

Abstract
The biological importance of microtubules in mitosis and cell division makes them an interesting target for the development of anticancer agents. Small molecules such as benzo[b]furans are attractive as inhibitors of tubulin polymerization. Thus, a new class of inhibitors of tubulin polymerization based on the 2-(3′,4′,5′-trimethoxybenzoyl)-benzo[b]furan molecular skeleton, with electron-donating (Me, OMe or OH) or electron-withdrawing (F, Cl and Br) substituents on the benzene ring, was synthesized and evaluated for antiproliferative activity, inhibition of tubulin polymerization and cell cycle effects. Adding a methyl group at the C-3 position resulted in increased activity. The most promising compound in this series was 2-(3′,4′,5′-trimethoxybenzoyl)-3-methyl-6-ethoxy-benzo[b]furan, which inhibits cancer cell growth at nanomolar concentrations and interacts strongly with tubulin by binding to the colchicine site.
Keywords: Benzo[b]furan derivatives, Antitubulin agents, Combretastatin-A4, Colchicine, Antiproliferative agents
1. Introduction
The microtubule system of eukaryotic cells plays important roles in regulating cell architecture, and it has an essential role in cell division, since microtubules are a key component of the mitotic spindle.1 Microtubules are a dynamic cellular compartment in both neoplastic and normal cells. This dynamicity is characterized by the continuous turnover of αβ-tubulin heterodimers in the polymeric microtubules. They are involved in a variety of essential cellular processes, such as regulation of motility, cell signaling, formation and maintenance of cell shape, as well as transport of material within the cell.2 Numerous chemically diverse antimitotic agents, many of which are derived from natural products, have been found to interact specifically with tubulin.3
Among the microtubule depolymerizing agents, combretastatin A-4 (CA4, 1; Chart 1) is one of the more studied compounds. CA4, isolated from the bark of the South African tree Combretum caffrum,4 strongly inhibits the polymerization of tubulin by binding to the colchicine site.5 Because of its structural simplicity, a wide number of CA4 analogues have been developed and evaluated in SAR studies.6
Chart 1.

Inhibitors of tubulin polymerization.
Among synthetic small molecule tubulin inhibitors, replacement of the olefinic bridge of 1 with a carbonyl group furnished a benzophenone-type CA4 analogue named phenstatin (2a). This compound demonstrated interesting efficacy in a variety of tumor models, while retaining the characteristics of 1.7 The 2-aminobenzophenone derivative 2b also strongly inhibited cancer cell growth and tubulin polymerization and caused mitotic arrest, as did 2a.8
In an earlier publication, we reported a series of methoxy-substituted 2-(3′,4′,5′-trimethoxybenzoyl)-benzo[b]furan derivatives with general structure 3, with an amino or dimethylamino substituent at the 3-position of the benzo[b]furan skeleton (compounds 3a and 3b, respectively) as a new class of antimitotic agents.9a The concomitant presence of a methoxy group at the 6-position contributed to maximal activity. These compounds, along with the corresponding benzo[b]thiophene9b,c and indole9d derivatives, inhibited the growth of different cancer cell lines and tubulin polymerization by binding to the colchicine site of tubulin and caused G2–M phase arrest of the cell cycle. The higher potency of the 3-dimethylamino derivative 3b allowed us to verify that an intramolecular hydrogen bond between the unsubstituted 3-amino group and the carbonyl oxygen of the 2-trimethoxybenzoyl moiety is not required for activity. These findings prompted us to study this class of compounds in more detail. We herein describe the synthesis and structure–activity relationship of 2-(3′,4′,5′-trimethoxybenzoyl)-benzo[b]furan derivatives in the continuation of our search for new potent antitubulin agents. We should note that previous studies have yielded a limited series of tubulin inhibitors with the benzo[b]furan molecular skeleton as the core structure. One of these compounds has structure 3c, which incorporates the 3-(3,4,5-trimethoxybenzoyl)-6-methoxybenzo[b] furan ring system.
In the present investigation, the 3′,4′,5′-trimethoxyphenyl of the 2-benzoyl moiety was kept unchanged because it is the characteristic structural requirement for activity in a numerous inhibitors of tubulin polymerization, such as colchicine, CA4 and podophyllo-toxin.6 In the current studies we examined the importance of the 3-position of the benzo[b]furan skeleton by studying the effects of replacing the amino or dimethylamino substituents of compounds with general structure 3 with a hydrogen or a methyl group, to furnish derivatives with general structure 4. Additional structure modifications were focused on the C-4/C-7 positions, as well as on the 2-carbonyl linker of the benzo[b]furan skeleton.
In a first series (derivatives 4a–x), the 3-amino group of the benzo[b]furan system was replaced by a hydrogen (4a–n) or a methyl (4o–x). These molecules possessed either no substituent (4a and 4o) or a methoxy group at each of the four possible positions on the benzene ring (compounds 4b–e and 4p–s). In an effort to design compounds with improved polarity, we decided to introduce an additional hydroxy group at the C-7 position of compound 4d, to obtain the C-6 methoxy, C-7 hydroxy derivative 4i. An increase of the polarity in the 2-aroyl benzo[b]furan structure can be also obtained replacing the methoxy group of compounds 4b–e and 4p–s with a more hydrophilic hydroxyl moiety, to obtain the derivatives 4l–n and 4v–x. The hydroxyl moiety provides a site for the preparation of a phosphate prodrug, similar to what has been successfully done in the case of CA4 phosphate.10 Keeping the C-6 methoxy group intact, compounds 4j and 4k were prepared with the aim of evaluating the effect on biological activity of one (4j) or two (4k) additional methoxy substituents at the C-5 and C-5, 7 positions, respectively. Besides the C-5 methoxy moiety, the substituents examined included electron-donating methyl (4f) and electron-withdrawing chlorine and bromine (4g and 4h, respectively) groups. Finally, the 6-methoxy position (corresponding to the 4-methoxy group in the B-ring of CA4 and 2-aminobenzophenone 2b) of 4r was further studied by synthesizing the corresponding 6-ethoxy (4t) and 6-fluorine (4u) analogues.
Starting from compound 4d, additional analogues were generated by modification of the 2-carbonyl function, which was converted into thiocarbonyl (4y), reduced to carbinol (4z) and to methylene (4ab) or transformed into a methyl ether (4aa).
Finally, by the synthesis of tetracyclic compound 4ac, the phenyl of the 2-(3′,4′,5′-trimethoxy)benzoyl moiety and the 3-position of the benzo[b]furan skeleton were locked in a conformation in which these two ring were forced to be coplanar.
We should note that previous studies have yielded a limited series of tubulin inhibitors with the benzo[b]furan molecular skeleton as the core structure. These compounds have general structure 6, which incorporates the 3-(3,4,5-trimethoxybenzoyl)-6-methoxybenzo[b]furan ring system.11
2. Chemistry
The 2-(3′,4′,5′-trimethoxybenzoyl)benzo[b]furan derivatives 4a–l, 4n–w, 6m and 6x were synthesized by a ‘one-step’ condensation of the corresponding, variously substituted salicylaldehydes 5a-n or 2-hydroxyacetophenones 5o–x with 2-bromo-1-(3,4,5-trimethoxyphenyl)ethanone9b and anhydrous potassium carbonate in refluxing acetone (Scheme 1). For the tert-butyldimethylsilyl (TBDMS) analogues 5l, 5n and 5v–w, the cyclization was accomplished by the concomitant removal of TBDMS-protecting group, to afford derivatives 4l, 4n and 4v–w. The 6-hydroxy-2-aroyl benzo[b]furan derivatives 4m and 4x were obtained by cleavage of the 6-benzyloxy group of 6m and 6x, respectively, with a mixture of activated palladium on charcoal and ammonium formate.
Scheme 1.

Reagents and conditions: (a) (3,4,5-Trimethoxyphenyl)-2-bromo-ethanone, K2CO3, (CH3)2CO, rt; (b) Lawesson's reagent, THF, rt from 4d; (c) HCO2NH4,10% Pd/C, MeOH, rt from 6m and 6x; (d) NaBH4, MeOH, rt from 4d; (e) PTSA, MeOH-THF, rt; (f) Et3SiH, TFA, CH2Cl2, rt; (g) NBS, benzoyl peroxide, MeCN, rt from 4d; (h) Pd(Ph3P)4, KOAc, DMA, 130 °C. For compounds 5l, 5n and 5v–w, condition ‘a’ led to a cyclization with concomitant removal of TBDMS group, to afford 4l, 4n and 4v–w, respectively.
