

Abstract
Arylamine N-acetyltransferase 1 (NAT1) and 2 (NAT2) exhibit single nucleotide polymorphisms (SNPs) in human populations that modify drug and carcinogen metabolism. This paper updates the identity, location, and functional effects of these SNPs and then follows with emerging concepts for understanding why pharmacogenetic findings may not be replicated consistently. Using this paradigm as an example, laboratory-based mechanistic analyses can reveal complexities such that genetic polymorphisms become biologically and medically relevant when confounding factors are more fully understood and considered. As medical care moves to a more personalized approach, the implications of these confounding factors will be important in understanding the complexities of personalized medicine.
Keywords: N-acetyltransferase 1, N-acetyltransferase 2, single nucleotide polymorphisms, acetylator polymorphism, personalized medicine
1. Introduction
The N-acetylation polymorphism was first identified in patients administered isoniazid for the treatment of tuberculosis [1]. Subsequently, it was discovered that N-acetyltransferase (E.C. 2.3.1.5) also catalyzes the N-acetylation of a diverse array of aromatic amines drugs and carcinogens [2]. Two arylamine N-acetyltransferase isozymes, NAT1 and NAT2, have been identified in humans and comprehensive reviews have been reported recently [3–10]. Single nucleotide polymorphisms (SNPs) in the NAT1 and NAT2 genes have been identified and tested for their functional effects. This paper updates the identity, location, and functional effects of these SNPs and then follows with emerging concepts which can serve as a paradigm for understanding why pharmacogenetic findings may not be replicated consistently.
NAT1 and NAT2 are 290 amino acid products of intronless 870 base pair open reading frames located within 170 kb on chromosome 8p22 [3–10]. A pseudogene (NATP) resides between the NAT1 and NAT2 coding exons. NAT1 and NAT2 open reading frames are 87% identical and the NAT1 and NAT2 proteins differ only in 55 amino acids [8]. In the pharmacogenetic literature, combinations of SNPs are identified as alleles or haplotypes. A consensus N-acetyltransferase gene nomenclature has been established [11] with NAT1*4 and NAT2*4 designated as the reference alleles for NAT1 and NAT2, respectively. SNPs are identified by designating “A” of the ATG translation initiation codon as number 1. SNPs upstream of the site are designated by negative numbers and SNPs downstream of this site are designated by positive numbers. NAT1*4 and NAT2*4 are the most common NAT1 and NAT2 alleles reported in some but not all ethnic groups and thus it is not appropriate to designate them as “wild-type” [12]. An international nomenclature committee publishes an internet website for updates in NAT1 and NAT2 alleles at http://N-acetyltransferasenomenclature.louisville.edu [12].
As reviewed recently [10,13], NAT1 and NAT2 differ in transcriptional regulation. For example, NAT1*10 which possesses SNPs solely in the 3-UTR was associated with slightly elevated NAT1 activity levels in human bladder and colon tissues [14,15] and increased N-acetylation capacity in vivo [16]. One of the SNPs present in the 3′-UTR of NAT1*10 (1088T>A) alters a consensus polyadenylation signal (identified as polyA-1) consistent with the suggestion that increased activity may relate to a change in the polyadenylation and subsequent enhanced mRNA stability. However, several studies have failed to replicate the increased catalytic activity associated with NAT1*10 [17–19] and most published NAT1 expressed sequence tags possessing poly A tails reflect a polyadenylation site (polyA-2) located downstream of 1088T>A [10]. Further research on SNPs in the 3′UTR is needed to better understand their functional effects, which may be tissue-specific. Additional non-genetic factors such as substrate-dependent inhibition, drug interactions and cellular redox conditions also can modify N-acetyltransferase activities [20]. These factors may modify SNP effects and the relationships between inherited genotype and phenotype, particularly for NAT1 where functional effects for SNPs in the 3′UTR are poorly understood.
Knowledge of the functional effects of coding region SNPs has been enhanced substantially by molecular modeling and crystallization of N-acetyltransferase enzymes, initially a prokaryotic N-acetyltransferase [21] followed more recently by human NAT1 and NAT2 proteins [22]. Both NAT1 and NAT2 possess a functional Cys-His-Asp catalytic triad, which resembles that of cysteine proteases [21]. In a ping-pong bi-bi reaction mechanism, the catalytic Cys68 is acetylated by acetyl-coenzyme A (AcCoA), followed by acetyl group transfer to the substrate [23]. Docking experiments have revealed important insights into isozyme-specific substrate specificity [22]. The NAT1 catalytic pocket is about 40% smaller than that of NAT2 [22] and substrate selectivity is strongly influenced by the three key active site loop residues F125, Y127, and R129 [22–25]. The functional effects of SNPs in the NAT1 and NAT2 coding regions have been determined primarily by characterization of recombinant NAT1 and NAT2 allozymes. Our understanding of these experimental observations has been enriched by studies of molecular structure and modeling. They provide explanations for the relative capacity of NAT1 and NAT2 to catalyze not only N-acetylation, but also AcCoA-dependent O-acetylation of N-hydroxyarylamines and AcCoA-independent N,O-acetylation of N-hydroxy-N-acetylarylamines; reactions also catalyzed by NAT1 and NAT2 [10,26]. The effects of selected SNPs on structure and function are outlined below.
2. SNPs in NAT1 coding region
At least 15 SNPs in the NAT1 coding region have been identified in human populations (Table 1). The sites for many of these on the NAT1 protein are illustrated in Figure 1. Analysis of the structural features and functional effects of many of these NAT1 SNPs was reported recently [27] and is summarized below. Data in support of their effects on NAT1 protein expression Vmax and substrate Km following recombinant expression of NAT1 variants in mammalian cells are shown in Figure 2.
Table 1.
SNPs in human NAT1 coding region
| SNPa | rs identifierb | SNP500 Cancer IDc | Amino acid change | NAT1 allele/haplotype | Functional effectd |
|---|---|---|---|---|---|
| 21T>G | 4986992 | NAT1-18 | L7L | NAT1*27 | Synonymous |
| 97C>T | 56318881 | NAT1-11 | R33X | NAT1*19 | Decrease |
| 190C>T | 56379106 | NAT1-19 | R64W | NAT1*17 | Decrease |
| 363C>T | 8190858 | NAT1-26 | V121V | Synonymous | |
| 402T>C | P134P | NAT1*20 | Synonymous | ||
| 445G>A | 4987076 | NAT1-05 | V149I | NAT1*11 | |
| 459G>A | 4986990 | NAT1-06 | T153T | NAT1*11 | Synonymous |
| 559C>T | 5030839 | NAT1-10 | R187X | NAT1*15 | Decrease |
| 560G>A | 4986782 | NAT1-07 | R187Q | NAT1*14 | Decrease |
| 613A>G | M205V | NAT1*21 | None | ||
| 640T>G | 4986783 | S214A | NAT1*11 | ||
| 752A>T | D251V | NAT1*22 | Decrease | ||
| 777T>C | 4986991 | NAT1-15 | S259S | NAT1*23 | Synonymous |
| 781G>A | E261K | NAT1*24 | None | ||
| 787A>G | I263V | NAT1*25 | None |
Reference gene sequence published in Genbank Assession Number X17059. Adapted from http://N-acetyltransferasenomenclature.louisville.edu
Not all SNPs have been designated.
Accessed from http://snp500cancer.nci.nih.gov/snplist.cfm?gene_id=NAT1&mode=valid. This SNP nomenclature is distinct from the NAT1 allele (haplotype) nomenclature.
SNPs are non-synonymous unless otherwise indicated. Only functional effects supported with experimental data are provided.
Figure 1.
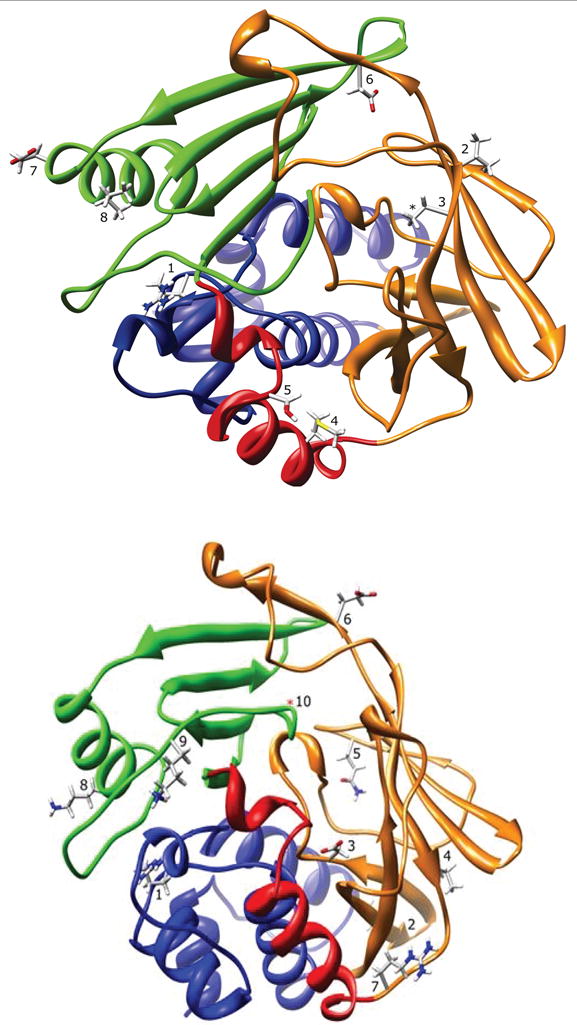
NAT1 (top) and NAT2 (bottom) ribbon diagrams. The ribbon is colored to indicate N-acetyltransferase protein domain I (blue), the interdomain region (red), domain II (orange), and domain III (green). Top: R64 (1), V149 (2), R187 (3), M205 (4), S214 (5), D251 (6), E261 (7), and I263 (8) are shown for NAT1. The 2PQT PDB file is missing coordinates for the arginine side-chain guanidine group at residue R187 (*). Bottom: R64 (1), I114 (2), D122 (3), L137 (4), Q145 (5), E167 (6), R197 (7), K268 (8), K282 (9), and G286 (10) are shown for NAT2. Adapted from [27,28].
