EC, Ford B, Pullman SL, Louis ED. Is essential tremor predominantly a kinetic or a postural tremor? A clinical and electrophysiological study. Mov Disord. 2002 Mar;17(2):313–316. doi: 10.1002/mds.10003. [DOI] [PubMed] [Google Scholar]
- 14.Louis ED, Ford B, Frucht S. Factors associated with increased risk of head tremor in essential tremor: a community-based study in northern Manhattan. Mov Disord. 2003 Apr;18(4):432–436. doi: 10.1002/mds.10395. [DOI] [PubMed] [Google Scholar]
- 15.Louis ED. Essential tremor. Lancet Neurol. 2005 Feb;4(2):100–110. doi: 10.1016/S1474-4422(05)00991-9. [DOI] [PubMed] [Google Scholar]
- 16.Jain S, Lo SE, Louis ED. Common misdiagnosis of a common neurological disorder: how are we misdiagnosing essential tremor? Arch Neurol. 2006 Aug;63(8):1100–1104. doi: 10.1001/archneur.63.8.1100. [DOI] [PubMed] [Google Scholar]
- 17.Stolze H, Petersen G, Raethjen J, Wenzelburger R, Deuschl G. The gait disorder of advanced essential tremor. Brain. 2001 Nov;124(Pt 11):2278–2286. doi: 10.1093/brain/124.11.2278. [DOI] [PubMed] [Google Scholar]
- 18.Helmchen C, Hagenow A, Miesner J, et al. Eye movement abnormalities in essential tremor may indicate cerebellar dysfunction. Brain. 2003 Jun;126(Pt 6):1319–1332. doi: 10.1093/brain/awg132. [DOI] [PubMed] [Google Scholar]
- 19.Gasparini M, Bonifati V, Fabrizio E, et al. Frontal lobe dysfunction in essential tremor: a preliminary study. J Neurol. 2001 May;248(5):399–402. doi: 10.1007/s004150170181. [DOI] [PubMed] [Google Scholar]
- 20.Troster AI, Woods SP, Fields JA, et al. Neuropsychological deficits in essential tremor: an expression of cerebello-thalamo-cortical pathophysiology? Eur J Neurol. 2002 Mar;9(2):143–151. doi: 10.1046/j.1468-1331.2002.00341.x. [DOI] [PubMed] [Google Scholar]
- 21.Benito-Leon J, Louis ED, Bermejo-Pareja F. Elderly-onset essential tremor is associated with dementia. Neurology. 2006 May 23;66(10):1500–1505. doi: 10.1212/01.wnl.0000216134.88617.de. [DOI] [PubMed] [Google Scholar]
- 22.Chatterjee A, Jurewicz EC, Applegate LM, Louis ED. Personality in essential tremor: further evidence of non-motor manifestations of the disease. J Neurol Neurosurg Psychiatry. 2004 Jul;75(7):958–961. doi: 10.1136/jnnp.2004.037176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lorenz D, Schwieger D, Moises H, Deuschl G. Quality of life and personality in essential tremor patients. Mov Disord. 2006 Aug;21(8):1114–1118. doi: 10.1002/mds.20884. [DOI] [PubMed] [Google Scholar]
- 24.Tan EK, Fook-Chong S, Lum SY, et al. Non-motor manifestations in essential tremor: use of a validated instrument to evaluate a wide spectrum of symptoms. Parkinsonism Relat Disord. 2005 Sep;11(6):375–380. doi: 10.1016/j.parkreldis.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 25.Miller KM, Okun MS, Fernandez HF, Jacobson CEt, Rodriguez RL, Bowers D. Depression symptoms in movement disorders: comparing Parkinson’s disease, dystonia, and essential tremor. Mov Disord. 2007 Apr 15;22(5):666–672. doi: 10.1002/mds.21376. [DOI] [PubMed] [Google Scholar]
- 26.Schneier FR, Barnes LF, Albert SM, Louis ED. Characteristics of social phobia among persons with essential tremor. J Clin Psychiatry. 2001 May;62(5):367–372. doi: 10.4088/jcp.v62n0511. [DOI] [PubMed] [Google Scholar]
- 27.Louis ED, Bromley SM, Jurewicz EC, Watner D. Olfactory dysfunction in essential tremor: a deficit unrelated to disease duration or severity. Neurology. 2002 Nov 26;59(10):1631–1633. doi: 10.1212/01.wnl.0000033798.85208.f2. [DOI] [PubMed] [Google Scholar]