The treatment of the 2-carbonyl moiety of 4d with Lawesson's reagent or its reduction with sodium borohydride furnished the thiocarbonyl or the carbinol derivatives 4y and 4z, respectively.12 This latter compound was further converted to the methyl ether 4aa by treatment with pyridinium p-toluensulfonate in a mixture of MeOH-THF or reduced to the methylene derivative 4ab with triethylsilane in trifluoroacetic acid. The cyclic ketone 4ac was synthesized by intramolecular cross-coupling of bromo derivative 7 catalyzed with palladium (0) tetrakistriphenylphosphine in N,N-dimethylacetamide at 130 °C. Derivative 7 was obtained by the chemoselective bromination of 4d with NBS in acetonitrile.13
3. Biological results and discussion
Table 1 summarizes the growth inhibitory effects of 2-(3′,4′,5′-trimethoxy)benzo[b]furan derivatives 4a–ac against murine leukemia (L1210), murine mammary carcinoma (FM3A), human T-lymphoblastoid (Molt/4 and CEM) and human cervix carcinoma (HeLa) cells, with CA4 (1), 3a and 3b as reference compounds. The 3-methyl-6-ethoxy derivative 4t possessed the highest potency, inhibiting the growth of L1210, FM3A, Molt/4, CEM and HeLa cells with IC50 values of 2.0, 2.8, 1.2, 2.8 and 6.3 nM,respectively. These values are similar to those obtained with CA4 and from 20- to 100-fold higher activity than those obtained with the 3-amino derivatives 3a and 3b. However, this marked improvement in activity relative to 3a–b may derive in part from the ethoxy substituent at C-6, since the analogue with a methoxyl at C-6 (4r) was approximately 10-fold less active than 4t in four of the cell lines.
Table 1.
In vitro inhibitory effects of compounds 3a–b, 4a–ac and CA4 against the proliferation of murine leukemia (L1210), murine mammary carcinoma (FM3A), human T-lymphocyte (Molt/4 and CEM) and human cervix carcinoma (HeLa) cells
| Compound | IC50 (nM)a | ||||
|---|---|---|---|---|---|
| L1210 | FM3A/0 | Molt4/C8 | CEM/0 | Hela | |
| 4a | >10,000 | >10,000 | 8000 ± 100 | >10,000 | 7900 ± 100 |
| 4b | 410 ± 23 | 550 ± 42 | 360 ± 10 | 870 ± 50 | 410 ± 16 |
| 4c | >10,000 | >10,000 | 9400 ± 500 | >10,000 | >10,000 |
| 4d | 90 ± 2 | 100 ± 10 | 59 ± 12 | 70 ± 21 | 110 ± 20 |
| 4e | >10,000 | >10,000 | 7800 ± 290 | >10,000 | >10,000 |
| 4f | 7100 ± 580 | 8400 ± 300 | 2500 ± 160 | 7200 ± 210 | >10 |
| 4g | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 |
| 4h | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 |
| 4i | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 |
| 4j | 1800 ± 400 | 1200 ± 400 | 770 ± 20 | 1900 ± 400 | 570 ± 110 |
| 4k | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 |
| 4l | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 |
| 4m | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 |
| 4n | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 |
| 4o | 2100 ± 200 | 2100 ± 10 | 1300 ± 80 | 2000 ± 50 | 890 ± 130 |
| 4p | 590 ± 40 | 570 ± 30 | 200 ± 70 | 490 ± 10 | 340 ± 30 |
| 4q | 2300 ± 400 | 4400 ± 390 | 1900 ± 100 | 400 ± 150 | 1900 ± 200 |
| 4r | 24 ± 0 | 27 ± 7 | 19 ± 2 | 20 ± 2 | 3.2 ± 0.6 |
| 4s | 1700 ± 400 | 1700 ± 100 | 400 ± 30 | 2400 ± 150 | 530 ± 28 |
| 4t | 2.0 ± 1.2 | 2.8 ± 1.0 | 1.2 ± 0.8 | 2.8 ± 0.6 | 6.3 ±2.7 |
| 4u | 3500 ± 200 | 3700 ± 100 | 2100 ± 0 | 2700 ± 200 | 2000 ± 0 |
| 4v | 480 ± 32 | 330 ± 10 | 84 ± 21 | 100 ± 10 | 130 ± 24 |
| 4w | 800 ± 31 | 430 ± 27 | 240 ± 30 | 430 ± 0 | 290 ± 10 |
| 4x | >10,000 | >10,000 | 1300 ± 90 | 4300 ± 300 | 260 ± 16 |
| 4y | 180 ± 0 | 320 ± 40 | 100 ± 10 | 220 ± 80 | 100 ± 25 |
| 4z | >10,000 | 7009 ± 530 | 3005 ± 120 | >10,000 | >10,000 |
| 4aa | >10,000 | >10,000 | >10,000 | >10,000 | 4400 ± 290 |
| 4ab | 2300 ± 400 | 2600 ± 600 | 770 ± 43 | 1500 ± 300 | 680 ± 10 |
| 4ac | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 |
| 3a | 430 ± 40 | 280 ± 160 | 140 ± 20 | 87 ± 22 | n.d. |
| 3b | 65 ± 5 | 59 ± 5 | 48 ± 4 | 78 ± 3 | n.d. |
| CA4 | 2.8 ± 1.1 | 42 ± 6.0 | 16 ± 1.4 | 1.9 ± 1.6 | 1.9 ± 1.6 |
n.d. = not determined.
IC50 = Compound concentration required to inhibit tumor cell proliferation by 50%. Data are expressed as the mean ± SE from the dose–response curves of at least three independent experiments.
Comparing the 6-methoxy derivatives 3a–b, 4d and 4r, the order of activity for the substituent at the 3-position of the benzo[b]furan moiety was methyl (4r) > hydrogen (4d) ≈ dimethylamino (3b) > amino (3a). With the four compounds IC50 values ranged from 3 to 27 nM for 4r, from 59 to 110 nM for 4d, from 48 to 78 for 3b, and from 87 to 430 nM for 3a. Comparing the activities of the carbonyl derivative 4d with those of derivatives 4y–z and 4aa–ab indicated that the 2-carbonyl moiety could only be replaced with a thiocarbonyl group (4y) among the bridging moieties examined.
Comparing the 3-unsubstituted derivatives 4a–e and 4l–m with their 3-methyl congeners 4o–s and 4w–x, the introduction of a methyl at the C-3 position of the benzo[b]furan system generally increased antiproliferative activity against all five cell lines. The antiproliferative activities were also dependent on the substitution pattern on the benzene part of the benzo[b]furan moiety, with the most favorable position being C-6 for a methoxy group (4r was more active than 4p, 4q or 4s, as was observed previously with the 3-aminobenzo[b]furan derivatives 3a–b), while C-4 was the best position for an hydroxyl group (4v more active than 4w or 4x). In the absence of the C-3 methyl group, the derivative with the C-6 methoxyl moiety (4d) was more active than analogues with the methoxy at other positions (4b, 4c and 4e). Additional methoxy groups on the benzene ring (4j and 4k) resulted in sharply reduced potency relative to 4d. The introduction of an additional hydroxyl group at the C-7 position of 4d, to furnish 4i, caused a substantial loss of activity.
In the series of 3-unsubstituted derivatives, the only compound with a methoxy moiety with significant activity was 4d and replacing the methoxy with the weaker electron-donating and more hydrophilic hydroxy group (compound 4m) resulted in a drastic loss of activity. With the 3-methyl analogues 4p–r, the substitution of the methoxy group at the C-4 or C-5 positions (4v and 4w) with the hydroxy function caused an increase in the antiproliferative activity. A dramatic loss of activity was only observed when the C-6 methoxy (4r) was replaced with an hydroxy group (4x).
For the inactive C-5 substituted derivative 4c, replacing the strong electron-donating methoxy function with the electron-withdrawing chlorine (4g) or bromine (4h) groups had no effect on activity, but replacement of the C-5 methoxy group with the moderately electron-donating methyl moiety (4f) slightly increased activity.
As noted above, the activity of 4r was increased 10-fold against L1210, FM3A, Molt/4 and CEM cells and 2-fold for HeLa cells when the C-6 methoxy group was replaced with an ethoxy moiety (4t). In contrast, an electron-withdrawing fluorine group at C-6 (4u) caused a dramatic reduction of antiproliferative activity.
