

Abstract
A one-step protocol for the conversion of carboxylic acids to thioesters, using Lawesson’s Reagent, has been developed.
Thioacids represent a well-established and versatile class of organic compounds. Due to their nucleophilicity, thioacids have found broad applications, for instance the synthesis of thiols and thioesters1 as well as in reactions with azides and nitrosulfonamides.2,3 The recognition that thioacids are not only exploitable nucleophiles, but also can function as overall acylating agents under mild conditions, has enabled the development of a number of new methods for the construction of simple amides, as well as amide bonds in the context of peptide ligations.4
In the course of a current program directed toward the synthesis of glycopeptides (and glycoproteins),5 we recently disclosed several strategies which use thioacids as the coupling partners.6 The value of such methods depends on the accessibility of the thioacids. We have now directed our efforts to the development of a new and practical protocol for the selective installation of thioacid functionality.
Among the current methods for synthesizing thioacids, three are most commonly used. One involves pre-formation of activated carboxylic acids, which are converted to thioacids by reaction with hydrosulfide (HS) anion. Another involves prior coupling of the carboxylic acid with either HSFmoc, HSTrt or HSTmob. Removal of the protecting group releases the target thioacid.8 Finally, Boc solid phase peptide synthesis (SPPS) with a thioester solid support can be applied to the preparation of large peptide thioacids.9 The major limitations of these approaches include the toxicity of H2S, required recourse to activating agents, current commercial unavailability of HSFmoc and HSTmob, and the inconvenience factors associated with a multistep protocol to accomplish a simple –OH to –SH interconversion. We describe herein the development of a new and efficient one-step synthesis of thioacids from the corresponding carboxylic acids, using Lawesson’s Reagent (LR).
Lawesson’s Reagent is commercially available, inexpensive, and widely used in organic synthesis, particularly for the transformation of a carbonyl functional group to its thiocarbonyl counterpart.10 Remarkably, there had been no systematic study investigating the preparation of thioacids by direct reaction between carboxylic acid and LR. As outlined in Scheme 1, we postulated that, perhaps, a carboxylic acid (1) could react with LR in a manner analogous to that observed in reaction with alcohols, through a ‘Wittig-like’ mechanism, to provide the corresponding thioacid (2).11
Scheme 1.

Proposed mechanism for thioacid formation with LR.
Accordingly, we initiated a model study with benzoic acid (3, Table 1). In practice, no thioacid was observed when 3 was treated with LR (0.55 equiv) at RT in DCM for 3 hrs (entry 1). Even when the reaction was run overnight (entry 2), only trace amounts of thiobenzoic acid 4 were observed. Since microwave (MW) treatment is often employed for the rapid preparation of thiocarbonyl compounds with LR,12 we wondered whether it might help to facilitate the desired transformation. Indeed, the conversion of 3→4 was accomplished efficiently within 10 min under MW conditions at 100 ºC, in 76% yield (entry 3). A preliminary screening of conditions revealed that solvents, such as DCM, CHCl3, and CH3CN, promote thioacid formation with high levels of efficiency (entries 3, 5, and 6).
Table 1.
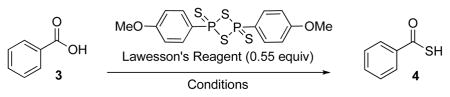 | ||
|---|---|---|
| entry | conditions | yield (%) |
| 1 | DCM, RT, 3h | NR* |
| 2 | DCM, RT, overnight | trace |
| 3 | DCM, microwave, 100 °C, 10 min | 76 |
| 4 | THF, microwave, 100 °C, 10 min | 46 |
| 5 | CHCl3, microwave, 100 °C, 10 min | 73 |
| 6 | CH3CN, microwave, 100 °C, 10 min | 77 |
NR = no reaction.
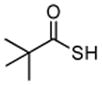
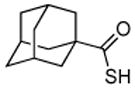
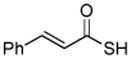
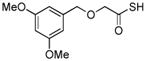
Encouraged by these preliminary results, we next explored the scope of the reaction (Table 2). We found that not only aliphatic thioacids (8 and 12), but also cinnamic thioacid (7) and hindered thioacids (5, 6 and 11) were produced under the conditions described above, in serviceable yields. Furthermore, excellent chemoselectivity was observed in the preparation of amino thioacids (9, 10 and 11). Thus, when 0.55 equivalents of LR were used, the Fmoc protecting group was retained. However, reaction of substrates 13 and 14, possessing more reactive alcohol and primary amide functionalities, respectively, did not yield the desired products (Scheme 2). While the specific reasons for this failure are presently unclear, conceivably, the primary alcohol and primary amides may be competing with the carboxylic acid functionality during the course of the reaction with LR. Alternatively, these functional groups might interdict mechanistic intermediates.