- 28.Ondo WG, Sutton L, Dat Vuong K, Lai D, Jankovic J. Hearing impairment in essential tremor. Neurology. 2003 Oct 28;61(8):1093–1097. doi: 10.1212/01.wnl.0000086376.40750.af. [DOI] [PubMed] [Google Scholar]
- 29.Louis ED, Ford B, Barnes LF. Clinical subtypes of essential tremor. Arch Neurol. 2000 Aug;57(8):1194–1198. doi: 10.1001/archneur.57.8.1194. [DOI] [PubMed] [Google Scholar]
- 30.Putzke JD, Whaley NR, Baba Y, Wszolek ZK, Uitti RJ. Essential tremor: predictors of disease progression in a clinical cohort. J Neurol Neurosurg Psychiatry. 2006 Nov;77(11):1235–1237. doi: 10.1136/jnnp.2006.086579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gironell A, Kulisevsky J, Barbanoj M, Lopez-Villegas D, Hernandez G, Pascual-Sedano B. A randomized placebo-controlled comparative trial of gabapentin and propranolol in essential tremor. Arch Neurol. 1999 Apr;56(4):475–480. doi: 10.1001/archneur.56.4.475. [DOI] [PubMed] [Google Scholar]
- 32.Louis ED, Vonsattel JP, Honig LS, et al. Essential tremor associated with pathologic changes in the cerebellum. Arch Neurol. 2006 Aug;63(8):1189–1193. doi: 10.1001/archneur.63.8.1189. [DOI] [PubMed] [Google Scholar]
- 33.Kalaitzakis ME, Graeber MB, Gentleman SM, Pearce RK. The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson’s disease: a critical analysis of alpha-synuclein staging. Neuropathol Appl Neurobiol. 2008 Jun;34(3):284–295. doi: 10.1111/j.1365-2990.2007.00923.x. [DOI] [PubMed] [Google Scholar]
- 34.Louis ED, Honig LS, Vonsattel JP, Maraganore DM, Borden S, Moskowitz CB. Essential tremor associated with focal nonnigral Lewy bodies: a clinicopathologic study. Arch Neurol. 2005 Jun;62(6):1004–1007. doi: 10.1001/archneur.62.6.1004. [DOI] [PubMed] [Google Scholar]
- 35.Olson L, Fuxe K. On the projections from the locus coeruleus noradrealine neurons: the cerebellar innervation. Brain Res. 1971 Apr 16;28(1):165–171. doi: 10.1016/0006-8993(71)90533-6. [DOI] [PubMed] [Google Scholar]
- 36.Sievers J, Berry M, Baumgarten H. The role of noradrenergic fibres in the control of post-natal cerebellar development. Brain Res. 1981 Feb 23;207(1):200–208. doi: 10.1016/0006-8993(81)90694-6. [DOI] [PubMed] [Google Scholar]
- 37.Deng H, Le W, Jankovic J. Genetics of essential tremor. Brain. 2007 Jun;130(Pt 6):1456–1464. doi: 10.1093/brain/awm018. [DOI] [PubMed] [Google Scholar]
- 38.Higgins JJ, Pho LT, Nee LE. A gene (ETM) for essential tremor maps to chromosome 2p22-p25. Mov Disord. 1997 Nov;12(6):859–864. doi: 10.1002/mds.870120605. [DOI] [PubMed] [Google Scholar]
- 39.Gulcher JR, Jonsson P, Kong A, et al. Mapping of a familial essential tremor gene, FET1, to chromosome 3q13. Nat Genet. 1997 Sep;17(1):84–87. doi: 10.1038/ng0997-84. [DOI] [PubMed] [Google Scholar]
- 40.Shatunov A, Sambuughin N, Jankovic J, et al. Genomewide scans in North American families reveal genetic linkage of essential tremor to a region on chromosome 6p23. Brain. 2006 Sep;129(Pt 9):2318–2331. doi: 10.1093/brain/awl120. [DOI] [PubMed] [Google Scholar]
- 41.Kovach MJ, Ruiz J, Kimonis K, et al. Genetic heterogeneity in autosomal dominant essential tremor. Genet Med. 2001 May–Jun;3(3):197–199. doi: 10.1097/00125817-200105000-00009. [DOI] [PubMed] [Google Scholar]
- 42.Louis ED, Ottman R. Study of possible factors associated with age of onset in essential tremor. Mov Disord. 2006 Nov;21(11):1980–1986. doi: 10.1002/mds.21102. [DOI] [PubMed] [Google Scholar]
- 43.Louis ED, Ottman R. How familial is familial tremor? The genetic epidemiology of essential tremor. Neurology. 1996 May;46(5):1200–1205. doi: 10.1212/wnl.46.5.1200. [DOI] [PubMed] [Google Scholar]