Linking the ortho-position of the phenyl of the 2-(3′,4′,5′-trimethoxybenzoyl)moiety, to the 3-position of benzo[b]furan moiety, to yield the conformationally constrained derivative 4ac, led to a completely inactive compound.
To confirm that the antiproliferative activities of these compounds were related to an interaction with the microtubule system, the most active compounds, 4b, 4d, 4p–r, 4t, 4v and 4w, along with the 6-fluoro derivative 4u, were evaluated for their in vitro inhibition of tubulin polymerization and for their inhibitory effects on the binding of [3H]colchicine to tubulin (in the latter assay, tubulin was examined at a concentration of 1 μM, while compounds and colchicine were at 5 μM).14,15 For comparison, CA4 was examined in contemporaneous experiments as a reference compound (Table 2). The benzo[b]furan derivatives 4d, 4r, 4t and 4v with IC50 values of 0.70, 0.55, 0.43 and 0.57 μM, respectively, exhibited antitubulin activity about two times greater than that of CA4 (1.0 μM), while 4b, 4q and 4w had IC50 values of 1.1–1.2 μM, essentially equivalent to that of CA4.
Table 2.
Inhibition of tubulin polymerization and colchicine binding by compounds 4b, 4d, 4p–r, 4t–w and CA4
| Compound | Tubulin assemblya IC50 ± SD (μM) | Colchicine bindingb % ± SD |
|---|---|---|
| 4b | 1.1 ± 0.1 | 76 ± 2 |
| 4d | 0.70 ± 0.08 | 79 ± 0 |
| 4p | 1.8 ± 0.2 | 66 ± 0 |
| 4q | 1.2 ± 0.1 | 65 ± 0 |
| 4r | 0.55 ± 0.03 | 97 ± 1 |
| 4t | 0.43 ± 0.06 | 96 ± 1 |
| 4u | 6.9 ± 0.8 | n.d. |
| 4v | 0.57 ± 0.03 | 85 ± 1 |
| 4w | 1.1 ± 0.1 | 69 ± 3 |
| CA4 (1) | 1.0 ± 0.1 | 99 ± 2 |
n.d.: not determined.
Inhibition of tubulin polymerization. Tubulin was at 10 μM.
Inhibition of [3H]colchicine binding. Tubulin, colchicine and tested compound were at 1, 5 and 5 μM,respectively.
The order of inhibitory action on tubulin assembly was 4t > 4r > 4v > 4d > CA4 > 4b > 4w > 4q > 4p ≫ 4u, which was consistent with the results of the antiproliferative assays, except that 4d was more potent than 4v and 4p was more potent than 4q. The most potent compound in this series was compound 4t, with an IC50 value of 0.43 μM, which correlates well with its having the greatest antiproliferative activity. Compounds 4b, 4q and 4w were as active as CA4 as inhibitors of tubulin assembly, although both compounds were less active in their effects on cell growth. Compound 4u showed weak antitubulin polymerization activity, which is consistent with its low antiproliferative activity.
In the colchicine binding studies, compounds 4r and 4t strongly inhibited the binding of [3H]colchicine to tubulin, since 97% and 96% inhibition, respectively, occurred with these agents and colchicine, both at 5 μM. These derivatives were as active as CA4, which in these experiments inhibited colchicine binding by 99%. These data indicate that 4r and 4t strongly bind to the colchicine site on tubulin.
Because molecules exhibiting effects on tubulin assembly should cause alteration of cell cycle parameters with preferential G2–M blockade, flow cytometry analysis was performed to determine the effect of the compounds 4b, 4d, 4p–r, 4t and 4u–w on K562 (human chronic myelogenous leukemia) cells. Cells were cultured for 24 h in the presence of each compound at the concentration able to inhibit 100% cell growth after 24 h (4b = 980 nM, 4d and 4p = 420 nM, 4q = 230 nM, 4r = 26 nM, 4t = 16 nM, 4u = 1.62 μM, 4v = 530 nM, 4w = 480 nM). Analysis of sub-G0–G1 (apoptotic peak, A), G0–G1, S, and G2–M peaks revealed that the compounds caused somewhat different effects on cell cycle distribution (Fig. 1). All tested compounds caused an increase in the proportion of cells in the G2–M peak relative to the untreated control, and compounds 4p and 4u also caused cells to accumulate in late S phase. For compounds 4b, 4p. 4u and 4w, the accumulation of cells in G2–M phase was accompanied by the appearance of a significant sub-G0–G1 peak (A = 20–44%) due to apoptosis.
Figure 1.
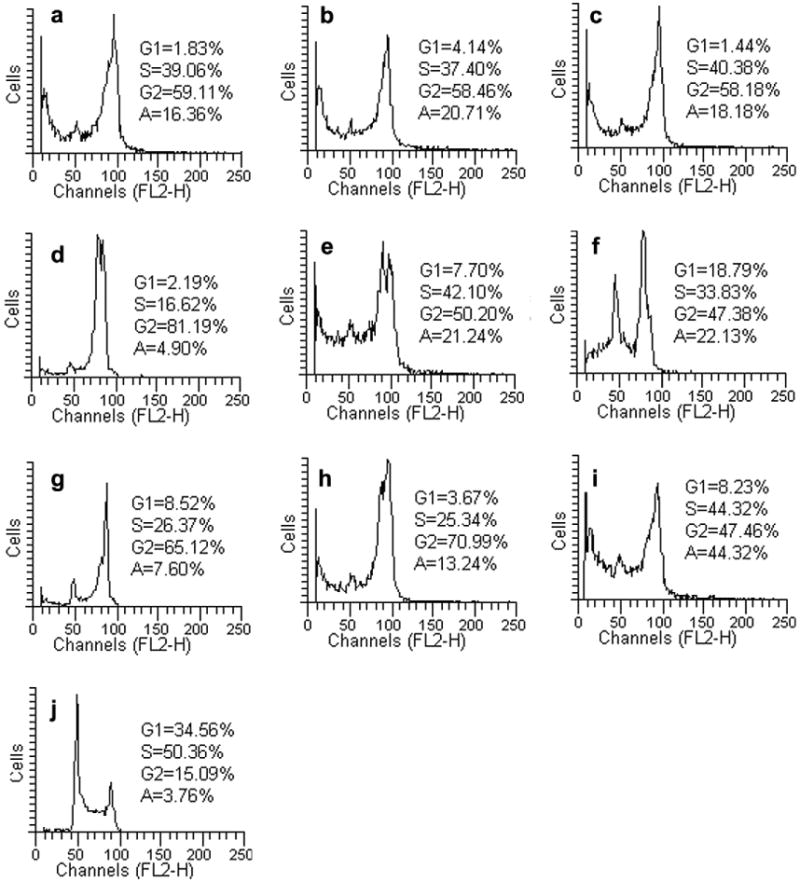
Effects of compounds 4d (a), 4b (b), 4q (c), 4r (d), 4p (e), 4w (f), 4v (g), 4t (h) and 4u (i) on DNA content/cell following the treatment of K562 cells for 24 h. Cell cycle distribution was analyzed by the standard propidium iodide procedure as described in Section 5. Control is reported in Figure 1 as panel j.
Molecular docking studies on this series of compounds in the colchicine site of tubulin were also performed. There was a comparable binding mode of these compounds to that of DAMA-colchicine co-crystallized in the tubulin structure used,16 with the trimethoxyphenyl moiety occupying the same pocket as the corresponding ring A of the colchicinoid (Fig. 2).
Figure 2.
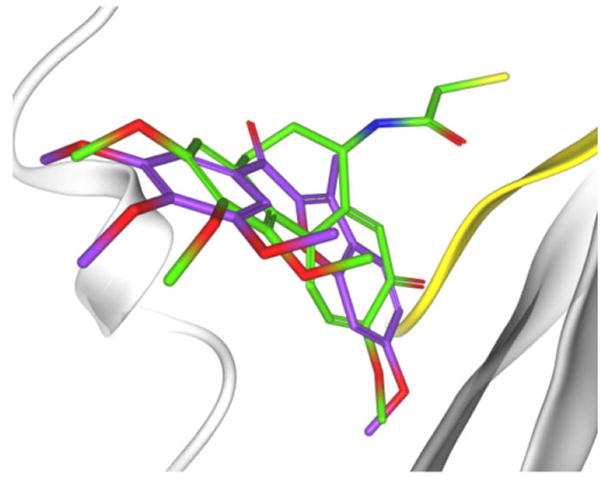
Putative binding mode of compound 4r (colored in purple); DAMA-colchicine colored in green.
