

Abstract
Galactose oxidase (GO) belongs to a class of proteins that self-catalyze assembly of their redox-active cofactors from active site amino acids. Generation of enzymatically active GO appears to require at least four sequential post-translational modifications: cleavage of a secretion signal sequence, copper-dependent cleavage of an N-terminal pro sequence, copper-dependent formation of a C228-Y272 thioether bond, and generation of the Y272 radical. The last two processes were investigated using a truncated protein (termed premat-GO) lacking the pro sequence and purified under copper-free conditions. Reactions of premat-GO with Cu(II) were investigated using optical, EPR, and resonance Raman spectroscopy, SDS-PAGE, and X-ray crystallography. Premat-GO reacted anaerobically with excess Cu(II) to efficiently form the thioether bond but not the Y272 radical. A potential C228-copper coordinated intermediate (λmax) 406 nm) in the processing reaction, which had not yet formed the C228-Y272 cross-link, was identified from the absorption spectrum. A copper-thiolate protein complex, with copper coordinated to C228, H496, and H581, was also observed in a 3 min anaerobic soak by X-ray crystallography, whereas a 24 h soak revealed the C228-Y272 thioether bond. In solution, addition of oxygenated buffer to premat-GO preincubated with excess Cu(II) generated the Y272 radical state. On the basis of these data, a mechanism for the formation of the C228-Y272 bond and tyrosyl radical generation is proposed. The 406 nm complex is demonstrated to be a catalytically competent processing intermediate under anaerobic conditions. We propose a potential mechanism which is in common with aerobic processing by Cu(II) until the step at which the second electron acceptor is required.
The range of chemistry available for enzyme catalysis may be extended by post-translational modifications of encoded amino acids, generating new intrinsic cofactor moieties (1). One general class of post-translationally modified cofactor involves protein-derived radicals, including tyrosyl, glycyl, thiyl, and tryptophanyl radicals (2-5). The second class includes proteins that undergo more extensive post-translational modifications derived by covalent modification of an amino acid, or involving peptide cleavage and modification, or new bond-forming reactions. Post-translational modification occurs either by a self-processing pathway or via the involvement of accessory enzymes, and both modes of modification may be utilized by an enzyme to facilitate new reactivity and/or provide additional structural functionality.
The most frequently modified residue appears to be tyrosine, which is transformed into a variety of novel structures, including cross-links and quinones. Often these novel cofactors are associated with a metal center and are catalytically active. In copper amine oxidases, an active site tyrosine is converted in a dioxygen- and copper-dependent autocatalytic reaction to a 2,4,5-trihydroxyphenylalanine quinone (6-9) while lysyl oxidase contains catalytically active lysyltyrosylquinone (10). KatG contains a M-Y-W cross-link adjacent to the heme cofactor (11) that forms autocatalytically and oxidatively (via compound I), during a turnover reaction (12); the presence of the cross-link appears to confer catalase activity to the catalase-peroxidase enzyme (13). Cytochrome c oxidase contains a Y-H bond at the CuB site (14, 15), where this histidine provides a copper ligand, and a tyrosyl radical at this location has been implicated in catalysis (16). A covalent bond between Y69 Cε and C161 S has been identified in the crystal structure of Mycobacterium tuberculosis sulfite reductase (NirA) at 2.8 Å resolution (17). The Y69-C161 cross-link is in the proximity of the siroheme redox cofactor, and mutagenesis studies suggest that it plays a role in catalysis. A cysteine-tyrosine cross-link was identified in the crystal structures of mouse (18) and rat (19) cysteine dioxygenase, where it is adjacent to the catalytic non-heme iron center and is proposed to play a role in enzymatic turnover. A Cys-Tyr bond also exists in glyoxal oxidase (20, 21). In most cases, the mechanism of formation of the post-translationally modified amino acid(s) has not been fully elucidated.