Figure 2.

Top: Effects of SNPs in the NAT1 coding region on recombinant NAT1 protein level. NAT1 protein and endogenous control α-tubulin levels were visualized by western blots, quantitated by densitometry, and presented in arbitrary units. REF refers to NAT1 4 (no SNPs). Each bar represents Mean ± S.E.M from three transfections in duplicate. NAT1-specific protein level for the variant allozymes was compared to NAT1 4 by one way ANOVA followed by Dunnett’s post-test. The NAT1 antibody did not detect the truncated NAT1 proteins from 97C>T(R33stop) and 559C>T(R187stop). **190C>T and 752A>T significantly (p<0.01) reduced NAT1 protein level over 40-fold and 560G>A significantly (p<0.01) reduced NAT1 protein level 4-fold. Middle: Effects of SNPs in the NAT1 coding region on p-aminobenzoic acid (PABA) N-acetyltransferase maximum velocities with PABA concentrations of 25 to 1000 μM in the presence of 1 mM AcCoA. PABA N-acetyltransferase activities were normalized to β-galactosidase activity. Each bar represents Mean ± S.E.M from three transfections in duplicate. Characters under bars indicate SNP(s) in corresponding haplotypes: REF, no SNP (reference NAT1*4) Activity for each variant allozymes was compared to reference NAT1 4 catalytic activity by one way ANOVA followed by Dunnett’s post-test. *significantly lower than NAT1 4 (p<0.01). PABA N-acetyltransferase catalytic activities for 97C>T, 190C>T, 559C>T and 752A>T were too low (<0.05 nmol/min/mg) to determine Vmax. Bottom: Effects of SNPs in the NAT1 coding region on PABA Km. The upper panel illustrates PABA Km determined with PABA concentrations of 25 to 1000 μM in the presence of 1 mM AcCoA. Each bar represents Mean ± S.E.M from three transfections in duplicate. REF, no SNP (reference NAT1*4). Km for each variant allozyme was compared to NAT1 4 by one way ANOVA followed by Dunnett’s post-test. *significantly greater than NAT1 4 (p<0.01). PABA N-acetyltransferase catalytic activities for 97C>T, 190C>T, 559C>T and 752A>T were too low (<0.05 nmol/min/mg) to determine Km. Adapted from [29].
2.1 NAT1-R64W (190C>T)
The hydrogen bonding interactions between positively charged R64 and negatively charged E38 are salt bridges which are stronger than normal hydrogen bonds. Replacing arginine with either glutamine or tryptophan at residue 64 reduces these interactions in turn altering conformation and dynamics of the first domain tertiary structure. Structural changes in this region could affect the positioning of the catalytic triad C68, and thereby alter its interactions with catalytic H107, and/or alter its acetylation status leading to increased proteasomal degradation [30]. Some hydrogen bonding interactions may be preserved with W64, but tryptophan is largely hydrophobic and does not have the hydrogen bonding capacity of arginine. The native interactions formed by R64 are likely important to the integrity of the tertiary structure. Functional studies of the R64W variant in yeast demonstrated reduced catalytic activity, reduced protein levels, and reduced thermostability [31,32]. Variant NAT1 proteins are more susceptible to proteasomal degradation due to altered acetyl-CoA binding capability, and reduced thermostability [30]. A loss of critical hydrogen bonding interactions in the R64W variant is consistent with reduced protein thermostability. A loss of structural integrity could lead to protein aggregation [33] or enhanced protein degradation.
2.2 NAT1-V149I (445G>A) and -S214A (640G>T)
V149I and S214A exist together as a result of SNPs in the NAT1*11 allele [11]. Functional effects of these two SNPs found in the NAT1*11 allele have been inconsistent. Recombinant expression of the NAT1*11 coding region in bacteria [34] and yeast [31,32] caused no change in catalytic activity, protein levels, or protein stability. Following recombinant expression in COS-1 cells, the NAT1*11 haplotype possessing both V149I and S214A resulted in elevated protein levels and catalytic activity in one study [29] but not another [17]. Red blood cells and leukocytes from individuals possessing the NAT1*11 allele resulted in equivalent and lower catalytic activities, respectively [35,36].
The side-chain of V149 is located on the surface of the second domain beta-barrel. Hydrophobic interactions are possible with P144 and I164, but because these residues are on adjacent beta strands, they should not contribute to secondary structure stability. Substitution of isoleucine for valine at 149 is conservative and thus inconsistent with structural changes. When recombinantly expressed in bacterial cells, the V149I variant resulted in acetylation rates of up to 2-fold higher without changes in protein level or thermostability [37].
The side-chain of residue S214 is located in the inter-domain region near the active site pocket and has no apparent molecular interactions with surrounding residues. Replacing serine with a smaller alanine residue should not modify active site access nor affect structure since the side-chain is near the surface and does not interact with other residues. Analysis of the NAT2 structure complexed with CoA demonstrates that Threonine 214 is involved in hydrogen bonding to CoA. Since the NAT1 S214 also may interact with CoA, substituting alanine for serine could affect this interaction. Since S214 is located adjacent to the active site and may be involved in AcCoA binding, S214A could play a role in the increased activity of the NAT1 protein via a mechanism that increases the steady state level of acetylated NAT1 enzyme.
2.3 NAT1-R187Q (560G>A)
The side-chain of R187 is located in the second domain beta barrel and is partially exposed both to the protein surface and the active site pocket. The arginine side-chain forms hydrogen bonds with the side chain of E182 and backbone of K188 in the second domain, and with the side-chain of C-terminal residue T289 in the third domain. These interactions help shape the active site pocket and stabilize the conformations of both the C-terminal tail and the second domain loop (amino acids 165–185). Changing this residue to glutamine may result in partial loss of these interactions, although the smaller glutamine residue may be capable of maintaining some of these hydrogen bonds. Loss of the R187 interactions could lead to a change in the dynamics or conformations of the C-terminal tail and/or the second domain loop resulting in destabilization of NAT1 structure. Changes in C-terminal tail conformation also may influence AcCoA binding and C68 acetylation in the active site. Since the active site is largely shaped by its interactions with both the C-terminal tail and the second domain loop, changes in the conformation and/or dynamics of either of these structures could influence substrate selectivity and catalytic activity.
Functional studies of the R187Q variant expressed in yeast demonstrated reduced catalytic activity, protein levels, and thermostability [31,32]. Reduced Vmax and increased substrate Km were observed following recombinant expression in bacteria [34] and mammalian cells [29]. These studies suggest that R187Q destabilizes the NAT1 structure and influences substrate binding by altering the size and shape of the active site. R187Q did not change AcCoA Km, indicating that altered interactions with the C-terminal tail do not significantly influence AcCoA binding [29].
2.4 NAT1-M205V (613A>G)
The side-chain of M205 is located in the interdomain region adjacent to the active site entrance and the second domain beta barrel. The side chain has no apparent interactions with surrounding residues, but is in close proximity to the backbone and side-chain of I106. Replacing the methionine with a valine maintains the hydrophobicity of residue 205, and should not introduce steric clashes with surrounding residues. Therefore, it is not expected to influence the position of catalytic triad residue H107. Since no interactions are lost and no clashes induced by replacing hydrophobic methionine with a smaller hydrophobic valine, no changes in protein stability or function are expected. Functional studies of the M205V variant in yeast [31,32] and mammalian cells [29] demonstrated no change in enzyme expression, catalytic activity, or thermostability. These findings are consistent with no alterations of important molecular interactions in the M205V variant.
2.5 NAT1-D251V (752A>T)
The side-chain of D251 is located on the third domain beta sheet and its side-chain is oriented into the protein core. The D251 side-chain forms hydrogen bonds with R166 on the second domain loop, with R242 of the third domain beta sheet, and with the backbone of N245 in the third domain. Although the interactions with R242 and N245 are not necessary for maintaining the stability of the beta sheet, hydrogen bonding with R242 may provide additional stability to loop-stabilizing hydrogen bonding interactions. The hydrogen bond between D251 and R166 influences the conformation of the second domain loop, providing support for its interaction with the backbone of V146. These interactions between the second domain loop and the third domain beta sheet contribute to protein stability. Replacing aspartate with a hydrophobic valine residue should reduce all of the D251 interactions, which could affect the dynamics and conformation of the second domain loop, thereby altering protein stability. Functional characterization of the D251V variant in yeast [31,32] and mammalian cells [29,30] demonstrated reduced protein stability, levels and catalytic activity.