The substituent in the position 6 of the benzofuran analogues, like the methoxy group on ring C of colchicinoids, falls into a hydrophobic region deep in the binding site (shown in green in Fig.3). Furthermore, a polar area on the tubulin surface corresponding to the carbonyl group of Thr179 (residue numbering derived from the crystal structure used) with potential for accepting a hydrogen bond is close to the C-4 position of the benzo[b]furan group (shown in purple in Fig. 3). This feature could be exploited by the hydroxyl group of 4v to establish a hydrogen bond with tubulin, which could compensate to the loss of the hydrophobic contact provided by the C-6 methoxy group, which is missing in 4v, and justify the activity of this compound in the tubulin polymerization assay. It should be mentioned that the position observed for the hydroxyl group of 4v is similar to the proposed binding location of the corresponding functional group of CA4, although in the case of 4v is the carbonyl group of Thr179 that is closer to the inhibitor (2.54 Å), while for CA4 the hydroxyl group is placed close to Val181 (see Fig. 1, Supplementary data).17
Figure 3.
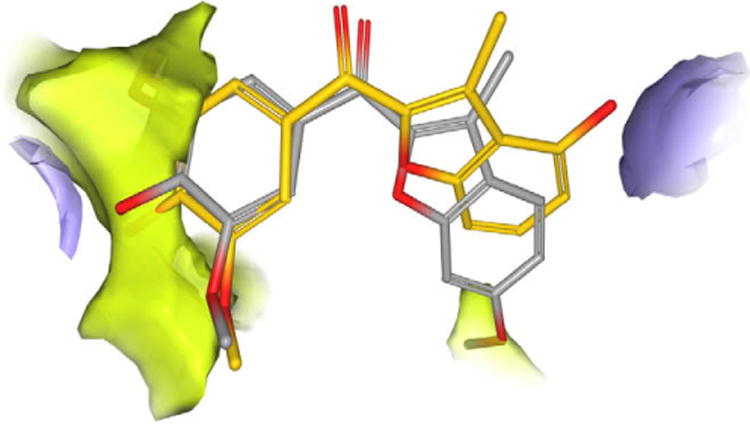
Putative binding mode of compound 4r (colored in gray) and 4v (colored in yellow). Lipophilic potential map represented in green; H-bond donor potential map represented in purple.
4. Conclusions
The SAR information indicated that the introduction of methyl at the C-3 position of the benzo[b]furan moiety resulted in increased activity compared with the corresponding 3-amino counterpart,9a thus revealing that this latter substituent is not essential for activity. Several compounds (4d, 4r, 4t and 4v) showed excellent activity as inhibitors of tubulin polymerization, and were more potent than CA4 in this assay. The interaction with tubulin leads to cell cycle arrest in the G2–M phase and to an apoptotic cell death. Our structure–activity relationship study indicates that a methoxy group located at the C-6 position of the benzo[b]-furan ring yields the most active compound. Changing its position from C-6 to C-4, C-5 or C-7 led to a reduction in potency. Replacement of methoxy by the more hydrophilic hydroxyl group turned out to be detrimental to potency in the case of the 2-unsubstituted benzo[b]furan series. The 2-(3′,4′,5′-trimethoxybenzoyl)-3-methyl-6-ethoxybenzo[b]furan derivative 4t was the most potent analogue, with IC50 values ranging from 1.2 to 6.3 nM, in the same range as the values obtained with CA4. Compound 4t also had excellent potency as an inhibitor of tubulin polymerization (IC50 = 0.43 μM). At the 2-position of the benzo[b]furan, the linker is much more effective as a carbonyl than as carbinol, methoxymethyl or simple methylene group. A thiocarbonyl linker, however, was compatible with good activity.
5. Experimental
5.1. Chemistry
5.1.1. Materials and methods
2-Hydroxybenzaldehyde (5a), 2-hydroxy-6-methoxybenzaldehyde (5b), 2-hydroxy-5-methoxybenzaldehyde (5c), 2-hydroxy-4-methoxybenzaldehyde (5d), 2-hydroxy-4-methoxybenzaldehyde (5e), 2-5-hydroxy-5-methylbenzaldehyde (5f), 2-hydroxy-5-chlorobenzaldehyde (5g), 2-hydroxy-5-bromobenzaldehyde (5h), 2-hydroxy-3-methoxybenzaldehyde (5i), 2-hydroxy-4-(benzyloxy) benzaldehyde (5m), 1-(2-hydroxyphenyl)ethanone (5o), 2-hydroxy-6-methoxyacetophenone (5p), 2-hydroxy-5-methoxyacetophenone (5q), 2-hydroxy-4-methoxyacetophenone (5r), 2-hydroxy-4-ethoxyacetophenone (5t), 2-hydroxy-4-fluoroacetophenone (5u) are commercially available and were used as received. For the preparation of 5j–l, 5n, 5s, 5v–x see Ref. 18.
1H NMR spectra were recorded on a Bruker AC 200 spectrometer. Chemical shifts (δ) are given in ppm upfield from tetramethylsilane as internal standard, and the spectra were recorded in appropriate deuterated solvents, as indicated. Melting points (mp) were determined on a Buchi-Tottoli apparatus and are uncorrected. All products reported showed 1H NMR spectra in agreement with the assigned structures. Elemental analyses were conducted by the Microanalytical Laboratory of the Chemistry Department of the University of Ferrara. Mass spectra were obtained by electrospray ionisation (ESI) in positive mode using a ESI Micromass ZMD 2000 mass spectrometer. All reactions were carried out under an inert atmosphere of dry nitrogen, unless otherwise described. Standard syringe techniques were applied for transferring dry solvents. Reaction courses and product mixtures were routinely monitored by TLC on silica gel (precoated F254 Merck plates) and visualized with aqueous KMnO4. Flash chromatography was performed using 230–400 mesh silica gel and the indicated solvent system. Organic solutions were dried over anhydrous Na2SO4. Calcium chloride was used in the distillation of DMF, and the distilled solvent was stored over molecular sieves (3 Å).
5.2. General procedure for the synthesis of 2-(3′,4′,5′-trimethoxybenzoyl)-3-amino benzofuranes (4a–l, 4n–w, 6m and 6x)
To a solution of the appropriate substituted salicylaldehyde 5a–n or 2-hydroxyacetophenone 5o–x (1 mmol) in dry acetone (15 mL) was added 2-bromo-1-(3,4,5-trimethoxyphenyl)ethanone (289 mg, 1 mmol) and anhydrous potassium carbonate (276 mg, 2 mmol) while stirring, and the reaction mixture was refluxed for 18 h. After cooling, the reaction mixture was evaporated, and the residue was dissolved in a mixture of dichloromethane (15 mL) and water (5 mL). The organic layer was washed with brine, dried and concentrated under reduced pressure to obtain a residue, which was purified by flash column chromatography. The final product was recrystallized from petroleum ether.
5.2.1. (Benzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (4a)
The residue was chromatographed with EtOAc/petroleum ether 3:7 as eluent to give 4a as a white solid, yield: 93%, mp 94–96 °C 1H NMR (CDCl3) δ: 3.95 (s, 6H), 3.96 (s, 3H), 7.36 (m, 3H), 7.50 (t, J = 8.0 Hz, 1H), 7.53 (s, 1H), 7.64 (d, J = 8.0 Hz, 1H), 7.76 (d, J = 8.0 Hz, 1H). Anal. Calcd for C18H16O5: C, 69.22; H, 5.16. Found: C, 69.01; H, 5.02.
5.2.2. (4-Methoxybenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (4b)
The residue was chromatographed with EtOAc/petroleum ether 3:7 as eluent to give 4b as a white solid, yield: 85%, mp 118–120 °C. 1H NMR (CDCl3) δ: 3.95 (s, 3H), 3.96 (s, 6H), 3.98 (s, 3H), 7.12 (m, 2H), 7.34 (s, 2H), 7.49 (s, 1H), 7.52 (d, J = 9.2 Hz, 1H). Anal. Calcd for C19H18O6: C, 66.66; H, 5.30. Found: C, 66.47; H, 5.12.
5.2.3. (5-Methoxybenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (4c)
The residue was chromatographed with EtOAc/petroleum ether 2:8 as eluent to give 4c as a white solid, yield: 83%, mp 154–156 °C. 1H NMR (CDCl3) δ: 3.88 (s, 3H), 3.96 (s, 6H), 3.97 (s, 3H), 6.70 (d, J = 7.6 Hz, 1H), 7.25 (m, 1H), 7.32 (s, 2H), 7.43 (d, J = 8.4 Hz, 1H), 7.62 (d, J = 1.0 Hz, 1H). Anal. Calcd for C19H18O6: C, 66.66; H, 5.30. Found: C, 66.38; H, 5.03.