Galactose oxidase (GO) was identified to contain a modified active site tyrosine residue (22), and the copper ligand Y272 is covalently linked to C228 via a thioether bond between the ring Cε1 atom and the Sγ atom. The modified tyrosine is oxidized to a tyrosyl radical in the enzymatically active state (22, 23). The crystal structure of Fusarium graminearum galactose oxidase provided the first structural description of a cofactor post-translationally derived from active site amino acids [PDB entry 1GOG (Figure 1A)] (22). The function of the C228-Y272 thioether bond has been attributed to lowering the tyrosine redox potential to 400 mV (24) from ∼1000 mV for an unmodified tyrosine residue (3). Post-translational modification of Y272 may also facilitate delocalization of the spin density of the tyrosyl free radical, although the effect on the distribution of spin density attributed to the cross-link varies in different studies (25, 26).
Figure 1.
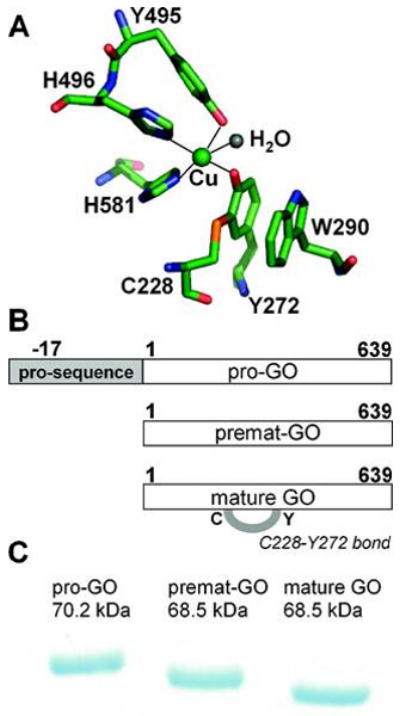
(A) Active site of mature galactose oxidase showing the C228–Y272 thioether bond (PDB entry 1GOG). (B) Schematic representation of unprocessed and mature galactose oxidase protein forms. (C) SDS–PAGE of unprocessed and mature galactose oxidase protein forms. The anomalous SDS–PAGE mobility of mature galactose oxidase (65.5 KDa) results from the presence of the covalent C228–Y272 cross-link.
Formation of the C228-Y272 cofactor in GO is only one of the maturation events leading to enzymatically active enzyme (68.5 kDa, no pro sequence, C228-Y272 bond formed). Two partially processed GO forms have been identified in growth media following heterologous expression of the complete Fusarium goa gene under reduced copper conditions. These are a precursor form, pro-GO (70.2 kDa, pro sequence present, no C228-Y272 bond), and a premat- GO form (68.5 kDa, no pro sequence, no C228-Y272 bond) (Figure 1B,C). The presence of these immature forms suggests that complete processing of GO requires at least four post-translational events: cleavage of the secretion signal, cleavage of an N-terminal pro sequence with associated structural rearrangements, formation of the C228-Y272 thioether bond, and a one-electron oxidation generating the tyrosyl radical (Y272·) (25, 27).
Formation of the C228-Y272 thioether bond and generation of the Y272 tyrosyl radical in pro-GO appear to occur via an autocatalytic mechanism that requires copper and dioxygen (27). Whittaker and Whittaker (28) reported that an immature form of GO lacking the pro sequence (a premat- GO form) processes aerobically with both Cu(I) and Cu(II), although processing is much faster with Cu(I) leading to the conclusion that in vivo processing involves Cu(I). Under anaerobic conditions, it was shown that Cu(II) leads to formation of a stable species with an absorption peak at 406 nm attributed to a Cu(II) thiolate-to-metal charge transfer band (28).
In this study, we have investigated events associated with formation of the thioether bond in recombinant GO lacking the pro sequence [premat GO (29)] following addition of Cu(II) under anaerobic and aerobic conditions. This study explores whether dioxygen is essential for both thioether bond formation and radical generation in GO (27). Processing has been followed by SDS-PAGE, optical, EPR, and resonance Raman spectroscopy, and X-ray crystallography. The studies reveal that the thioether bond can form even under anaerobic conditions which points to the involvement of electron acceptors other than dioxygen.