2.6 NAT1-E261K (781G>A)
The side-chain of E261 is located on the protein surface in the third domain helix. The E261 side-chain forms hydrogen bonds with the side chain of S259 in the coil between the third domain beta sheet and helix. It is unlikely that this interaction is required for stability of the helix or the third domain tertiary structure. Although hydrogen bonding with S259 may be lost, replacing glutamate with lysine at residue 261 should not cause major changes in dynamics or conformation, because the helix secondary structure is not dependent on this interaction and the third domain helix is stabilized by hydrophobic forces. Functional studies of the E261K variant in yeast [31,32] and mammalian [29,30] cells caused no reduction in protein levels, catalytic activity and thermostability. These data are consistent with observations that residue 261 is located on the protein surface, with a single interaction that makes no apparent contribution to the dynamics or structural conformations in that region.
2.7 NAT1-I263V (787A>G)
I263 is located on the third domain helix. Its side-chain is part of a hydrophobic core located at the interface between the third domain helix and beta sheet, and the C-terminal tail coil. Substitution of isoleucine for the smaller valine at position 263 preserves the hydrophobic interactions without introducing steric clashes. Because the hydrophobic forces are not disturbed, this substitution should not affect NAT1 structural dynamics or conformation. Functional studies of the I263V variant in yeast [31,32] and mammalian cells [29] showed no change in protein level or catalytic activity. The yeast cell studies also demonstrated no reduction in protein thermostability [31,32].
3. Relationships between NAT1 allele/haplotypes, genotypes, and phenotypes
Deduction of NAT1 phenotypes has remained problematic because the relationship between NAT1 genotype and phenotype is poorly understood and in contrast with NAT2 (discussed later) is more strongly influenced by factors other than coding region SNPs. This also follows from the fact that SNPs in the NAT1 coding region, in contrast to those in the NAT2 coding region, are relatively uncommon. We presently recommend that individuals homozygous or heterozygous for “slow” acetylator alleles be identified as slow acetylator phenotypes. This includes individuals who possess 97C>T, 190C>T, 559C>T, 560G>A, and 752A>T and therefore allele/haplotypes NAT1*14A,B; NAT1*17, NAT1*19, and NAT1*22. NAT1*10 is a fairly common allele with SNPs in the 3-UTR but none in the coding region. The functional effects of these SNPs as described earlier are not well understood. NAT1*10 may be designated by some investigators as a “rapid” acetylator phenotype because of the data that supports this designation [14–16]. NAT1*11 is also sometime designated as a “rapid” [29,37]or “slow” [35] allele. Because of the ambiguity involving the NAT1*10 and NAT1*11 phenotypes, we recommend designating them as “at risk” alleles or haplotypes until their phenotypes are clarified.
4. SNPS in NAT2 coding region
Over 25 SNPs in the NAT2 coding region have been identified in human populations (Table 2). The locations for many of these on the NAT2 protein are illustrated in Figure 1. A brief description and possible explanation for the functional effects follows below, supported with data illustrating their effects on NAT2 protein expression, Vmax and substrate Km (Figure 3). Analysis the of structural features and functional effects of many of these NAT2 SNPs was reported recently [28] and is summarized below.
Table 2.
SNPs in human NAT2 coding region
| SNPa | rs identifierb | SNP500 Cancer IDc | Amino acid change | NAT2 allele/haplotype | Functional effectd |
|---|---|---|---|---|---|
| 34T>C | Y12H | ||||
| 70T>A | L24I | NAT2*5L | |||
| 111T>C | NAT2-20 | F37F | NAT2*6D | Synonymous | |
| 190C>T | 1805158 | NAT2-03 | R64W | NAT2*19 | Decrease |
| 191G>A | 1801279 | NAT2-04 | R64Q | NAT1*14 cluster and others | Decrease |
| 282C>T | 1041983 | NAT2-05 | Y94Y | NAT2*13 cluster and others | Synonymous |
| 341C>T | 1801280 | NAT2-06 | I114T | NAT2*5 cluster | Decrease |
| 345C>T | D115D | NAT2*6L | Synonymous | ||
| 364G>A | 4986996 | NAT2-09 | D112N | NAT2*12D | Decrease |
| 403C>G | 12720065 | NAT2-19 | L135V | NAT2*12H | |
| 411A>T | 4986997 | NAT2-10 | L137F | NAT2*5I | |
| 434A>C | Q145P | NAT2*17 | Decrease | ||
| 472A>C | I158L | ||||
| 481C>T | 1799929 | NAT2-07 | L161L | Many | Synonymous |
| 499G>A | E167K | NAT2*10 | |||
| 518A>G | K173R | ||||
| 578C>T | T193M | NAT2*12E; *13B | |||
| 589C>T | R197X | ||||
| 590G>A | 1799930 | NAT2-08 | R197Q | NAT2*6 cluster and others | Decrease |
| 609G>T | E203D | NAT2*12G | |||
| 622T>C | Y208H | NAT2*12F | |||
| 641C>T | T214I | ||||
| 683C>T | P228L | NAT2*14H | |||
| 759C>T | 56011192 | NAT2-17 | V253V | NAT2*5F | Synonymous |
| 766A>G | K256E | NAT2*6H | |||
| 803A>G | 1208 | NAT2-01 | K268R | NAT2*12 cluster and others | No effect |
| 809T>C | I270T | ||||
| 838G>A | V280M | NAT2*5M; *6I; *6K | |||
| 845A>C | 56054745 | NAT2-18 | K282T | NAT2*18 | Substrate-dependent decrease |
| 857G>A | 1799931 | NAT2-02 | G286E | NAT2*7 cluster | Substrate-dependent decrease |
| 859Del | S287frameshift | NAT2*11B |
Reference gene sequence published in Genbank Assession Number X14672. Most common SNPs are bolded. Adapted from http://N-acetyltransferasenomenclature.louisville.edu and [39,40].
Not all SNPs have been designated.
Accessed from http://snp500cancer.nci.nih.gov/snplist.cfm?gene_id=NAT2&mode=valid. This SNP nomenclature is distinct from the NAT2 allele (haplotype) nomenclature.
SNPs are non-synonymous unless otherwise indicated. Only functional effects supported with experimental data are provided.
Figure 3.

Top: Effects of SNPs in the NAT2 coding region on recombinant NAT2 protein level. NAT2 protein and endogenous control α-tubulin levels were visualized by western blots, quantitated by densitometry, and presented in arbitrary units. REF refers to NAT2 4 (no SNPs). Mock: COS-1 cells transfected with the mock pcDNA5/FRT plasmid. Each bar represents Mean± S.E.M from three transfections in duplicate. NAT2-specific protein level for the variant allozymes was compared to NAT2 4 by one way ANOVA followed by Dunnett’s post-test.Mock: *significantly different (p<0.001). Middle: Effect of SNPs in the NAT2 coding region on sulfamethazine (SMZ) N-acetyltransferase maximum velocities with SMZ concentrations of 10 to 5000 μM in the presence of 1 mM AcCoA. SMZ N-acetyltransferase activities were normalized to β-galactosidase activity. Each bar represents Mean ± S.E.M from three transfections in duplicate. REF, no SNP (reference NAT2*4). Activity for each variant allozymes was compared to reference NAT2 4 catalytic activity by one way ANOVA followed by Dunnett’s post-test. *significantly lower than NAT2 4 (p<0.001). Bottom: Effect of SNPs in the NAT2 coding region on SMZ N-acetyltransferase Km with SMZ concentrations of 10 to 5000 μM in the presence of 1 mM AcCoA. SMZ N-acetyltransferase activities were normalized to β-galactosidase activity. Each bar represents Mean ± S.E.M from three transfections in duplicate. REF, no SNP (reference NAT2*4). Km for each variant allozymes was compared to reference NAT2 4 catalytic activity by one way ANOVA followed by Dunnett’s post-test. *significantly lower than NAT2 4 (p<0.001). Adapted from [41].
4.1 NAT2-R64Q (191G>A) and -R64W (190C>T)
The side-chain of R64 bonds twice to E38 and twice to N41 in a stretch of the first domain coil (V35-G51) that is mostly absent of secondary structure. As for NAT1, the hydrogen bonding interactions between positively charged R64 and negatively charged E38 are salt bridges, and are stronger than normal hydrogen bonds. Substitution of arginine with either glutamine or tryptophan at residue 64 causes loss of these interactions which contribute to the conformation and dynamics of the first domain tertiary structure. Functional studies of the R64Q variant in bacterial [42,43], yeast [44], and mammalian cells [41] demonstrated reduced activity and protein levels due to reduced protein stability. These data suggest that the R64 interactions are necessary for structural stability. Since glutamine is less bulky than arginine, the loss of enzyme stability cannot be attributed to steric clashes. Functional studies of the R64W variant in yeast demonstrated reduced protein activity and protein levels due to reduced protein stability [45]. A loss of critical hydrogen bond and salt bridge interactions in the R64W variant is consistent with reduced protein thermostability.
4.2 NAT2-I114T (341T>C)
The I114 side chain shares hydrophobic interactions with residues L21 and L24 in the first domain, and F84 and V112 in the second domain. Changing this hydrophobic residue to a polar hydrophilic threonine residue may alter hydrophobic interactions at the interface between the second domain beta barrel and the first domain helix. However, because of the peripheral location of I114, and the surrounding protein structure that is highly organized into secondary and tertiary structures, it is unlikely that a reduction of hydrophobic forces in this region will result in major structural changes. I114T causes large reductions in catalytic activity when recombinantly expressed in bacteria [42,43] and yeast [44]. Recombinant expression of the I114T variant in mammalian cells did not result in changes in protein stability or apparent kinetic parameters, but led to a reduction of active enzyme possibly due to enhanced protein degradation [46]. These functional data are consistent with a structural change that increases protein aggregation and/or targeting for degradation without altering the protein’s stability.