5.2.4. (6-Methoxybenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (4d)
The residue was chromatographed with EtOAc/petroleum ether 3:7 as eluent to give 4d as a white solid, yield: 89%, mp 115–117 °C. 1H NMR (CDCl3) δ: 3.99 (s, 3H), 3.94 (s, 6H), 3.95 (s, 3H), 6.97 (dd, J = 8.8 and 2.2 Hz, 1H), 7.01 (d, J = 2.4 Hz, 1H), 7.29 (s, 2H), 7.48 (s, 1H), 7.60 (d, J = 8.8 Hz, 1H). Anal. Calcd for C19H18O6: C, 66.66; H, 5.30. Found: C, 66.42; H, 5.14.
5.2.5. (7-Methoxybenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (4e)
The residue was chromatographed with EtOAc/petroleum ether 3:7 as eluent to give 4e as a yellow solid, yield: 95%, mp 116–118 °C 1H NMR (CDCl3) δ: 3.95 (s, 3H), 3.96 (s, 6H), 4.03 (s, 3H), 6.96 (d, J = 7.6 and 2.0 Hz, 1H), 7.27 (t, J = 7.6 Hz, 1H), 7.33 (d, J = 7.6 Hz, 1H), 7.44 (s, 2H), 7.57 (s, 1H). Anal. Calcd for C19H18O6: C, 66.66; H, 5.30. Found: C, 66.39; H, 5.09.
5.2.6. (3,4,5-Trimethoxyphenyl)(5-methylbenzofuran-2-yl)methanone (4f)
The residue was chromatographed with EtOAc/petroleum ether 3:7 as eluent to give 4f as a white solid, yield: 88%, mp 106–107 °C 1H NMR (CDCl3) δ: 2.47 (s, 3H), 3.94 (s, 6H), 3.96 (s, 3H), 7.30 (d, J = 9.2 Hz, 1H), 7.33 (s, 2H), 7.47 (s, 1H), 7.52 (m, 2H). Anal. Calcd for C19H18O5: C, 69.93; H, 5.56. Found: C, 69.77; H, 5.33.
5.2.7. (5-Chlorobenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (4g)
The residue was chromatographed with EtOAc/petroleum ether 2:8 as eluent to give 4g as a white solid, yield: 72%, mp 136–138 °C. 1H NMR (CDCl3) δ: 3.95 (s, 6H), 3.97 (s, 3H), 7.33 (s, 2H), 7.44 (m, 2H), 7.56 (d, J = 8.8 Hz, 1H), 7.72 (d, J = 2.0 Hz, 1H). Anal. Calcd for C18H15ClO5: 62.35; H, 4.36; Cl, 10.22. Found: 62.06; H, 4.14; Cl, 10.01.
5.2.8. (5-Bromobenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (4h)
The residue was chromatographed with EtOAc/petroleum ether 2:8 as eluent to give 4h as a white solid, yield: 83%, mp 144–146 °C. 1H NMR (CDCl3) δ: 3.94 (s, 6H), 3.96 (s, 3H), 7.33 (s, 2H), 7.47 (s, 1H), 7.50 (d, J = 8.8 Hz, 1H), 7.56 (dd, J = 8.8 and 2.0 Hz, 1H), 7.88 (d, J = 2.0 Hz, 1H). Anal. Calcd for C18H15BrO5: C, 55.26; H, 3.86; Br, 20.42. Found: C, 55.02; H, 3.68; Br, 20.17.
5.2.9. (7-Hydroxy-6-methoxybenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (4i)
The residue was chromatographed with EtOAc/petroleum ether 4:6 as eluent to give 4i as a white solid, yield: 83%, mp 128–130 °C. 1H NMR (CDCl3) δ: 3.91 (s, 3H), 3.93 (s, 6H), 3.95 (s, 3H), 6.97 (d, J = 8.6 Hz, 1H), 7.28 (s, 2H), 7.32 (d, J = 8.6 Hz, 1H), 7.50 (s, 1H), 10.2 (s, 1H). Anal. Calcd for C19H18O7: C, 63.68; H, 5.06. Found: C, 63.50; H, 4.88.
5.2.10. (5,6-Dimethoxybenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (4j)
The residue was chromatographed with EtOAc/petroleum ether 4:6 as eluent to give 4j as a yellow solid, yield: 87%, mp 145–147 °C. 1H NMR (CDCl3) δ: 3.94 (s, 6H), 3.95 (s, 6H), 3.98 (s, 3H), 7.08 (s, 1H), 7.11 (s, 1H), 7.29 (s, 2H), 7.47 (s, 1H). Anal. Calcd for C20H20O7: C, 64.51; H, 5.41. Found: C, 64.40; H, 5.20.
5.2.11. (5,6,7-Trimethoxybenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (4k)
The residue was chromatographed with EtOAc/petroleum ether 4:6 as eluent to give 4k as a yellow solid, yield: 73%, mp 100–102 °C. 1H NMR (CDCl3) δ: 3.92 (s, 3H), 3.93 (s, 3H), 3.95 (s, 3H), 3.96 (s, 3H), 3.97 (s, 3H), 4.24 (s, 3H), 6.84 (s, 1H), 7.40 (s, 2H), 7.51 (s, 1H). Anal. Calcd for C21H22O8: C, 62.68; H, 5.51. Found: C, 62.44; H, 5.38.
5.2.12. (5-Hydroxybenzofuran-2-yl)(3,4,5-trimethoxyphenyl)-methanone (4l)
The residue was chromatographed with EtOAc/petroleum ether 4:6 as eluent to give 4l as a yellow solid, yield: 80%, mp 155–157 °C. 1H NMR (CDCl3) δ: 3.94 (s, 3H), 3.96 (s, 6H), 7.06 (dd, J = 8.8 and 2.4 Hz, 1H), 7.11 (d, J = 2.4 Hz, 1H), 7.33 (s, 2H), 7.44 (s, 1H), 7.48 (d, J = 8.8 Hz, 1H), 7.52 (s, 1H). Anal. Calcd for C18H16O6: C, 65.85; H, 4.91. Found: C, 65.68; H, 4.67.
5.2.13. (7-Hydroxybenzofuran-2-yl)(3,4,5-trimethoxyphenyl)-methanone (4n)
The residue was chromatographed with EtOAc/petroleum ether 3:7 as eluent to give 4n as a yellow solid, yield: 74%, mp 166–168 °C. 1H NMR (CDCl3) δ: 3.94 (s, 3H), 3.96 (s, 6H), 7.07 (d, J = 7.6 and 2.0 Hz, 1H), 7.22 (t, J = 7.6 Hz, 1H), 7.28 (m, 3H), 7.53 (s, 2H). Anal. Calcd for C18H16O6: C, 65.85; H, 4.91. Found: C, 65.59; H, 4.39.
5.2.14. (3,4,5-Trimethoxyphenyl)(3-methylbenzofuran-2-yl)-methanone (4o)
The residue was chromatographed with EtOAc/petroleum ether 1:9 as eluent to give 40 as a white solid, yield: 85%, mp 108–110 °C. 1HNMR (CDCl3) δ: 2.65 (s, 3H), 3.94 (s, 6H), 3.96 (s, 3H), 7.36 (t, J = 7.2 Hz, 1H), 7.50 (s, 2H), 7.52 (m, 2H), 7.70 (dd, J = 8.0 and 1.0 Hz, 1H). Anal. Calcd for C19H18O5: C, 69.93; H, 5.56. Found: C, 69.68; H, 5.38.
5.2.15. (4-Methoxy-3-methylbenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (4p)
The residue was chromatographed with EtOAc/petroleum ether 2:8 as eluent to give 4p as a white solid, yield: 90%, mp 107–109 °C. 1H NMR (CDCl3) δ: 2.78 (s, 3H), 3.9 (s, 3H), 3.95 (s, 6H), 3.96 (s, 3H), 6.65 (d, J = 7.6 Hz, 1H), 7.13 (d, J = 7.6 Hz, 1H), 7.37 (m, 3H). Anal. Calcd for C20H20O6: C, 67.41; H, 5.66. Found: C, 67.20; H, 5.38.