Experimental Procedures
Biochemical Methods
For metal-free conditions, plasticware was soaked in 0.1 M EDTA1 and glassware was soaked in a 1:1 nitric acid/sulfuric acid mixture, followed by extensive rinsing with doubly deionized or MilliQ water. Copper-free solutions were prepared using either batch or column Chelex (Bio-Rad, Hercules, CA) resin treatment. Metal samples were of high purity (99.99% trace metal basis). Cu(II) stock solutions (10 mM) were prepared by dissolving CuSO4 in 50 mM Tris (pH 8.0). Addition of small aliquots of this reagent to protein solutions did not alter the experimental pH. Tetrakis(acetonitrile) Cu(I) hexafluorophosphate was prepared according to ref 28 by dissolving it in anaerobic acetonitrile immediately prior to use. SDS-PAGE was performed using either hand-cast 8% Tris-HCl gels or precast 7% Tris-acetate gels (Invitrogen, Carlsbad, CA). Anaerobic PAGE samples were prepared by transferring anaerobic protein aliquots via a gastight syringe to an equal volume of SDS-PAGE sample buffer (2×) in a PCR tube in a thermocycler at 100 °C. To determine the time course of formation of the C228-Y272 bond, the stained gel was scanned using a Hewlett-Packard flatbed scanner equipped with HP Precision Scan Pro software. Densitometry analysis of the scanned protein bands was performed via AIDA (RayTest).
Expression and Purification of Premat-GO
The coding sequence for mature F. graminearum galactose oxidase, lacking the pro sequence (30), was fused in-frame with the vector-encoded R-mating factor signal sequence in pPICZRB (Invitrogen) (29). Pichia pastoris X-33 transformed with the expression vector was cultured according to standard protocols (Invitrogen). Yeast cultures were grown in a buffered glycerol-complex medium at 300 rpm and 30 °C for 24 h. Using metal-free conditions, protein expression was then induced by transferring the cells to buffered methanolcomplex medium with incubation at 300 rpm and 25 °C. Methanol was added to 0.5% of the culture volume every 24 h for 48 h.
Protein purification steps were performed using metal-free buffers and equipment. All buffer solutions included 1 mM EDTA. The culture medium was centrifuged, and the cell-free supernatant was dialyzed against 1 mM EDTA and then loaded on a cellulose phosphate column equilibrated with 10 mM potassium phosphate (pH 7.3) and 1 mM EDTA. The protein-bound resin was washed with 10 mM potassium phosphate (pH 7.3) and 1 mM EDTA and eluted with a linear gradient to 0.5 M potassium phosphate (pH 7.3) and 1 mM EDTA. The selected GO-containing fractions were concentrated by ultrafiltration (Amicon) and dialyzed into EDTA-free 50 mM Pipes (pH 6.8). The protein concentration was determined by measuring the absorbance at 280 nm (ε) 104900 M-1 cm-1) (31). The quality of the purified premat- GO was assessed by SDS-PAGE, the optical spectrum of the copper-free protein, the rate of formation of the Y272 radical (k445), and the proportion of Y272· formed on addition of an aerobic excess of Cu(II).
Enzyme Activity Assay
The specific activity of GO-containing samples was measured using a coupled assay medium containing d-galactose (600 mM), 2,22-azinobis(3- ethylbenzothiazoline-6-sulfonic acid), diammonium salt (ABTS, 0.8 mM), and horseradish peroxidase (20 units/mL) in 100 mM sodium phosphate (pH 7.0). Typically, 50 μL of the enzyme solution was added to 950 μL of the assay mix at 25 °C. The rate of change of absorbance at 415 nm (ε415) 31300 M-1 cm-1) was monitored on a Shimadzu UV- 2401 PC spectrophotometer. One unit of activity is defined as the amount of enzyme required to form 1 μmol of H2O2/min (32).
ICP-MS Analysis
Determination of the total copper content of galactose oxidase samples in 50 mM Pipes (pH 6.8) was performed by ICP-MS. Doubly deionized water and a commercial copper standard were used as controls.