4.3 NAT2-D122N (364G>A)
D122 is part of the catalytic triad and completely buried in the protein. It forms hydrogen bonds to the side chains of N72, H107, S125, Y190, and the backbone of G124. These multiple hydrogen bonds likely contribute to the stability of the active site loop conformation and the interaction between the first and second domains. Because D122 is a catalytic triad residue, any change will adversely affect the function of the catalytic triad [47]. Recombinant expression of the D122N variant in mammalian cells resulted in undetectable levels of catalytic activity with reduced protein probably due to protein degradation pathways [48]. Disruption of the catalytic triad likely also affects enzyme acetylation, thereby increasing proteasomal degradation [30].
4.4 NAT2-L137F (411A>T)
The side-chain of L137 is oriented toward the interior of the second domain beta-barrel. L137 shares hydrophobic interactions with I120, L152, W159, F192, and L194. The L137F substitution is likely to affect the beta barrel structure due to steric clashes that result from replacing leucine with a larger phenylalanine residue. It is also possible that aromatic interactions between the F137 and W159 side chains alter the folding of the second domain. Functional studies of the L137F variant in mammalian cells demonstrated reduction in protein levels with no change in protein stability, possibly the result of proteasomal degradation [48]. These data are consistent with a change in secondary structure enhancing protein degradation.
4.5 NAT2-Q145P (434A>C)
The side-chain of Q145 forms hydrogen bonds with the backbone of W132, Q133 and Q145 on an adjacent strand in the second domain. Q145P is likely to result in disruption of secondary structure due to the loss of stabilizing hydrogen bonding interactions and the introduction of a rigid proline residue [49]. Since W132 and Q133 are part of the coil that becomes the active site loop, altering their backbone interactions with residue 145 may affect the conformation of the active site loop and thereby alter enzymatic activity and/or substrate selectivity. Functional studies of the Q145P variant in yeast demonstrated reduced or undetectable catalytic activity due to reduced protein levels, although the protein stability was not affected [44]. This reduction in protein is probably due to enhanced protein degradation that is triggered by these structural changes.
4.6 NAT2-E167K (499G>A)
E167 is part of the unstructured “loop” that plays a role in stabilizing mammalian N-acetyltransferases [38]. The side-chain of E167 forms weak hydrogen bonds with K185 which is in an adjacent strand of the second domain loop. Removing this interaction may affect the dynamics or conformation of the loop, which largely lacks defined secondary structure and is therefore more susceptible to dynamic and conformational changes. Functional studies of the E167K variant in mammalian cells demonstrated a reduction in catalytic activity due to reduced protein levels, with no reductions in protein thermostability [41]. It is possible that the E167K variant has small structural changes that cause protein aggregation and/or increased degradation.
4.7 NAT2-R197Q (590G>A)
The R197 side-chain is located near the protein surface of the second domain near the inter-domain helix. Electrostatic interactions are likely between R197 side-chain and E195, and with the lone pairs of electrons on the M105 side-chain sulfur. Replacement of the positively charged arginine with a neutral glutamine residue results in loss of these electrostatic interactions. Steric forces or van der Waals forces are also involved in the close interactions of E195 with R197. Functional studies of the R197Q variant in bacteria, yeast, and mammalian cells demonstrated reduced NAT2 activity and protein levels due to reduced protein thermostability [41–44]. These results are consistent with loss of the relatively weak electrostatic interactions of R197 with E195 and M105.
4.8 NAT2-K268R (803A>G)
The side-chain of residue 268 is located on the protein surface of the third domain alpha helix and has no interactions with surrounding residues or symmetry related crystal neighbors. Replacing K268 with arginine is not expected to affect the alpha helical structure. Functional studies of the K268R variant recombinantly expressed in bacterial [42,43], yeast [44,50] and mammalian cells [41] caused no changes in protein expression, protein stability, or catalytic activity.
4.9 NAT2-G286E (857G>A)
G286 is located on the C-terminal tail in the third domain directly adjacent to the active site and does not directly interact with other residues. Replacement of glycine with a much larger glutamate at residue 286 could significantly alter the conformation of the C-terminal tail adjacent to the active site opening due to steric clashes with nearby residues and loss of the highly flexible glycine residue. Since the C-terminal tail has an important role in defining the size and shape of the active site cavity, the G286E variant protein is likely to have altered active site access and altered substrate selectivity. Such a significant change to a C-terminal residue adjacent to the active site is also likely to affect AcCoA binding and C68 acetylation. The NAT2 crystal structure (PDB ID# 2PFR) has CoA bound in its active site with hydrogen bonding between CoA and S287. The C-terminal tail conformational change that may accompany the G286E variant would likely influence this interaction between S287 and CoA.
Differences between NAT1 and NAT2 in the size of the active site [22], and in substrate selectivities and/or catalytic activities [51] are likely influenced by the difference in bulkiness of the smaller NAT2 G286 residue compared to the larger NAT1 R286 residue side chain, which is oriented toward the active site opening. The addition of a bulky glutamate side-chain at residue 286 in the G286E variant should alter substrate selectivity and/or catalytic activity.
Functional studies of the G286E variant in mammalian cells demonstrated reduced activity for some substrates but not for others, and reduced protein and thermostability [41]. The observation that the G286E residue change could alter the size and shape of the active site pocket is consistent with the substrate-dependent activity changes observed experimentally [41]. In addition, the finding that the G286E alters the Km for AcCoA [41] is consistent with a conformational change in the C-terminal tail which may alter interactions with AcCoA. It is possible that reduced protein acetylation contributes to the overall reduction of protein levels through proteasomal degradation [30].
5. Relationships between NAT2 allele/haplotypes, genotypes, and phenotypes
Following recombinant expression in yeast, 190C>T (R64W), 191G>A (R64Q), 341T>C (I114T), 434A>C (Q145P) and 590G>A (R197Q) reduce NAT2 catalytic activities, whereas 111T>C (F37F), 282C>T (Y94Y), 481C>T (L161L), 759C>T (V253V), and 803A>G (K268R) do not [43,44]. The effects of 845A>C (K282T) and 857G>A (G286E) differ with substrate, as they reduce catalytic N- and O-acetyltransferase activities towards some substrates and not others. The reduction in NAT2 protein appeared to be related to instability of the protein for 190C>T (R64W), 191G>A (R64W), 590G>A (R197Q) and 857G>A (G286E), whereas 341T>C (I114T), 411A>T (L137F), and 499G>A (E167K) did not appear to increase NAT2 instability [44,45]. Following recombinant expression in mammalian cells, the SNPs which reduced NAT2 activities each did so by reductions in expression of recombinant NAT2 protein but not recombinant NAT2 mRNA [41]. 857G>A (G286E) changed apparent Km, reducing it towards SMZ and increasing it towards AcCoA.
As previously reviewed [52] deduction of NAT2 phenotypes is assigned based on co-dominant expression of rapid and slow acetylator NAT2 alleles or haplotypes. Individuals homozygous for rapid NAT2 acetylator alleles are deduced as rapid acetylators, individuals homozygous for slow acetylator NAT2 alleles are deduced as slow acetylators, and individuals possessing one rapid and one slow NAT2 allele are deduced as intermediate acetylators.
6. Associations with increased toxicity and disease incidence
Associations between acetylator polymorphism and drug toxicity or disease frequency are often inconsistent and not replicated. A confounding issue is the poor understanding of the many factors, in addition to SNPs in the coding region, which modify N-acetyltransferase capacity. Some examples are discussed below:
There are striking differences in substrate selectivity between NAT1 and NAT2 [53, 54]. For example, O-acetylation of heterocyclic amines is catalyzed to a considerably greater extent by NAT2 than NAT1 [54–57]. Depending upon the tissue, acetylation capacity is determined by both NAT1 and NAT2 phenotypes. The role of either NAT1 or NAT2 phenotype on drug efficacy or toxicity and on disease frequency is dependent upon the relative specificity of the drug or chemical for NAT1 versus NAT2. There also is linkage disequilibrium between NAT1 and NAT2 haplotypes/alleles [58,59].
NAT1 and NAT2 differ in their relative capacity to catalyze N-acetylation versus O-acetylation. Many heterocyclic amine carcinogens exhibit steric hindrance of the exocyclic amine resulting in absence or very low capacity for N-acetylation [55–57]. Thus, many aromatic amines undergo both N- and O-acetylation whereas many heterocyclic amines undergo O- but not N-acetylation.