5.2.16. (5-Methoxy-3-methylbenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (4q)
The residue was chromatographed with EtOAc/petroleum ether 2:8 as eluent to give 4q as a white solid, yield: 81%, mp 122–124 °C 1H NMR (CDCl3) δ: 2.62 (s, 3H), 3.90 (s, 3H), 3.93 (s, 6H), 3.96 (s, 3H), 7.07 (m, 1H), 7.12 (d, J = 8.6 Hz, 1H), 7.42 (m, 3H). Anal. Calcd for C20H20O6: C, 67.41; H, 5.66. Found: C, 67.12; H, 5.24.
5.2.17. (6-Methoxy-3-methylbenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (4r)
The residue was chromatographed with EtOAc/petroleum ether 2:8 as eluent to give 4r as a white solid, yield: 77%, mp 101–103 °C. 1H NMR (CDCl3) δ: 2.61 (s, 3H), 3.89 (s, 3H), 3.94 (s, 6H), 3.96 (s, 3H), 6.99 (m, 2H), 7.39 (s, 2H), 7.59 (d, J = 9.6 Hz, 1H). Anal. Calcd for C20H20O6: C, 67.41; H, 5.66. Found: C, 67.19; H, 5.44.
5.2.18. (7-Methoxy-3-methylbenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (4s)
The residue was chromatographed with EtOAc/petroleum ether 2:8 as eluent to give 4s as a white solid, yield: 71%, mp 143–145 °C. 1H NMR (CDCl3) δ: 2.66 (s, 3H), 3.95 (s, 3H), 3.97 (s, 6H), 4.00 (s, 3H), 6.97 (dd, J = 6.0 and 2.8 Hz, 1H), 7.27 (m, 2H), 7.53 (s, 2H). Anal. Calcd for C20H20O6: C, 67.41; H, 5.66. Found: C, 67.27; H, 5.49.
5.2.19. (6-Ethoxy-3-methylbenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (4t)
The residue was chromatographed with EtOAc/petroleum ether 1.5:8.5 as eluent to give 4t as a yellow solid, yield: 81%, mp 98–100 °C. 1H NMR (CDCl3) δ: 1.41 (t, J = 7.2 Hz, 3H), 2.60 (s, 3H), 3.93 (s, 6H), 3.95 (s, 3H), 4.12 (q, J = 7.2 Hz, 2H), 6.97 (m, 2H), 7.38 (s, 2H), 7.55 (d, J = 9.2 Hz, 1H). Anal. Calcd for C21H22O6: C, 68.10; H, 5.99. Found: C, 67.89; H, 5.79.
5.2.20. (6-Fluoro-3-methylbenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (4u)
The residue was chromatographed with EtOAc/petroleum ether 1:9 as eluent to give 4u as a brown solid, yield: 79%, mp 130–132 °C. 1H NMR (CDCl3) δ: 2.62 (s, 3H), 3.93 (s, 6H), 3.96 (s, 3H), 7.08 (dd, J = 9.0 and 2.6 Hz, 1H),), 7.16 (dd, J = 9.4 and 2.2 Hz, 1H), 7.38 (s, 2H), 7.66 (dd, J = 8.8 and 5.2 Hz, 1H). Anal. Calcd for C19H17FO5: C, 66.27; H, 4.98. Found: C, 66.03; H, 4.76.
5.2.21. (4-Hydroxy-3-methylbenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (4v)
The residue was chromatographed with EtOAc/petroleum ether 3:7 as eluent to give 4v as a yellow solid, yield: 65%, mp 203–205 °C. 1H NMR (CDCl3) δ: 2.82 (s, 3H), 3.93 (s, 3H), 3.96 (s, 6H), 5.83 (bs, 1H), 6.59 (d, J = 8.2 Hz, 1H), 7.11 (d, J = 8.2 Hz, 1H), 7.29 (m, 1H), 7.39 (s, 2H). Anal. Calcd for C19H18O6: C, 66.66; H, 5.30. Found: C, 66.48; H, 5.11.
5.2.22. (5-Hydroxy-3-methylbenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (4w)
The residue was chromatographed with EtOAc/petroleum ether 4:6 as eluent to give 4w as a yellow solid, yield: 53%, mp 181–183 °C. 1H NMR (CDCl3) δ: 2.59 (s, 3H), 3.93 (s, 3H), 3.96 (s, 6H), 7.01 (m, 1H), 7.06 (d, J = 6.4 Hz, 1H), 7.38 (d, J = 6.4 Hz, 1H), 7.41 (s, 1H), 7.42 (s, 2H). Anal. Calcd for C19H18O6: C, 66.66; H, 5.30. Found: C, 66.50; H, 5.16.
5.2.23. (6-(Benzyloxy)benzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (6m)
The residue was chromatographed with EtOAc/petroleum ether 4:6 as eluent to give 6m as a pink solid, yield: 61%, mp 182–184 °C. 1H NMR (CDCl3) δ: 3.94 (s, 3H), 3.96 (s, 6H), 4.35 (s, 2H), 6.51 (d, J = 2.4 Hz, 1H), 7.10 (dd, J = 8.6 and 2.4 Hz, 1H), 7.28 (m, 5H), 7.29 (s, 2H), 7.47 (s, 1H), 7.61 (d, J = 8.6 Hz, 1H). ESI-MS m/z 419.1 [M+1]+.
5.2.24. (6-(Benzyloxy)-3-methylbenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (6x)
The residue was chromatographed with EtOAc/petroleum ether 2:8 as eluent to give 6x as a white solid, yield: 68%, mp 110–112 °C. 1H NMR (CDCl3) δ: 2.61 (s, 3H), 3.93 (s, 3H), 3.95 (s, 6H), 5.15 (s, 2H), 7.07 (m, 2H), 7.37 (s, 2H), 7.44 (m, 5H), 7.56 (d, J = 8.6 Hz, 1H). ESI-MS m/z 433.2 [M+1]+.
5.3. General procedure for the synthesis of 4m and 4x
A suspension of benzyloxy derivative 6m or 6x (1 mmol) ammonium formate (630 mg, 10 mmol) and activated Pd on charcoal (10%, 150 mg) in a mixture of THF/MeOH (1:1, 20 mL) was heated to reflux for 1 h. The catalyst was filtered over Celite, the solvent removed under reduced pressure and the product purified by column chromatography (silica gel, EtOAc/petroleum ether 1:1).
5.3.1. (6-Hydroxybenzofuran-2-yl)(3,4,5-trimethoxyphenyl)-methanone (4m)
The residue was chromatographed with EtOAc/petroleum ether 1:1 as eluent to give 4m as a yellow solid, yield: 46%, mp 141–143 °C. 1H NMR (CDCl3) δ: 3.84 (s, 3H), 3.88 (s, 6H), 6.87 (dd, J = 8.6 and 2.0 Hz, 1H), 6.92 (s, 1H), 7.23 (s, 2H), 7.66 (d, J = 8.4 Hz, 1H), 7.77 (s, 1H), 10.1 (s, 1H). Anal. Calcd for C18H16O6: C, 65.85; H, 4.91. Found: C, 65.58; H, 4.72.
5.3.2. (6-Hydroxy-3-methylbenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (4x)
The residue was chromatographed with EtOAc/petroleum ether 1:1 as eluent to give 4x as a yellow solid, yield: 46%, mp 211–213 °C. 1H NMR (CDCl3) δ: 2.08 (s, 3H), 3.77 (s, 3H), 3.85 (s, 6H), 6.86 (dd, J = 8.6 and 2.0 Hz, 1H), 6.99 (d, J = 2.0 Hz, 1H), 7.29 (s, 2H), 7.62 (d, J = 8.6 Hz, 1H), 10.1 (bs, 1H). Anal. Calcd for C19H18O6: C, 66.66; H, 5.30. Found: C, 66.38; H, 5.08.
5.3.3. Synthesis of (6-methoxybenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanethione (4y)
To a magnetically stirred solution of 4d (1 mmol) in anhydrous THF at room temperature was added Lawesson's reagent (1.1 mmol), and the mixture was stirred for 1 h. The solvent was evaporated in vacuo, and the residue was dissolved in dichloromethane (15 mL). The organic extract was washed with water (5 mL), dried over Na2SO4 and concentrated in vacuo. The residue purified by flash chromatography (ethyl acetate/petroleum ether 3:7) furnished 4y as a white powder, yield: 96%, mp 143–145 °C. 1H NMR (CDCl3) δ: 3.90 (s, 3H), 3.92 (s, 6H), 3.94 (s, 3H), 6.94 (dd, J = 8.8 and 2.4 Hz, 1H), 7.10 (s, 2H), 7.12 (d, J = 2.4 Hz, 1H), 7.37 (s, 1H), 7.62 (d, J = 8.8 Hz, 1H). Anal. Calcd for C19H18O5S: C, 63.67; H, 5.06; S, 8.95. Found: C, 63.48; H, 4.88; S, 8.74.