Spectroscopy
Optical spectra were recorded using either a Cary 6000i, Shimadzu UV-2401 PC, or Perkin-Elmer Lambda 18 instrument. Circular dichroism (CD) spectra were collected at room temperature using a Jasco J-710 spectropolarimeter. Resonance Raman (rR) spectra were obtained using a Coherent Innova 400 argon ion laser and a Spex triplemate spectrophotometer equipped with a CCD detector.
X-Band EPR spectra were recorded by S. Fairhurst (John Innes Centre, Norwich, U.K.) using a Bruker Elexys spectrometer equipped with an Oxford cryostat. Anaerobic premat-GO samples in buffer containing 0.1 M NaCl were incubated with either 0.8 equiv of Cu(II) followed by a second aliquot to 3.5 equiv or a single addition of 3.5 equiv of Cu(II). The incubated samples (150 μL) were mixed under anaerobic conditions with 10 μL of EDTA (0.23 M) and transferred to quartz EPR tubes followed by immediate flash freezing in liquid nitrogen. Experimental conditions for detection of Cu(II) in anaerobic protein samples were as follows: 60 K, 9.47 GHz, 1 mW power, and two accumulations over a field sweep of 600-4600 G. Spectral baseline correction was applied using X-Sophe (Bruker). Quantification of the Cu(II) content of a protein sample was determined by comparison of the integrated value of its EPR spectrum to a standard curve of similar data constructed over a concentration range of 0-400 μM CuSO4 in 50 mM Pipes buffer (pH 6.8), 0.1 M NaCl, and 10 mM EDTA. Experimental conditions for the detection of a potential protein radical were as follows: 60 K, 9.42 GHz, 0.01 mW power, and two accumulations over a field sweep of 600-4600 G.
Aerobic Addition of Excess Cu(II) to Premat-GO
Aerobic experiments were conducted under ambient laboratory conditions. The optical spectrum of premat-GO in 50 mM Pipes (pH 6.8) and 0.1 M NaCl was recorded. Cu(II) was added to a level of 3.5 mol/mol of premat-GO, and the optical spectrum was recorded over time at 25 °C. SDS-PAGE samples were taken before and after the experiment.
Anaerobic Studies
All non-protein solutions were made anaerobic by purging them with oxygen-scrubbed argon or nitrogen. Protein solutions were deoxygenated by gently blowing oxygen-scrubbed argon over the protein surface while stirring them periodically, for 20 min. Anaerobic manipulations were performed in either a nitrogen-purged glovebag (I2R, Terre Haute, IN) or a glovebox (Belle Technology, Portesham, U.K.). Reduced methyl viologen (33) remained blue for more than 3 h, confirming glovebox anaerobicity. To ensure that anaerobic processing was not due to residual dioxygen, solution experiments were also performed in the presence of a glucose/glucose oxidase system to deplete any dissolved O2 (34). There was essentially no difference in the observed rate of formation of the thioether-containing mature GO during the 30 min incubation under standard or enzymatically purged conditions. Subsequent experiments were thus conducted without glucose or glucose oxidase.
Anaerobic Addition of a Stoichiometric Amount of Cu(II) to Premat-GO
Prior to the addition of Cu(II), a sample of premat-GO in 50 mM Pipes (pH 6.8) was subjected to SDS-PAGE. Oxygen-free Cu(II) (104 μM, 0.8 mol/mol of protein) was added to premat-GO (132 μm) in a quartz cuvette sealed with a serum cap (Sigma-Aldrich, Dorset, U.K.), and the optical spectrum was recorded over time at 25 °C. The resulting complex was further analyzed by CD and resonance Raman spectroscopies and SDS-PAGE.
Anaerobic Addition of Excess Cu(II) to Premat-GO
The optical spectrum of premat-GO in 50 mM Pipes (pH 6.8), 0.1 M NaCl, and 10 mM CH3CN was recorded. Cu(II) was added to a level of 3.5 mol/mol of premat-GO, and the optical spectrum was recorded over time at 25 °C until spectral changes were complete. SDS-PAGE samples were taken before and after the experiment, and EPR samples were prepared.