NAT1 and NAT2 differ in tissue specificity. NAT1 appears to be widely expressed in virtually all human tissues examined [60]. In contrast, NAT2 is predominantly expressed in liver, small intestine and colon, although NAT2 mRNA is detectable in most human tissues at basal levels [61]. As recently reviewed [5,9,10], the NAT1 gene consists of at least nine exons. The entire open reading frame is contained in exon 9 while the first eight exons are located in the 5′-untranslated region (UTR). The majority of NAT1 transcripts arise from two promoters now identified as NATb and NATa. NATb promotes the production of the major transcript detected in most human tissues. The presence of a functional Sp1 element in NATb is consistent with its widespread tissue distribution. NATa appears to promote a smaller set of transcripts in a restricted set of tissues, with high levels in liver, lung, kidney and trachea and lower levels in prostate and testes. The activity of this alternative promoter could be regulated in a different manner than NATa and may be more relevant in certain diseases. The structure of the NAT2 gene is less complex. The entire open reading frame is located in exon 2, and with a single non-coding exon in the 5′-UTR [61]. The NAT2 promoter does not contain Sp1, consistent with its more restrictive tissue distribution. Although SNPs have been identified in the NAT2 promoter sequence [62–64] their functional consequences have yet to be determined. Additional identification and characterization of NAT2 promoter elements is necessary in order to understand these functional effects. Urinary bladder cancer is associated with aromatic amine carcinogens such as 4-aminobiphenyl present in cigarette smoke. NAT2-catalyzed N-acetylation competes with P450-catalyzed N-hydroxylation in the liver, thus accounting for associations between slow acetylator NAT2 phenotype and urinary bladder cancer in chemical dye workers [65] and smokers [66]. In contrast, NAT2-catalyzed O-acetylation of N-hydroxy-heterocyclic amine carcinogens within the colon accounts for associations between rapid acetylator NAT2 phenotype and colorectal cancer in people who frequently eat well done meat [67–73]. Whereas NAT1-catalyzed O-acetylation may be an activation route in colon, NAT1-catalyzed N-acetylation represents “first-pass” detoxification in the skin when exposures to aromatic amines occur via the dermal route [74].
The nature of the arylamine exposure is important. For example, many arylamines undergo both N- and O-acetylation via NAT1 and/or NAT2, whereas most heterocyclic amine carcinogens undergo only O-acetylation. In addition, N-acetylation of an arylmonoamine such as 4-aminobiphenyl. may decrease urinary bladder cancer risk, whereas N-acetylation of an aryldiamine such as benzidine may enhance N-hydroxylation of the second exocyclic amine and thus increase urinary bladder cancer risk [52,75,76].
All SNPs do not produce same effect implying heterogeneity within the “slow” acetylator phenotype. This has significant implications in for both drug toxicity and chemical carcinogenesis. For example, isoniazid -induced hepatotoxicity is higher in NAT2 slow acetylators [77,78] and is particularly highest in “super slow” acetylators [77,80]. Individuals with slow acetylator NAT2 phenotype experienced more severe hepatotoxicity than rapid acetylators and less risk was observed in individuals possessing NAT2*7 haplotypes [78]. English chemical dye workers with documented exposure to aromatic amine carcinogens showed a striking association between urinary bladder cancer and slow acetylator phenotypes [65]. The NAT2 phenotype data was not separated into two phenotypes (rapid and slow), but rather into eight phenotypes of metabolic ratios. Five of these ratios correspond to rapid acetylators, and the other three correspond to various degrees of slow acetylator phenotype. As reviewed previously [80], urinary bladder cancer risk increased as NAT2 metabolic ratio (phenotype) decreased and the risk was markedly increased in the “slowest” NAT2 phenotype. Four studies found that urinary bladder cancer risk was highest in individuals possessing NAT2*5 haplotypes [81–84]. The 341T>C (I114T) SNP associated with NAT2*5 alleles or haplotypes yields very large reductions in NAT2 protein and activity [42–44] resulting from protein degradation [46]. NAT2*5 alleles also were associated with increased risk for breast cancer in women smokers [85]. These results suggest that NAT2 slow acetylator phenotype is not homogeneous, but rather that multiple slow acetylator phenotypes exist resulting from different mechanisms inferred by various SNPs and haplotypes. If the SNP increases N-acetyltransferase protein degradation and thus reduces protein level, then the functional effects should not vary with drug or chemical. In contrast, if the SNP alters N-acetyltransferase affinity, then the functional effect might be observed with some drugs/chemicals and not others and at some exposure levels not others. Evidence for such an exposure effect was recently reported [86].
The frequency of SNPs in NAT1 and NAT2 varies between and within various race or ethnic groups. This was comprehensively shown recently for NAT2 [87]. This reinforces the importance of proper selection of control groups for pharmacogenetic and epidemiological studies investigating the role of NAT1 and/or NAT2 SNPs on disease or drug effect. Current genotyping methods simulataneously analyze both the maternal and the paternal and alleles. Ambiguous haplotype and/or genotype construction of this data leads to misclassification errors that also impair ability and power to observe SNP modifier effects on disease or drug effect [88,89]. A table illustrating NAT2 diplotypes and genotypes recently was published to assist laboratories in making these conversions [89].
There is lack of understanding and appreciation for transcriptional and post-translational regulation of NAT1 and NAT2. Recent studies show that oxidative/nitroxidative stress in lung epithelial cells, due to air pollution and/or inflammation, could contribute to local and/or systemic dysfunctions by altering functions of pulmonary N-acetyltransferase enzymes [90]. As recently reviewed [20], environmental factors alter expression and much more research is needed.
7. Conclusions
Although much has been learned regarding the identity, location, and functional effects of SNPs in NAT1 and NAT2, additional research is needed. Emerging concepts regarding the functional effects of SNPs in NAT1 and NAT2 suggest pharmacogenetic effects that are not replicated may ultimately be comfirmed when all factors affecting the phenotype are better understood.
8. Expert opinion
An increased understanding of pharmacogenetic principles is leading to advances in personalized drug treatment (higher efficacy and lower toxicity) together with more individualized risk assessments to disease and/or toxicities associated with carcinogen exposures. The latter falls within the discipline of “molecular epidemiology” and emerging concepts suggest that confounding variables can result in lack of replication across studies leading to conclusions that genetic polymorphisms are not sufficiently biologically or medically relevant to merit attention. Using arylamine N-acetyltransferase as a paradigm, laboratory-based mechanistic analyses are revealing additional complexities leading to the alternative conclusion that genetic polymorphisms are biologically and medically relevant when the confounding factors are more fully understood and considered. As medical care moves to a more personalized approach, the implications of these confounding factors will be very important in understanding the complexities of personalized medicine.
Acknowledgments
The author acknowledges the essential contributions of all investigators in the arylamine N-acetyltransferase field, particularly those who I have worked with and generated data presented in this paper. Also acknowledged are the relevant research grants from the National Cancer Institute (R01-CA034627), the National Institute of Environmental Health Sciences (P30-ES014443) and the National Institute of Child Health and Development (U10-HD045934).
Footnotes
Job Titles: Peter K. Knoefel Endowed Chair of Pharmacology, Distinguished University Scholar, Professor and Chairman of the Department of Pharmacology and Toxicology, Leader, Cancer Prevention and Control Program, James Graham Brown Cancer Center, and Special Assistant to the Provost for Strategic Planning, University of Louisville.