5.3.4. Synthesis of (6-methoxybenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanol (4z)
To a cooled solution of 4d (1 mmol) in dioxane (5 mL) was added NaBH4 (1 mmol), and then the mixture was allowed to stir at room temperature under nitrogen for 2 h. The solvent was concentrated under reduced pressure, and aqueous hydrochloric acid (1 M, 2 mL) was added to the residue. The mixture was extracted with dichloromethane (2 × 10 mL) and the organic layer washed with water (2 × 5 mL) and brine (5 mL), dried with Na2SO4 and concentrated under reduced pressure to give a light yellow oil residue. Purification by flash column chromatography (petroleum ether/ethyl acetate 1:1) gave 4z as a white solid, which was recrystallized from petroleum ether. Yield: 84%, mp 148–150 °C. 1H NMR (CDCl3) δ: 3.84 (s, 6H), 3.86 (s, 6H), 5.87 (bs, 1H), 6.47 (s, 1H), 6.57 (bs, 1H), 6.73 (s, 2H), 6.84 (dd, J = 8.6 and 2.4 Hz, 1H), 7.00 (d, J = 2.4 Hz, 1H), 7.37 (d, J = 8.6 Hz, 1H). Anal. Calcd for C19H20O6: C, 66.27; H, 5.85. Found: C, 66.04; H, 5.67.
5.3.5. Preparation of 6-methoxy-2-(methoxy(3,4,5-trimethoxyphenyl)methyl)benzofuran (4aa)
A mixture of 4z (86 mg, 0.26 mmol) and pyridinium p-toluensulfonate (125 mg, 0.5 mmol) in a mixture of THF-MeOH (10 mL, 0.5:9.5) was stirred for 15 h in a sealed tube. The reaction mixture was evaporated under vacuum, the crude mixture was dissolved with dichloromethane (5 mL) and the organic phase was washed with water (1 mL) and brine (1 mL), dried over Na2SO4 and concentrated under reduced pressure. The residue was chromatographed with EtOAc/petroleum ether 3:7 as eluent to give 4aa as a white solid, yield: 96%, mp 148–150 °C 1H NMR (CDCl3) δ: 3.46 (s, 3H), 3.84 (s, 3H), 3.85 (s, 3H), 3.87 (s, 6H), 5.29 (bs, 1H), 6.46 (d, J = 1.0 Hz, 1H), 6.71 (s, 2H), 6.84 (dd, J = 8.4 and 2.2 Hz, 1H), 7.01 (d, J = 2.2 Hz, 1H), 7.35 (d, J = 8.6 Hz, 1H). Anal. Calcd for C20H22O6: C, 67.03; H, 6.19. Found: C, 66.88; H, 6.07.
5.3.6. Preparation of 2-(3,4,5-trimethoxybenzyl)-6-methoxybenzofuran (4ab)
To a magnetically stirred suspension of 4z (0.5 mmol) in dichloromethane (3 mL) at room temperature was added trifluoroacetic acid (410 μL, 5.5 mmol) and then triethylsilane (890 μL, 5.5 mmol). After 15 min, the solvent was evaporated in vacuo, and the residue was dissolved in dichloromethane (20 mL). The organic layer was washed with an aqueous saturated solution of NaHCO3 (5 mL) and brine (5 mL), dried (Na2SO4) and concentrated in vacuo. Flash chromatography using ethyl acetate–petroleum ether 1:1, furnished 4ab as a brown oil, yield: 94%. 1H NMR (CDCl3) δ: 3.83 (s, 6H), 3.84 (s, 6H), 4.01 (s, 2H), 6.34 (d, J = 1.0 Hz, 1H), 6.51 (s, 2H), 6.82 (dd, J = 8.4 and 2.4 Hz, 1H), 6.98 (d, J = 2.4 Hz, 1H), 7.37 (d, J = 8.5 Hz, 1H). Anal. Calcd for C19H20O5: C, 69.50; H, 6.14. Found: C, 69.31; H, 5.95.
5.3.7. Synthesis of (2-bromo-3,4,5-trimethoxyphenyl)(6-methoxybenzofuran-2-yl)methanone (7)
To a solution of 4d (342 mg, 1 mmol.) in acetonitrile (10 mL) was added a mixture of benzoyl peroxide (48 mg, 0.2 mmol) and NBS (214 mg, 1.2 mmol). The mixture was heated under reflux for 2 h. The solvent was removed under reduced pressure, and the residue dissolved in EtOAc (15 mL), which was washed successively with a saturated solution of NaHCO3 (5 mL), water (5 mL) and brine (5 mL), dried (Na2SO4) and concentrated in vacuo. Purification by flash column chromatography (petroleum ether/ethyl acetate 7:3) gave 7 as a white solid. Yield: 65%, mp 144–145 °C. 1H NMR (DMSO-d6) δ: 3.83 (s, 6H), 3.85 (s, 3H), 3.86 (s, 3H), 6.99 (dd, J = 8.8 and 2.2 Hz, 1H), 7.15 (s, 1H), 7.36 (d, J = 2.4 Hz, 1H), 7.54 (s, 1H), 7.68 (d, J = 8.8 Hz, 1H). ESI-MS m/z 421.0/423.0 [M+1]+/[M+3]+.
5.3.8. Synthesis of 3,8,9,10-tetramethoxy-6H-benzo[b]indeno[1,2-d]furan-6-one (4ac)
A mixture of bromo derivative 7 (160 mg, 0.38 mmol), Pd (Ph3P)4 (12 mg) and potassium acetate (40 mg, 0.42 mmol) in 2 mL of N,N-dimethylacetamide was heated at 160 °C for 5 h. The solvent was then removed under reduced pressure and the residue purified by flash chromatography on silica gel using EtOAc/petroleum ether 2:8 as eluent. Yield: 46%, brown solid, mp 124–125 °C. 1H NMR (DMSO-d6) δ: 3.88 (s, 6H), 3.93 (s, 3H), 4.04 (s, 3H), 6.92 (s, 1H), 7.00 (m, 2H), 7.73 (d, J = 9.2 Hz, 1H). Anal. Calcd for C19H16O6: C, 67.05; H, 4.74. Found: C, 66.89; H, 4.59.
5.4. Cell growth inhibitory activity
Murine leukemia L1210, murine mammary carcinoma FM3A and human T-lymphocyte Molt 4 and CEM and human cervix carcinoma (HeLa) cells were suspended at 300,000-500,000 cells/mL of culture medium, and 100 μL of a cell suspension was added to 100 μL of an appropriate dilution of the test compounds in wells of 96-well microtiter plates. After incubation at 37 °C for two days, cell number was determined using a Coulter counter. The IC50 value was defined as the compound concentration required to inhibit cell proliferation by 50%.
5.5. Effects on tubulin polymerization and on colchicine binding to tubulin
Bovine brain tubulin was purified as described previously.19 To evaluate the effect of the compounds on tubulin assembly in vitro,14 varying concentrations were preincubated with 10 μM tubulin in glutamate buffer at 30 °C and then cooled to 0 °C. After addition of GTP, the mixtures were transferred to 0 °C cuvettes in a recording spectrophotometer and warmed to 30 °C, and the assembly of tubulin was observed turbidimetrically. The IC50 value was defined as the compound concentration that inhibited the extent of assembly by 50% after a 20 min incubation. The ability of the test compounds to inhibit colchicine binding to tubulin was measured as described,15 except that the reaction mixtures contained 1 μM tubulin, 5 μM [3H]colchicine and 5 μM test compound.
5.6. Flow cytometric analysis of cell cycle distribution
For details, see Ref. 9a.
5.7. Molecular modeling
All molecular modeling studies were performed on a MacPro dual 2.66 GHz Xeon running Ubuntu 8. The tubulin structure was downloaded from the PDB data bank (http://www.rcsb.org/ - PDB code: 1SA0). Docking simulations were performed using PLANTS20 and results examined using ZODIAC.21 H-Bond and lipophilicity maps were created using ZODIAC.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Alberto Casolari and Paolo Orlandini for the technical assistance.
Footnotes
Supplementary data: Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmc.2009.08.027.