Anaerobic Addition of EDTA and Cu(II) to Premat-GO
Under anaerobic conditions, aliquots of premat-GO were incubated with either a stoichiometric or a 3.5-fold excess of EDTA to protein. Following mixing, Cu(II) was added to a 3.5-fold excess over protein and incubated for 30 min prior to samples being subjected to SDS-PAGE analysis.
Addition of Cu(I) to Premat-GO
Tetrakis(acetonitrile) Cu(I) hexafluorophosphate was weighed into a microcentrifuge tube and exposed to the anaerobic environment of the glovebox for 2 h. It was then dissolved in acetonitrile and added to the protein solution to give a 1:1 stoichiometry and the solution mixed. A 50 μL aliquot was removed, diluted 4-fold with oxygen-saturated buffer, and incubated for 5 min, while the remaining volume was left under anaerobic conditions. Samples of the anaerobic and aerobic reaction mixtures were then analyzed by SDS-PAGE.
Crystallization of Premat-GO and Anaerobic Copper Soaks of Premat-GO Crystals
Premat-GO was exchanged into 50 mM MES (pH 6.3) by repeated concentration and dilution using a spin concentrator (Microcon) pretreated with 10 mM EDTA. Sitting drop crystallizations were set up in a nitrogen-purged glovebox (Belle Technology, Portesham, U.K.) using 4–24% PEG 8000, 100 mM MES (pH 5.8-6.3), and 200 mM calcium acetate. Dissolved oxygen was removed from mother liquor solutions when nitrogen gas was bubbled through them for 1 h. All well solutions were treated with Chelex resin (Bio-Rad) for at least 1 h prior to the addition of protein. Either 2 or 7 μL samples of premat- GO (2-5 mg/mL) were mixed with an equal volume of well solution in sitting drops, and the crystals grew within 1 week. Crystals were washed three times in oxygen-free mother liquor, prior to being soaked in oxygen-free Cu(II) solutions for 3 min (mother liquor, 2 or 10 mM copper acetate) or 24 h (mother liquor, 2 mM copper acetate). After being soaked, crystals were washed briefly in cryoprotectant (mother liquor, 20% PEG 400) and flash-frozen in liquid nitrogen.
X-ray diffraction data were collected at Daresbury Synchrotron Radiation Source (Cheshire, U.K.) stations 14.1 (λ) 1.488 Å) and 14.2 (λ) 0.978 Å) equipped with an ADSC Quantum IV charge-coupled device detector. Data were processed in DENZO (35) and scaled and merged in SCALEPACK (35). Subsequent calculations were carried out using the CCP4 suite (36). The crystal structure of mature GO expressed in P. pastoris (29), essentially identical to the structure of the Fusarium GO (22), was used as a starting model. Electron density map inspection and model rebuilding were carried out using O (37). Refinement was carried out in CNS (38), using rigid body, simulated annealing, positional, and individual temperature factor refinement procedures. CNS was also used to calculate 2Fo-Fc, Fo - Fc, and Fo - Fo electron density maps. Waters were inserted into the model, using the PEAKMAX, WATPEAK (36), and the WATER-PEAKPIK routine in O (37). Refinement and model building were continued until the R-factors had converged and no further model building could be achieved.
Results
Production of Unprocessed Premat-GO
Premat-GO, a recombinant form of galactose oxidase without the pro sequence (29), was expressed and purified under strict copper-free conditions (27). It is virtually impossible to express a metalloprotein totally lacking metal, but the purified protein was confirmed to be predominantly metal-free and unprocessed by copper analysis, by ICP-MS, which confirmed the lack of detectable copper in premat-GO and by specific activity, which was only 1% of that for the mature enzyme. Premat-GO migrated on a SDS-PAGE gel with a mobility corresponding to 68.5 kDa, confirming the absence of the C228-Y272 thioether bond. The migration of mature GO via SDS-PAGE corresponds to an apparent size of ∼65.5 kDa, which differs from its molecular mass determined from the primary structure (68.5 kDa) (Figure 1C). The C228-Y272 thioether bond prevents the protein from being fully linearized in SDS, resulting in more rapid migration (39, 40). Similar anomalous SDS-PAGE behavior was observed with NirA sulfite reductase (17) and cysteine dioxygenase (41). The anomalous migration behavior of the mature enzyme provides an important tool for investigating formation of the C228-Y272 bond in solutions of galactose oxidase.