References
- 1.Weber WW. The Acetylator Genes and Drug Response. Oxford University Press; New York: 1987. **Excellent overview, particularly with respect to history of the discovery. [Google Scholar]
- 2.Weber WW, Hein DW. N-acetylation pharmacogenetics. Pharmacol Rev. 1985;37:25–79. **Excellent overview, particularly with respect to clinical and toxicological consequences. [PubMed] [Google Scholar]
- 3.Boukouvala S, Fakis G. Arylamine N-acetyltransferases: what we learn from genes and genomes. Drug Metab Rev. 2005;37:511–564. doi: 10.1080/03602530500251204. **Excellent overview, particularly with respect to genomic organization. [DOI] [PubMed] [Google Scholar]
- 4.Westwood IM, Kawamura A, Fullam E, Russell AJ, Davies SG, Sim E. Structure and mechanism of arylamine N-acetyltransferases. Curr Top Med Chem. 2006;6:1641–1654. doi: 10.2174/156802606778108979. [DOI] [PubMed] [Google Scholar]
- 5.Minchin RF, Hanna PE, Dupret JM, Wagner CR, Rodrigues-Lima F, Butcher NJ. Arylamine N-acetyltransferase I. Int J Biochem Cell Biol. 2007;39:1999–2005. doi: 10.1016/j.biocel.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 6.Sim E, Westwood I, Fullam E. Arylamine N-acetyltransferases. Expert Opin Drug Metab Toxicol. 2007;3:169–184. doi: 10.1517/17425255.3.2.169. [DOI] [PubMed] [Google Scholar]
- 7.Agundez JA. Polymorphisms of human N-acetyltransferases and cancer risk. Curr Drug Metab. 2008;9:520–531. doi: 10.2174/138920008784892083. [DOI] [PubMed] [Google Scholar]
- 8.Grant DM. Structures of human arylamine N-acetyltransferases. Curr Drug Metab. 2008;9:465–470. doi: 10.2174/138920008784892029. *Excellent overview, particularly with respect to NAT1 and NAT2 structure. [DOI] [PubMed] [Google Scholar]
- 9.Sim E, Walters K, Boukouvala S. Arylamine N-acetyltransferases: from structure to function. Drug Metab Rev. 2008;40:479–510. doi: 10.1080/03602530802186603. *Excellent overview, particularly with respect to N-acetyltransferase structure and function. [DOI] [PubMed] [Google Scholar]
- 10.Sim E, Lack N, Wang CJ, Long H, Westwood I, Fullam E, Kawamura A. Arylamine N-acetyltransferases: structural and functional implications of polymorphisms. Toxicology. 2008;254:170–183. doi: 10.1016/j.tox.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 11.Vatsis KP, Weber WW, Bell DA, Dupret JM, Evans DA, Grant DM, Hein DW, Lin HJ, Meyer UA, Relling MV, et al. Nomenclature for N-acetyltransferases. Pharmacogenetics. 1995;5:1–17. doi: 10.1097/00008571-199502000-00001. **Initial N-acetyltransferase nomenclature paper. [DOI] [PubMed] [Google Scholar]
- 12.Hein DW, Boukouvala S, Grant DM, Minchin RF, Sim E. Changes in consensus arylamine N-acetyltransferase gene nomenclature. Pharmacogenet Genomics. 2008;18:367–368. doi: 10.1097/FPC.0b013e3282f60db0. *Most updated N-acetyltransferase gene nomenclature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butcher NJ, Tiang J, Minchin RF. Regulation of arylamine N-acetyltransferases. Curr Drug Metab. 2008;9:498–504. doi: 10.2174/138920008784892128. *Excellent overview, particularly with respect to transcriptional regulation. [DOI] [PubMed] [Google Scholar]
- 14.Badawi AF, Hirvonen A, Bell DA, Lang NP, Kadlubar FF. Role of aromatic amine acetyltransferases, NAT1 and NAT2, in carcinogen-DNA adduct formation in the human urinary bladder. Cancer Res. 1995;55:5230–5237. [PubMed] [Google Scholar]
- 15.Bell DA, Badawi AF, Lang NP, Ilett KF, Kadlubar FF, Hirvonen A. Polymorphism in the N-acetyltransferase 1 (NAT1) polyadenylation signal: association of NAT1*10 allele with higher N-acetylation activity in bladder and colon tissue. Cancer Res. 1995;55:5226–5229. [PubMed] [Google Scholar]
- 16.Hein DW, McQueen CA, Grant DM, Goodfellow GH, Kadlubar FF, Weber WW. Pharmacogenetics of the arylamine N-acetyltransferases: a symposium in honor of Wendell W. Weber. Drug Metab Dispos. 2000;28:1425–1432. [PubMed] [Google Scholar]
- 17.de Leon JH, Vatsis KP, Weber WW. Characterization of naturally occurring and recombinant human N-acetyltransferase variants encoded by NAT1. Mol Pharmacol. 2000;58:288–299. doi: 10.1124/mol.58.2.288. [DOI] [PubMed] [Google Scholar]
- 18.Payton MA, Sim E. Genotyping human arylamine N-acetyltransferase type 1 (NAT1): the identification of two novel allelic variants. Biochem Pharmacol. 1998;55:361–366. doi: 10.1016/s0006-2952(97)00478-4. [DOI] [PubMed] [Google Scholar]
- 19.Bruhn C, Brockmoller J, Cascorbi I, Roots I, Borchert HH. Correlation between genotype and phenotype of the human arylamine N-acetyltransferase type 1 (NAT1) Biochem Pharmacol. 1999;58:1759–1764. doi: 10.1016/s0006-2952(99)00269-5. [DOI] [PubMed] [Google Scholar]
- 20.Rodrigues-Lima F, Dairou J, Dupret JM. Effect of environmental substances on the activity of arylamine N-acetyltransferases. Curr Drug Metab. 2008;9:505–509. doi: 10.2174/138920008784892092. *Excellent overview, particularly with respect to environmental regulation. [DOI] [PubMed] [Google Scholar]
- 21.Sinclair JC, Sandy J, Delgoda R, Sim E, Noble ME. Structure of arylamine N-acetyltransferase reveals a catalytic triad. Nat Struct Biol. 2000;7:560–564. doi: 10.1038/76783. **First report of N-acetyltransferase crystal structure. [DOI] [PubMed] [Google Scholar]
- 22.Wu H, Dombrovsky L, Tempel W, Martin F, Loppnau P, Goodfellow GH, Grant DM, Plotnikov AN. Structural basis of substrate-binding specificity of human arylamine N-acetyltransferases. J Biol Chem. 2007;282:30189–30197. doi: 10.1074/jbc.M704138200. **First report of human N-acetyltransferase crystal structures. [DOI] [PubMed] [Google Scholar]
- 23.Dupret JM, Grant DM. Site-directed mutagenesis of recombinant human arylamine N-acetyltransferase expressed in Escherichia coli. Evidence for direct involvement of Cys68 in the catalytic mechanism of polymorphic human NAT2. J Biol Chem. 1992;267:7381–7385. *Key early mechanism paper for N-acetylation. [PubMed] [Google Scholar]
- 24.Goodfellow GH, Dupret JM, Grant DM. Identification of amino acids imparting acceptor substrate selectivity to human arylamine acetyltransferases NAT1 and NAT2. Biochem J. 2000;348(Pt 1):159–166. [PMC free article] [PubMed] [Google Scholar]
- 25.Liu L, Von Vett A, Zhang N, Walters KJ, Wagner CR, Hanna PE. Arylamine N-acetyltransferases: characterization of the substrate specificities and molecular interactions of environmental arylamines with human NAT1 and NAT2. Chem Res Toxicol. 2007;20:1300–1308. doi: 10.1021/tx7001614. [DOI] [PubMed] [Google Scholar]
- 26.Hein DW. Acetylator genotype and arylamine-induced carcinogenesis. Biochim Biophys Acta. 1988;948:37–66. doi: 10.1016/0304-419x(88)90004-2. [DOI] [PubMed] [Google Scholar]
- 27.Walraven JM, Trent JO, Hein DW. Structure-function analyses of single nucleotide polymorphisms in human N-acetyltransferase 1. Drug Metab Rev. 2008;40:169–184. doi: 10.1080/03602530701852917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walraven JM, Zang Y, Trent JO, Hein DW. Structure/function evaluations of single nucleotide polymorphisms in human N-acetyltransferase 2. Curr Drug Metab. 2008;9:471–486. doi: 10.2174/138920008784892065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu Y, Hein DW. Functional effects of single nucleotide polymorphisms in the coding region of human N-acetyltransferase 1. Pharmacogenomics J. 2008;8:339–348. doi: 10.1038/sj.tpj.6500483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Butcher NJ, Arulpragasam A, Minchin RF. Proteasomal degradation of N-acetyltransferase 1 is prevented by acetylation of the active site cysteine: a mechanism for the slow acetylator phenotype and substrate-dependent down-regulation. J Biol Chem. 2004;279:22131–22137. doi: 10.1074/jbc.M312858200. *Conclusive evidence for substrate-dependent down-regulation of N-acetyltransferase. [DOI] [PubMed] [Google Scholar]
- 31.Fretland AJ, Doll MA, Leff MA, Hein DW. Functional characterization of nucleotide polymorphisms in the coding region of N-acetyltransferase 1. Pharmacogenetics. 2001;11:511–520. doi: 10.1097/00008571-200108000-00006. [DOI] [PubMed] [Google Scholar]
- 32.Fretland AJ, Doll MA, Zhu Y, Smith L, Leff MA, Hein DW. Effect of nucleotide substitutions in N-acetyltransferase-1 on N-acetylation (deactivation) and O-acetylation (activation) of arylamine carcinogens: implications for cancer predisposition. Cancer Detect Prev. 2002;26:10–14. doi: 10.1016/s0361-090x(02)00005-3. [DOI] [PubMed] [Google Scholar]