- 44.Tanner CM, Goldman SM, Lyons KE, et al. Essential tremor in twins: an assessment of genetic vs environmental determinants of etiology. Neurology. 2001 Oct 23;57(8):1389–1391. doi: 10.1212/wnl.57.8.1389. [DOI] [PubMed] [Google Scholar]
- 45.Lorenz D, Frederiksen H, Moises H, Kopper F, Deuschl G, Christensen K. High concordance for essential tremor in monozygotic twins of old age. Neurology. 2004 Jan 27;62(2):208–211. doi: 10.1212/01.wnl.0000103236.26934.41. [DOI] [PubMed] [Google Scholar]
- 46.Larsson T, Sjogren T. Essential tremor: a clinical and genetic population study. Acta Psychiatr Scand Suppl. 1960;36(144):1–176. [PubMed] [Google Scholar]
- 47.Louis ED. Environmental Epidemiology of Essential Tremor. Neuroepidemiology. 2008 Aug 21;31(3):139–149. doi: 10.1159/000151523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Critchley M, Greenfield JG. Olivo-pontocerebellar atrophy. Brain. 1948;71:343–364. [PubMed] [Google Scholar]
- 49.Adams RDVM. Princples of Neurology. 4. New York: McGraw-Hill; 1989. [Google Scholar]
- 50.Critchley M. Observations of essential (heredofamilial) tremor. Brain. 1949;72:113–139. doi: 10.1093/brain/72.2.113. [DOI] [PubMed] [Google Scholar]
- 51.Benito-Leon JLE, Bermejo-Pareja F. Risk of incident Parkinson’s disease and Parkinsonism in essential tremor: A population-based study. Neurology. 2008;70(Suppl 1):A191. doi: 10.1136/jnnp.2008.147223. [DOI] [PubMed] [Google Scholar]
- 52.Deuschl G, Bain P, Brin M. Consensus statement of the Movement Disorder Society on Tremor. Ad Hoc Scientific Committee. Mov Disord. 1998;13 (Suppl 3):2–23. doi: 10.1002/mds.870131303. [DOI] [PubMed] [Google Scholar]
- 53.Bain P, Brin M, Deuschl G, et al. Criteria for the diagnosis of essential tremor. Neurology. 2000;54(11 Suppl 4):S7. [PubMed] [Google Scholar]
- 54.Louis ED, Ford B, Lee H, Andrews H. Does a screening questionnaire for essential tremor agree with the physician’s examination? Neurology. 1998 May;50(5):1351–1357. doi: 10.1212/wnl.50.5.1351. [DOI] [PubMed] [Google Scholar]
- 55.Inzelberg R, Mazarib A, Masarwa M, Abuful A, Strugatsky R, Friedland RF. Essential tremor prevalence is low in Arabic villages in Israel: door-to-door neurological examinations. J Neurol. 2006 Dec;253(12):1557–1560. doi: 10.1007/s00415-006-0253-5. [DOI] [PubMed] [Google Scholar]
- 56.Sur H, Ilhan S, Erdogan H, Ozturk E, Tasdemir M, Boru UT. Prevalence of essential tremor: A door-to-door survey in Sile, Istanbul, Turkey. Parkinsonism Relat Disord. 2008 May 11; doi: 10.1016/j.parkreldis.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 57.Farrer M, Gwinn-Hardy K, Muenter M, et al. A chromosome 4p haplotype segregating with Parkinson’s disease and postural tremor. Hum Mol Genet. 1999 Jan;8(1):81–85. doi: 10.1093/hmg/8.1.81. [DOI] [PubMed] [Google Scholar]
- 58.Bergareche A, De La Puente E, Lopez De Munain A, et al. Prevalence of essential tremor: a door-to-door survey in bidasoa, spain. Neuroepidemiology. 2001 May;20(2):125–128. doi: 10.1159/000054771. [DOI] [PubMed] [Google Scholar]
- 59.Dogu O, Sevim S, Louis ED, Kaleagasi H, Aral M. Reduced body mass index in patients with essential tremor: a population-based study in the province of Mersin, Turkey. Arch Neurol. 2004 Mar;61(3):386–389. doi: 10.1001/archneur.61.3.386. [DOI] [PubMed] [Google Scholar]
- 60.Louis ED, Benito-Leon J, Ottman R, Bermejo-Pareja F. A population-based study of mortality in essential tremor. Neurology. 2007 Nov 20;69(21):1982–1989. doi: 10.1212/01.wnl.0000279339.87987.d7. [DOI] [PubMed] [Google Scholar]