Aerobic C228-Y272 Cross-Link and Tyrosyl Radical Formation in Premat-GO with Excess Cu(II)
Processing of pro-GO to the mature enzymatically active Cu(II)-Y272 radical form occurs on addition of Cu(II) and O2 (27) with significant structural rearrangement upon pro sequence cleavage to form the mature enzyme active site (42). Precursor forms of GO lacking the pro region (premat-GO) have been shown to be processed with either Cu(I) (28) or Cu(II) (28, 29). Under ambient laboratory conditions, the aerobic addition of a 3.5-fold excess of Cu(II) to copperfree premat-GO resulted in the immediate appearance of absorbance peaks at 445 and 800 nm (Figure 2A) corresponding to the distinctive electronic absorption spectrum of oxidized [Cu(II)-Y·] mature galactose oxidase.
Figure 2.
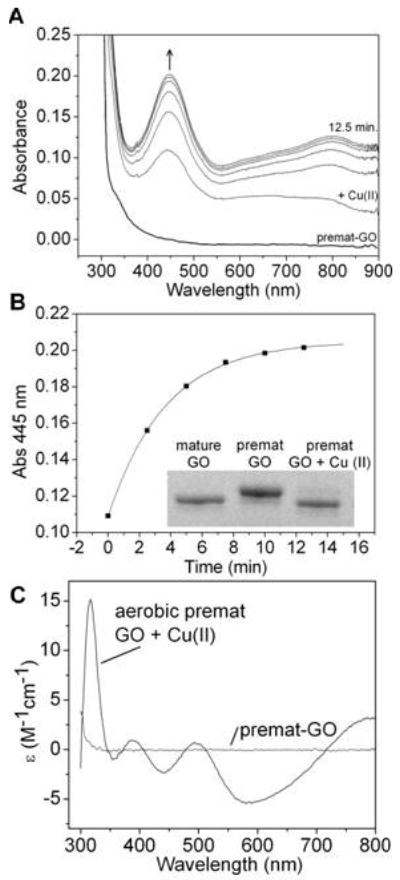
Aerobic biogenesis of premat-GO. (A) Addition of a 3.5-fold excess of Cu(II) (175 μm) to premat-GO (50.4 μm) in 50 mM Pipes and 0.1 M NaCl (pH 6.8) at 25 °C recorded at 2.5 min intervals. The spectrum of premat-GO prior to copper addition is also shown. (B) Appearance of the 445 nm absorbance band of newly formed oxidized mature GO (k445 = 0.21 ± 0.03 min-1) fitted to a single exponential. The inset shows a 7% Tris-acetate SDS–PAGE analysis of formation of the C228–Y272 bond. (C) CD spectrum of the oxidized mature GO produced following treatment with an aerobic excess of Cu(II). The CD spectrum of copper-free premat-GO is also shown.