References and notes
- 1.(a) Honore S, Pasquier E, Braguer D. Cell Mol Life Sci. 2005;62:3039. doi: 10.1007/s00018-005-5330-x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hearn BR, Shaw SJ, Myles DC. Comp Med Chem II. 2007;7:81. [Google Scholar]; (c) Pasquier E, Andrè N, Braguer D. Curr Cancer Drug Target. 2007;7:566. doi: 10.2174/156800907781662266. [DOI] [PubMed] [Google Scholar]
- 2.(a) Pellegrinelli F, Budman DR. Cancer Invest. 2005;23:264. doi: 10.1081/cnv-200055970. [DOI] [PubMed] [Google Scholar]; (b) Chaplin DJ, Horsman MR, Siemann DW. Curr Opin Invest Drugs. 2006;7:522. [PubMed] [Google Scholar]; (c) Nam NH. Curr Med Chem. 2003;10:1697. doi: 10.2174/0929867033457151. [DOI] [PubMed] [Google Scholar]
- 3.(a) Li Q, Sham HL. Exp Opin Ther Pat. 2002;12:1663. [Google Scholar]; (b) Beckers T, Mahboobi S. Drugs Future. 2003;28:767. [Google Scholar]; (c) Ducki S. Anti-cancer Agents Med Chem. 2009;9:336. doi: 10.2174/1871520610909030336. [DOI] [PubMed] [Google Scholar]
- 4.Pettit GR, Singh SB, Hamel E, Lin CM, Alberts DS, Garcia-Kendall D. Experientia. 1989;45:209. doi: 10.1007/BF01954881. [DOI] [PubMed] [Google Scholar]
- 5.Lin CM, Ho HH, Pettit GR, Hamel E. Biochemistry. 1989;28:6984. doi: 10.1021/bi00443a031. [DOI] [PubMed] [Google Scholar]
- 6.(a) Tron GC, Pirali T, Sorba G, Pagliai F, Busacca S, Genazzani AA. J Med Chem. 2006;49:3033. doi: 10.1021/jm0512903. [DOI] [PubMed] [Google Scholar]; (b) Mahindroo N, Liou JP, Chang JY, Hsieh HP. Exp Opin Ther Pat. 2006;16:647. [Google Scholar]; (c) Jordan MA, Wilson L. Nat Rev Cancer. 2004;4:253. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]; (d) Chaudhary A, Pandeya SN, Kumar P, Sharma PP, Gupta S, Soni N, Verma KK, Bhardwaj G. Mini-Rev Med Chem. 2007;7:1186. doi: 10.2174/138955707782795647. [DOI] [PubMed] [Google Scholar]; (e) Hsie HP, Liou JP, Mahindroo N. Curr Pharm Des. 2005;11:1655. doi: 10.2174/1381612053764751. [DOI] [PubMed] [Google Scholar]
- 7.Pettit GR, Toki B, Herald DL, Verdier-Pinard P, Boyd MR, Hamel E, Pettit RK. J Med Chem. 1998;41:1688. doi: 10.1021/jm970644q. [DOI] [PubMed] [Google Scholar]
- 8.Liou JP, Chang CW, Song JW, Yang YN, Yeh CF, Tseng HY, Lo YK, Chang YL, Chang CM, Hsieh HP. J Med Chem. 2002;45:2556. doi: 10.1021/jm010365+. [DOI] [PubMed] [Google Scholar]
- 9.(a) Romagnoli R, Baraldi PG, Sarkar T, Carrion MD, Cruz-Lopez O, Lopez-Cara C, Tolomeo M, Grimaudo S, Di Cristina A, Pipitone MR, Balzarini J, Gambari R, Ilaria L, Saletti R, Brancale A, Hamel E. Bioorg Med Chem. 2008;16:8419. doi: 10.1016/j.bmc.2008.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Romagnoli R, Baraldi PG, Carrion MD, Lopez Cara C, Preti D, Fruttarolo F, Pavani MG, Tabrizi MA, Tolomeo M, Grimaudo S, Di Antonella C, Balzarini J, Hadfield JA, Brancale A, Hamel E. J Med Chem. 2007;50:2273. doi: 10.1021/jm070050f. [DOI] [PubMed] [Google Scholar]; (c) Romagnoli R, Baraldi PG, Jung MK, Iaconinoto MA, Carrion MD, Preti D, Tabrizi MA, Fruttarlo F, De Clercq E, Balzarini J, Hamel E. Bioorg Med Chem Lett. 2005;15:4048. doi: 10.1016/j.bmcl.2005.06.022. [DOI] [PubMed] [Google Scholar]; (d) Romagnoli R, Baraldi PG, Sarkar T, Carrion MD, Lopez-Cara C, Cruz-Lopez O, Preti D, Tabrizi MA, Tolomeo M, Grimaudo S, Di Cristina A, Zonta N, Balzarini J, Brancale A, Hsieh HP, Hamel E. J Med Chem. 2008;51:1464. doi: 10.1021/jm7011547. [DOI] [PubMed] [Google Scholar]
- 10.(a) Young SL, Chaplin DJ. Exp Opin Invest Drugs. 2004;13:1171. doi: 10.1517/13543784.13.9.1171. [DOI] [PubMed] [Google Scholar]; (b) Pettit GR, Temple C, Jr, Narayanan VL, Varma R, Boyd MR, Rener GA, Bansal N. Anti-Cancer Drug Des. 1995;10:299. [PubMed] [Google Scholar]
- 11.Flynn BL, Hamel E, Jung MK. J Med Chem. 2002;45:2670. doi: 10.1021/jm020077t. [DOI] [PubMed] [Google Scholar]
- 12.Dupeyre G, Chabot GG, Thoret S, Cachet X, Seguin J, Guénard D, Tillequin F, Scherman D, Koch M, Michel S. Bioorg Med Chem. 2006;14:4410. doi: 10.1016/j.bmc.2006.02.037. [DOI] [PubMed] [Google Scholar]
- 13.Kozikowski AP, Ma D. Tetrahedron Lett. 1991;32:3317. [Google Scholar]
- 14.Hamel E. Cell Biochem Biophys. 2003;38:1. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
- 15.Verdier-Pinard P, Lai JY, Yoo HD, Yu J, Marquez B, Nagle DG, Nambu M, White JD, Falck JR, Gerwick WH, Day BW, Hamel E. Mol Pharmacol. 1998;53:62. doi: 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]
- 16.Ravelli RBG, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M. Nature. 2004;428:198. doi: 10.1038/nature02393. [DOI] [PubMed] [Google Scholar]
- 17.Kong Y, Grembecka J, Edler MC, Hamel E, Mooberry SL, Sabat M, Rieger J, Brown ML. Chem Biol. 2005;12:1007. doi: 10.1016/j.chembiol.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 18.For the preparation of 5j see: Foyer R, Rene L, Cavier R, Lemoine J. Eur J Med Chem. 1977;12:455.For the preparation of 5k see: Fukuda Y, Furuta H, Kusama Y, Ebisu H, Oomori Y, Terashima S. J Med Chem. 1999;42:1448. doi: 10.1021/jm980668c.Compound 5l was synthesized following the procedure: Wang X, Porco JA., Jr Angew Chem, Int Ed. 2006;45:6607.Compound 5n was prepared following the procedure: Kemoto H, Miyata J, Yoshida M, Raku N, Fukumoto K. J Org Chem. 1997;62:7850.For the synthesis of 5s see: Kagawa H, Shigematsu A, Ohta S, Harigaya Y. Chem Pharm Bull. 2005;53:547. doi: 10.1248/cpb.53.547.For the synthesis of 5v: Zhang X, Sui Z. Synthesis. 2006:2568.For the preparation of 5w see: Aggarwal R, Giles RGF, Green IR, Oosthuizen FJ, Taylor CP. Org Biomol Chem. 2005;3:263. doi: 10.1039/b414213f.For the synthesis of 5x see: Bennett CJ, Caldwell ST, McPhail DB, Morrice PC, Duthie GG, Hartley RC. Bioorg Med Chem. 2004;12:2079. doi: 10.1016/j.bmc.2004.02.031.
- 19.Hamel E, Lin CM. Biochemistry. 1984;23:4173. doi: 10.1021/bi00313a026. [DOI] [PubMed] [Google Scholar]
- 20.Korb O, Stützle T, Exner TE. Swarm Intelligence. 2007;1:115. [Google Scholar]
- 21.Zonta N, Grimstead IJ, Avis NJ, Brancale A. J Mol Mod. 2009;15:193. doi: 10.1007/s00894-008-0387-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.