- 33.Liu F, Zhang N, Zhou X, Hanna PE, Wagner CR, Koepp DM, Walters KJ. Arylamine N-acetyltransferase aggregation and constitutive ubiquitylation. J Mol Biol. 2006;361:482–492. doi: 10.1016/j.jmb.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 34.Hughes NC, Janezic SA, McQueen KL, Jewett MA, Castranio T, Bell DA, Grant DM. Identification and characterization of variant alleles of human acetyltransferase NAT1 with defective function using p-aminosalicylate as an in-vivo and in-vitro probe. Pharmacogenetics. 1998;8:55–66. doi: 10.1097/00008571-199802000-00008. [DOI] [PubMed] [Google Scholar]
- 35.Bruhn C, Brockmoller J, Cascorbi I, Roots I, Borchert HH. Correlation between genotype and phenotype of the human arylamine N-acetyltransferase type 1 (NAT1) Biochem Pharmacol. 1999;58:1759–1764. doi: 10.1016/s0006-2952(99)00269-5. [DOI] [PubMed] [Google Scholar]
- 36.Zhangwei X, Jianming X, Qiao M, Xinhua X. N-Acetyltransferase-1 gene polymorphisms and correlation between genotype and its activity in a central Chinese Han population. Clin Chim Acta. 2006;371:85–91. doi: 10.1016/j.cca.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 37.Doll MA, Jiang W, Deitz AC, Rustan TD, Hein DW. Identification of a novel allele at the human NAT1 acetyltransferase locus. Biochem Biophys Res Commun. 1997;233:584–591. doi: 10.1006/bbrc.1997.6501. [DOI] [PubMed] [Google Scholar]
- 38.Walraven JM, Trent JO, Hein DW. Computational and experimental analyses of mammalian arylamine N-acetyltransferase structure and function. Drug Metab Dispos. 2007;35:1001–1007. doi: 10.1124/dmd.107.015040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luca F, Bubba G, Basile M, Brdicka R, Michalodimitrakis E, Rickards O, Vershubsky G, Quintana-Murci L, Kozlov AI, Novelletto A. Multiple advantageous amino acid variants in the NAT2 gene in human populations. PLoS ONE. 2008;3:e3136. doi: 10.1371/journal.pone.0003136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matimba A, Del-Favero J, Van Broeckhoven C, Masimirembwa C. Novel variants of major drug-metabolising enzyme genes in diverse African populations and their predicted functional effects. Hum Genomics. 2009;3:169–190. doi: 10.1186/1479-7364-3-2-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zang Y, Doll MA, Zhao S, States JC, Hein DW. Functional characterization of single-nucleotide polymorphisms and haplotypes of human N-acetyltransferase 2. Carcinogenesis. 2007;28:1665–1671. doi: 10.1093/carcin/bgm085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hein DW, Ferguson RJ, Doll MA, Rustan TD, Gray K. Molecular genetics of human polymorphic N-acetyltransferase: enzymatic analysis of 15 recombinant wild-type, mutant, and chimeric NAT2 allozymes. Hum Mol Genet. 1994;3:729–734. doi: 10.1093/hmg/3.5.729. *First comprehensive functional analysis of NAT2 SNPs. [DOI] [PubMed] [Google Scholar]
- 43.Hein DW, Doll MA, Rustan TD, Ferguson RJ. Metabolic activation of N-hydroxyarylamines and N-hydroxyarylamides by 16 recombinant human NAT2 allozymes: effects of 7 specific NAT2 nucleic acid substitutions. Cancer Res. 1995;55:3531–3536. [PubMed] [Google Scholar]
- 44.Fretland AJ, Leff MA, Doll MA, Hein DW. Functional characterization of human N-acetyltransferase 2 (NAT2) single nucleotide polymorphisms. Pharmacogenetics. 2001;11:207–215. doi: 10.1097/00008571-200104000-00004. [DOI] [PubMed] [Google Scholar]
- 45.Zhu Y, Doll MA, Hein DW. Functional genomics of C190T single nucleotide polymorphism in human N-acetyltransferase 2. Biol Chem. 2002;383:983–987. doi: 10.1515/BC.2002.105. [DOI] [PubMed] [Google Scholar]
- 46.Zang Y, Zhao S, Doll MA, States JC, Hein DW. The T341C (Ile114Thr) polymorphism of N-acetyltransferase 2 yields slow acetylator phenotype by enhanced protein degradation. Pharmacogenetics. 2004;14:717–723. doi: 10.1097/00008571-200411000-00002. *Mechanism for slow NAT2 phenotype. [DOI] [PubMed] [Google Scholar]
- 47.Sandy J, Mushtaq A, Holton SJ, Schartau P, Noble ME, Sim E. Investigation of the catalytic triad of arylamine N-acetyltransferases: essential residues required for acetyl transfer to arylamines. Biochem J. 2005;390:115–123. doi: 10.1042/BJ20050277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zang Y, Zhao S, Doll MA, Christopher States J, Hein DW. Functional characterization of the A411T (L137F) and G364A (D122N) genetic polymorphisms in human N-acetyltransferase 2. Pharmacogenet Genomics. 2007;17:37–45. doi: 10.1097/01.fpc.0000236325.73186.2c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li SC, Goto NK, Williams KA, Deber CM. Alpha-helical, but not beta-sheet, propensity of proline is determined by peptide environment. Proc Natl Acad Sci U S A. 1996;93:6676–6681. doi: 10.1073/pnas.93.13.6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hein DW, Fretland AJ, Doll MA. Effects of single nucleotide polymorphisms in human N-acetyltransferase 2 on metabolic activation (O-acetylation) of heterocyclic amine carcinogens. Int J Cancer. 2006;119:1208–1211. doi: 10.1002/ijc.21957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grant DM, Hughes NC, Janezic SA, Goodfellow GH, Chen HJ, Gaedigk A, Yu VL, Grewal R. Human acetyltransferase polymorphisms. Mutat Res. 1997;376:61–70. doi: 10.1016/s0027-5107(97)00026-2. [DOI] [PubMed] [Google Scholar]
- 52.Hein DW. N-acetyltransferase 2 genetic polymorphism: effects of carcinogen and haplotype on urinary bladder cancer risk. Oncogene. 2006;25:1649–1658. doi: 10.1038/sj.onc.1209374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grant DM, Blum M, Beer M, Meyer UA. Monomorphic and polymorphic human arylamine N-acetyltransferases: a comparison of liver isozymes and expressed products of two cloned genes. Mol Pharmacol. 1991;39:184–191. **Initial characterization of NAT1 and NAT2 substrate specificity. [PubMed] [Google Scholar]
- 54.Hein DW, Doll MA, Rustan TD, Gray K, Feng Y, Ferguson RJ, Grant DM. Metabolic activation and deactivation of arylamine carcinogens by recombinant human NAT1 and polymorphic NAT2 acetyltransferases. Carcinogenesis. 1993;14:1633–1638. doi: 10.1093/carcin/14.8.1633. [DOI] [PubMed] [Google Scholar]
- 55.Minchin RF, Reeves PT, Teitel CH, McManus ME, Mojarrabi B, Ilett KF, Kadlubar FF. N-and O-acetylation of aromatic and heterocyclic amine carcinogens by human monomorphic and polymorphic acetyltransferases expressed in COS-1 cells. Biochem Biophys Res Commun. 1992;185:839–844. doi: 10.1016/0006-291x(92)91703-s. [DOI] [PubMed] [Google Scholar]
- 56.Hein DW, Rustan TD, Ferguson RJ, Doll MA, Gray K. Metabolic activation of aromatic and heterocyclic N-hydroxyarylamines by wild-type and mutant recombinant human NAT1 and NAT2 acetyltransferases. Arch Toxicol. 1994;68:129–133. doi: 10.1007/s002040050045. [DOI] [PubMed] [Google Scholar]
- 57.Lau EY, Felton JS, Lightstone FC. Insights into the o-acetylation reaction of hydroxylated heterocyclic amines by human arylamine N-acetyltransferases: a computational study. Chem Res Toxicol. 2006;19:1182–1190. doi: 10.1021/tx0600999. [DOI] [PubMed] [Google Scholar]
- 58.Patin E, Harmant C, Kidd KK, Kidd J, Froment A, Mehdi SQ, Sica L, Heyer E, Quintana-Murci L. Sub-Saharan African coding sequence variation and haplotype diversity at the NAT2 gene. Hum Mutat. 2006;27:720. doi: 10.1002/humu.9438. [DOI] [PubMed] [Google Scholar]
- 59.Sabbagh A, Langaney A, Darlu P, Gerard N, Krishnamoorthy R, Poloni ES. Worldwide distribution of NAT2 diversity: implications for NAT2 evolutionary history. BMC Genet. 2008;9:21. doi: 10.1186/1471-2156-9-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Husain A, Zhang X, Doll MA, States JC, Barker DF, Hein DW. Functional analysis of the human N-acetyltransferase 1 major promoter: quantitation of tissue expression and identification of critical sequence elements. Drug Metab Dispos. 2007;35:1649–1656. doi: 10.1124/dmd.107.016485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Husain A, Zhang X, Doll MA, States JC, Barker DF, Hein DW. Identification of N-acetyltransferase 2 (NAT2) transcription start sites and quantitation of NAT2-specific mRNA in human tissues. Drug Metab Dispos. 2007;35:721–727. doi: 10.1124/dmd.106.014621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yuliwulandari R, Sachrowardi Q, Nishida N, Takasu M, Batubara L, Susmiarsih TP, Rochani JT, Wikaningrum R, Miyashita R, Miyagawa T, Sofro AS, Tokunaga K. Polymorphisms of promoter and coding regions of the arylamine N-acetyltransferase 2 (NAT2) gene in the Indonesian population: proposal for a new nomenclature. J Hum Genet. 2008;53:201–209. doi: 10.1007/s10038-007-0237-z. [DOI] [PubMed] [Google Scholar]
- 63.Moslehi R, Chatterjee N, Church TR, Chen J, Yeager M, Weissfeld J, Hein DW, Hayes RB. Cigarette smoking, N-acetyltransferase genes and the risk of advanced colorectal adenoma. Pharmacogenomics. 2006;7:819–829. doi: 10.2217/14622416.7.6.819. [DOI] [PubMed] [Google Scholar]