Table 2.
 | |||
|---|---|---|---|
 |
78% | 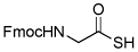 |
82% |
 |
80% | 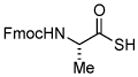 |
83% |
 |
75% | 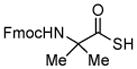 |
80% |
 |
84% | 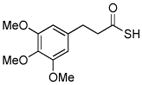 |
94% |
Scheme 2.
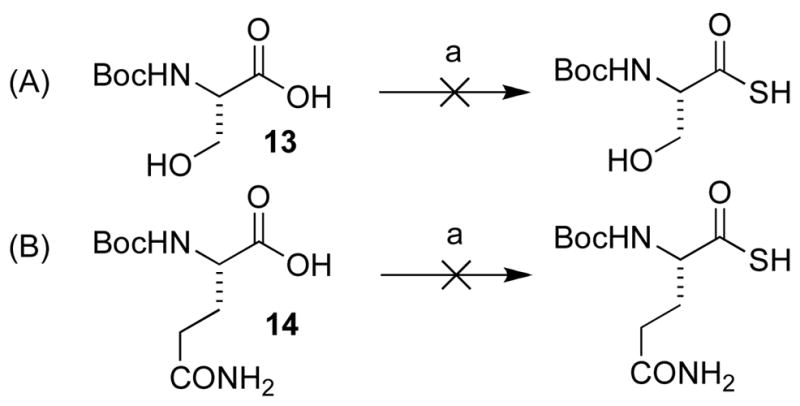
Reagents and conditions: a) DCM, LR, MW, 100 °C, 10 min.
With this new method in hand, we sought to explore its potential applications to the preparation of synthetically valuable thioesters and peptidic thioacids. As illustrated in Scheme 3, an aspartic thioester (16) was efficiently prepared from aspartic acid (15) through a ‘two-step’ procedure, in 72% overall yield. When dipeptide 17 was subjected to LR at RT overnight,13 the corresponding thioacid 18 was obtained, with its internal amide bond intact. However, significant epimerization (possibly resulting from in situ oxazolone formation) was observed.
Scheme 3.

Reagents and conditions: a) DCM, MW, LR 100 °C; b) BnBr, K2CO3, THF/MeOH, rt; (72%, 2 steps); c) LR, DCM, rt, 24h; (68%, 4–5:1 dr).
In summary, a practical new method for the synthesis of thioacids with LR has been developed. This method has been found to be generally useful for the preparation of a wide variety of thioacids. The reaction demonstrates good chemoselectivity for a number of functional groups, such as arenes, olefins, carbamates, esters and amides. It has been successfully demonstrated in the context of the preparation of thioester 16. Although our preliminary results indicate that epimerization may be a problem for the preparation of peptidic thioacids such as 18, solutions to this issue are being pursued. Application of these new capabilities will be described in due course.
Supplementary Material
Acknowledgments
Support was provided by the NIH (CA103823 to SJD). P.N. thanks the National Institutes of Health for a Ruth L. Kirschstein postdoctoral fellowship (CA125934-02). We thank Prof. W. F. Berkowitz for helpful discussions. Special thanks to Ms. Rebecca Wilson for editorial consultation. We acknowledge Ms. Dana Ryan for assistance with the preparation of the manuscript. We thank Dr. George Sukenick, Ms. Hui Fang, and Ms. Sylvi Rusli of the Sloan-Kettering Institute’s NMR core facility for mass spectral and NMR spectroscopic analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mapp AK, Dervan PB. Tetrahedron Lett. 2000;41:9451–9454. [Google Scholar]
- 2.Barlett KN, Kolakowski RV, Katukojvala S, Williams LJ. Org Lett. 2006;8:823–826. doi: 10.1021/ol052671d. [DOI] [PubMed] [Google Scholar]
- 3.Crich D, Sana K, Guo S. Org Lett. 2007;9:4423–4426. doi: 10.1021/ol701583t. [DOI] [PubMed] [Google Scholar]
- 4.(a) Crich D, Sharma I. Angew Chem, Int Ed. 2009;48:2355–2358. doi: 10.1002/anie.200805782. [DOI] [PubMed] [Google Scholar]; (b) Crich D, Sasaki K. Org Lett. 2009;11:3514–3517. doi: 10.1021/ol901370y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Merkx R, van Haren MJ, Rijkers DTS, Liskamp RMJ. J Org Chem. 2007;72:4574–4577. doi: 10.1021/jo0704513. [DOI] [PubMed] [Google Scholar]; (d) Kolakowski RV, Shangguan N, Sauers RR, Williams LJ. J Am Chem Soc. 2006;128:5695–5702. doi: 10.1021/ja057533y. [DOI] [PubMed] [Google Scholar]; (e) Barlett KN, Kolakowski RV, Katukojvala S, Williams LJ. Org Lett. 2006;8:823–826. doi: 10.1021/ol052671d. [DOI] [PubMed] [Google Scholar]; (f) Tam JP, Lu YA, Liu CF, Shao J. Proc Natl Acad Sci USA. 1995;92:12485–12489. doi: 10.1073/pnas.92.26.12485. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Dawson PE, Muir TW, Clark-Lewis I, Kent SBH. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]; (h) Yamashiro D, Li CH. Int J Pept Protein Res. 1988;31:322–334. doi: 10.1111/j.1399-3011.1988.tb00040.x. [DOI] [PubMed] [Google Scholar]; (i) Blake J, Yamashiro D, Ramasharma K, Li CH. Int J Pept Protein Res. 1986;28:468–476. doi: 10.1111/j.1399-3011.1986.tb03281.x. [DOI] [PubMed] [Google Scholar]; (j) Blake J, Li CH. Proc Natl Acad Sci USA. 1981;78:4055–4058. doi: 10.1073/pnas.78.7.4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagorny P, Fasching B, Li X, Chen G, Aussedat B, Danishefsky SJ. J Am Chem Soc. 2009;131:5792–5799. doi: 10.1021/ja809554x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Stockdill JL, Wu X, Danishefsky SJ. Tetrahedron Lett. 2009;50:5152–5155. doi: 10.1016/j.tetlet.2009.06.129. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu X, Yuan Y, Li X, Danishefsky SJ. Tetrahedron Lett. 2009;50:4666–4669. doi: 10.1016/j.tetlet.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yuan Y, Zhu J, Li X, Wu X, Danishefsky SJ. Tetrahedron Lett. 2009;50:2329–2333. doi: 10.1016/j.tetlet.2009.02.205. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Rao Y, Li X, Danishefsky SJ. J Am Chem Soc. 2009;131 doi: 10.1021/ja906005j. ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cronyn MW, Jiu J. J Am Chem Soc. 1952;74:4726. [Google Scholar]
- 8.(a) Crich D, Bowers AA. Org Lett. 2007;9:5323–5325. doi: 10.1021/ol702570x. [DOI] [PubMed] [Google Scholar]; (b) Vetter S. Synth Commun. 1998;28:3219–3223. [Google Scholar]
- 9.(a) Zhang X, Lu X, Liu C. Tetrahedron Lett. 2008;49:6122–6125. [Google Scholar]; (b) Gaertner H, Villain M, Botti P, Canne L. Tetrahedron Lett. 2004;45:2239–2241. [Google Scholar]
- 10.(a) Ozturk T, Ertas E, Mert O. Chem Rev. 2007;107:5210–5278. doi: 10.1021/cr040650b. [DOI] [PubMed] [Google Scholar]; (b) Cava MlP, Levinson MI. Tetrahedron. 1985;41:5061–5087. [Google Scholar]; (c) Schmidt A, Merkel L, Eisfeld W. Eur J Org Chem. 2005;10:2124–2130. [Google Scholar]
- 11.Nishio T. Chem Commun. 1989;4:205–206. [Google Scholar]
- 12.(a) Varma RS, Kumar D. Org Lett. 1999;1:697–700. doi: 10.1021/ol990629a. [DOI] [PubMed] [Google Scholar]; (b) Filippi JJ, Fernandez X, Lizzani-Cuvelier L, Loiseau AM. Tetrahedron Lett. 2003;44:6647–6650. [Google Scholar]
- 13.When the reaction was carried out under the microwave conditions, extensive decomposition of the starting material, 17, and a low yield of 18 were observed. It is highly possible that the internal amide group may be competing with the carboxylic acid functionality during the course of the reaction with LR under the microwave conditions. However, the corresponding reaction was found to proceed at room temperature, albeit at a slower rate. There is a possibility that the superior reactivity of 17 at room temperature is facilitated by the intermediacy of the oxazolone. Further studies, which will shed some light into the origins of this effect, will be reported in a due course.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.