- 64.McKay JD, Hashibe M, Hung RJ, Wakefield J, Gaborieau V, Szeszenia-Dabrowska N, Zaridze D, Lissowska J, Rudnai P, Fabianova E, Mates D, Foretova L, Janout V, Bencko V, Chabrier A, Hall J, Boffetta P, Canzian F, Brennan P. Sequence variants of NAT1 and NAT2 and other xenometabolic genes and risk of lung and aerodigestive tract cancers in Central Europe. Cancer Epidemiol Biomarkers Prev. 2008;17:141–147. doi: 10.1158/1055-9965.EPI-07-0553. [DOI] [PubMed] [Google Scholar]
- 65.Cartwright RA, Glashan RW, Rogers HJ, Ahmad RA, Barham-Hall D, Higgins E, Kahn MA. Role of N-acetyltransferase phenotypes in bladder carcinogenesis: a pharmacogenetic epidemiological approach to bladder cancer. Lancet. 1982;2:842–845. doi: 10.1016/s0140-6736(82)90810-8. **First conclusive report of the role of NAT2 phenotype in cancer susceptibility. [DOI] [PubMed] [Google Scholar]
- 66.Garcia-Closas M, Malats N, Silverman D, Dosemeci M, Kogevinas M, Hein DW, Tardon A, Serra C, Carrato A, Garcia-Closas R, Lloreta J, Castano-Vinyals G, Yeager M, Welch R, Chanock S, Chatterjee N, Wacholder S, Samanic C, Tora M, Fernandez F, Real FX, Rothman N. NAT2 slow acetylation, GSTM1 null genotype, and risk of bladder cancer: results from the Spanish Bladder Cancer Study and meta-analyses. Lancet. 2005;366:649–659. doi: 10.1016/S0140-6736(05)67137-1. **Convincing evidence for the role of NAT2 phenotype in cancer susceptibility. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lang NP, Butler MA, Massengill J, Lawson M, Stotts RC, Hauer-Jensen M, Kadlubar FF. Rapid metabolic phenotypes for acetyltransferase and cytochrome P4501A2 and putative exposure to food-borne heterocyclic amines increase the risk for colorectal cancer or polyps. Cancer Epidemiol Biomarkers Prev. 1994;3:675–682. [PubMed] [Google Scholar]
- 68.Chen J, Stampfer MJ, Hough HL, Garcia-Closas M, Willett WC, Hennekens CH, Kelsey KT, Hunter DJ. A prospective study of N-acetyltransferase genotype, red meat intake, and risk of colorectal cancer. Cancer Res. 1998;58:3307–3311. [PubMed] [Google Scholar]
- 69.Le Marchand L, Hankin JH, Wilkens LR, Pierce LM, Franke A, Kolonel LN, Seifried A, Custer LJ, Chang W, Lum-Jones A, Donlon T. Combined effects of well-done red meat, smoking, and rapid N-acetyltransferase 2 and CYP1A2 phenotypes in increasing colorectal cancer risk. Cancer Epidemiol Biomarkers Prev. 2001;10:1259–1266. [PubMed] [Google Scholar]
- 70.Lilla C, Verla-Tebit E, Risch A, Jager B, Hoffmeister M, Brenner H, Chang-Claude J. Effect of NAT1 and NAT2 genetic polymorphisms on colorectal cancer risk associated with exposure to tobacco smoke and meat consumption. Cancer Epidemiol Biomarkers Prev. 2006;15:99–107. doi: 10.1158/1055-9965.EPI-05-0618. [DOI] [PubMed] [Google Scholar]
- 71.Ognjanovic S, Yamamoto J, Maskarinec G, Le Marchand L. NAT2, meat consumption and colorectal cancer incidence: an ecological study among 27 countries. Cancer Causes Control. 2006;17:1175–1182. doi: 10.1007/s10552-006-0061-3. [DOI] [PubMed] [Google Scholar]
- 72.Cotterchio M, Boucher BA, Manno M, Gallinger S, Okey AB, Harper PA. Red meat intake, doneness, polymorphisms in genes that encode carcinogen-metabolizing enzymes, and colorectal cancer risk. Cancer Epidemiol Biomarkers Prev. 2008;17:3098–3107. doi: 10.1158/1055-9965.EPI-08-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shin A, Shrubsole MJ, Rice JM, Cai Q, Doll MA, Long J, Smalley WE, Shyr Y, Sinha R, Ness RM, Hein DW, Zheng W. Meat intake, heterocyclic amine exposure, and metabolizing enzyme polymorphisms in relation to colorectal polyp risk. Cancer Epidemiol Biomarkers Prev. 2008;17:320–329. doi: 10.1158/1055-9965.EPI-07-0615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Goebel C, Hewitt NJ, Kunze G, Wenker M, Hein DW, Beck H, Skare J. Skin metabolism of aminophenols: human keratinocytes as a suitable in vitro model to qualitatively predict the dermal transformation of 4-amino-2-hydroxytoluene in vivo. Toxicol Appl Pharmacol. 2009;235:114–123. doi: 10.1016/j.taap.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 75.Hayes RB, Bi W, Rothman N, Broly F, Caporaso N, Feng P, You X, Yin S, Woosley RL, Meyer UA. N-acetylation phenotype and genotype and risk of bladder cancer in benzidine-exposed workers. Carcinogenesis. 1993;14:675–678. doi: 10.1093/carcin/14.4.675. [DOI] [PubMed] [Google Scholar]
- 76.Carreon T, Ruder AM, Schulte PA, Hayes RB, Rothman N, Waters M, Grant DJ, Boissy R, Bell DA, Kadlubar FF, Hemstreet GP, 3rd, Yin S, Lemasters GK. NAT2 slow acetylation and bladder cancer in workers exposed to benzidine. Int J Cancer. 2006;118:161–168. doi: 10.1002/ijc.21308. [DOI] [PubMed] [Google Scholar]
- 77.Huang YS, Chern HD, Su WJ, Wu JC, Lai SL, Yang SY, Chang FY, Lee SD. Polymorphism of the N-acetyltransferase 2 gene as a susceptibility risk factor for antituberculosis drug-induced hepatitis. Hepatology. 2002;35:883–889. doi: 10.1053/jhep.2002.32102. [DOI] [PubMed] [Google Scholar]
- 78.Cho HJ, Koh WJ, Ryu YJ, Ki CS, Nam MH, Kim JW, Lee SY. Genetic polymorphisms of NAT2 and CYP2E1 associated with antituberculosis drug-induced hepatotoxicity in Korean patients with pulmonary tuberculosis. Tuberculosis (Edinb) 2007;87:551–556. doi: 10.1016/j.tube.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 79.Makarova SI. Human N-acetyltransferases and drug-induced hepatotoxicity. Curr Drug Metab. 2008;9:538–545. doi: 10.2174/138920008784892047. [DOI] [PubMed] [Google Scholar]
- 80.Hein DW. Molecular genetics and function of NAT1 and NAT2: role in aromatic amine metabolism and carcinogenesis. Mutat Res. 2002;506–507:65–77. doi: 10.1016/s0027-5107(02)00153-7. [DOI] [PubMed] [Google Scholar]
- 81.Brockmoller J, Cascorbi I, Kerb R, Roots I. Combined analysis of inherited polymorphisms in arylamine N-acetyltransferase 2, glutathione S-transferases M1 and T1, microsomal epoxide hydrolase, and cytochrome P450 enzymes as modulators of bladder cancer risk. Cancer Res. 1996;56:3915–3925. [PubMed] [Google Scholar]
- 82.Okkels H, Sigsgaard T, Wolf H, Autrup H. Arylamine N-acetyltransferase 1 (NAT1) and 2 (NAT2) polymorphisms in susceptibility to bladder cancer: the influence of smoking. Cancer Epidemiol Biomarkers Prev. 1997;6:225–231. [PubMed] [Google Scholar]
- 83.Filiadis IF, Georgiou I, Alamanos Y, Kranas V, Giannakopoulos X, Lolis D. Genotypes of N-acetyltransferase-2 and risk of bladder cancer: a case-control study. J Urol. 1999;161:1672–1675. [PubMed] [Google Scholar]
- 84.El Desoky ES, AbdelSalam YM, Salama RH, El Akkad MA, Atanasova S, von Ahsen N, Armstrong VW, Oellerich M. NAT2*5/*5 genotype (341T>C) is a potential risk factor for schistosomiasis-associated bladder cancer in Egyptians. Ther Drug Monit. 2005;27:297–304. doi: 10.1097/01.ftd.0000164197.95494.aa. [DOI] [PubMed] [Google Scholar]
- 85.van der Hel OL, Peeters PH, Hein DW, Doll MA, Grobbee DE, Kromhout D, Bueno de Mesquita HB. NAT2 slow acetylation and GSTM1 null genotypes may increase postmenopausal breast cancer risk in long-term smoking women. Pharmacogenetics. 2003;13:399–407. doi: 10.1097/00008571-200307000-00005. [DOI] [PubMed] [Google Scholar]
- 86.Lubin JH, Kogevinas M, Silverman D, Malats N, Garcia-Closas M, Tardon A, Hein DW, Garcia-Closas R, Serra C, Dosemeci M, Carrato A, Rothman N. Evidence for an intensity-dependent interaction of NAT2 acetylation genotype and cigarette smoking in the Spanish Bladder Cancer Study. Int J Epidemiol. 2007;36:236–241. doi: 10.1093/ije/dym043. [DOI] [PubMed] [Google Scholar]
- 87.Garcia-Martin E. Interethnic and intraethnic variability of NAT2 single nucleotide polymorphisms. Curr Drug Metab. 2008;9:487–497. doi: 10.2174/138920008784892155. [DOI] [PubMed] [Google Scholar]
- 88.Deitz AC, Rothman N, Rebbeck TR, Hayes RB, Chow WH, Zheng W, Hein DW, Garcia-Closas M. Impact of misclassification in genotype-exposure interaction studies: example of N-acetyltransferase 2 (NAT2), smoking, and bladder cancer. Cancer Epidemiol Biomarkers Prev. 2004;13:1543–1546. [PubMed] [Google Scholar]
- 89.Agundez JA, Golka K, Martinez C, Selinski S, Blaszkewicz M, Garcia-Martin E. Unraveling ambiguous NAT2 genotyping data. Clin Chem. 2008;54:1390–1394. doi: 10.1373/clinchem.2008.105569. [DOI] [PubMed] [Google Scholar]
- 90.Dairou J, Petit E, Ragunathan N, Baeza-Squiban A, Marano F, Dupret JM, Rodrigues-Lima F. Arylamine N-acetyltransferase activity in bronchial epithelial cells and its inhibition by cellular oxidants. Toxicol Appl Pharmacol. 2009 doi: 10.1016/j.taap.2009.02.010. 2009 Feb 24. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]