The rate constant k445 corresponding to Y272· formation was 0.21 (0.03 min-1 fit to a single exponential (Figure 2B). The CD spectrum of premat-GO (400-800 nm) was featureless, as expected for copper-free protein. Upon addition of Cu(II), a spectrum indicative of mature oxidized GO with positive CD bands at 317, 388, and 495 nm and negative bands at 440 and 588 nm rapidly developed (43) (Figure 2C). SDS-PAGE confirmed the thioether bond, absent prior to copper addition, had been completely formed (Figure 2B, inset). Densitometry measurements on stained gel bands from samples taken during the reaction provided a rate constant (k) for formation of the C228-Y272 bond 0.3 (0.05 min-1. The rate constants for cofactor formation and radical generation are comparable, suggesting that the rate-limiting step in formation of oxidized mature GO is formation of the thioether bond. To examine whether the heating step during SDS-PAGE sample preparation enhanced the rate of thioether bond formation, the processing reaction was otherwise quenched by GO precipitation with cold trichloroacetic acid to 10%. There were no apparent differences in rate constants obtained from samples prepared by boiling or by acid precipitation. Oxygen uptake was observed polarographically and resulted in ∼1.3 O2 molecules being consumed. This is in good agreement with the 1.5 O2 molecules predicted for Cu(II) processing of GO. The Cu(II) would accept the first electron for thioether bond formation, resulting in Cu(I). Three further oxidative electron transfer steps involve the second electron required for thioether bond formation, one electron for Cu(I) reoxidation, and one electron for radical formation. Cumulatively, these steps require 1.5 O2 molecules. The qualitative formation of hydrogen peroxide was detected using Amplex red following aerobic processing under excess copper conditions (data not shown). The resulting oxidized mature GO was enzymatically active with a typical specific activity of 560 (20 μmol min-1 mg-1 corresponding to 70% of the specific activity measured for mature GO expressed and purified using copper-supplemented conditions.
We were interested to know whether O2 was essential for both thioether bond formation and radical generation and so studied premat-GO processing under anaerobic conditions.
Anaerobic Cu(II)-Premat-GO Complex
The addition of a slightly substoichiometric amount (0.8 equiv) of Cu(II) to premat-GO under anaerobic conditions generated a yellow complex with an intense absorption band (λmax) 406 nm; k) 0.036 (0.001 min-1) and a weaker band around 600 nm (Figure 3 and inset). SDS-PAGE showed the absence of the C228-Y272 cross-link (data not shown) in the yellow anaerobic premat-GO-copper complex. The 406 nm feature likely corresponds to the transient feature around 410 nm observed during aerobic excess copper processing of pro- GO (27) and has also been reported by Whittaker and Whittaker (28).
Figure 3.
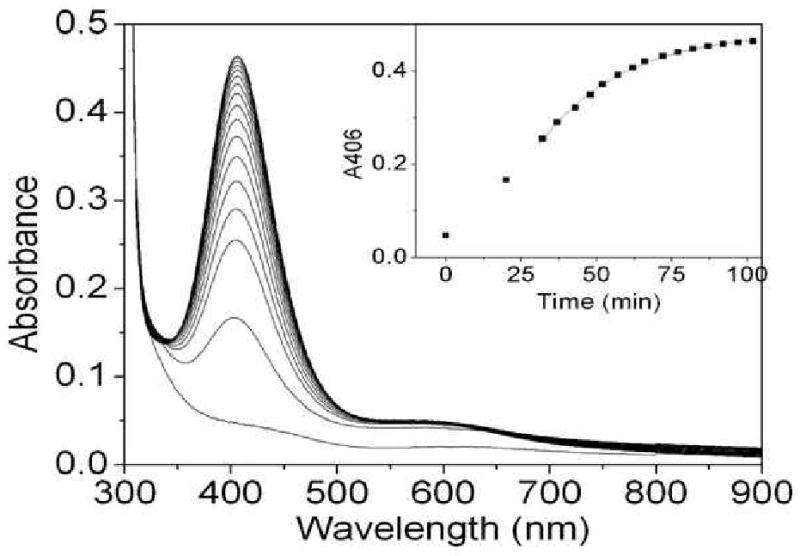
Formation of the anaerobic Cu(II)–premat-GO complex. Cu(II) (0.8 equiv, 104 μM) was added under anaerobic conditions to premat-GO (130 μm) in 50 mM Pipes (pH 6.8) at 25 °C. The inset shows the formation of the absorption band at 406 nm vs time.
The CD spectrum of the yellow anaerobic Cu(II)-premat- GO complex (λmax) 406 nm) had two bands at 434 and 748 nm (Figure 4A). A small positive CD band at 314 nm likely corresponds to a histidine copper ligand N f Cu(II) transition (44). Vibrational frequencies observed at 257 and 343 cm-1 in the resonance Raman spectrum (Figure 4B) have been assigned as a Cu-S stretch (28). These results support the identification of the 406 nm species as a Cu(II)-thiolate complex.
Figure 4.
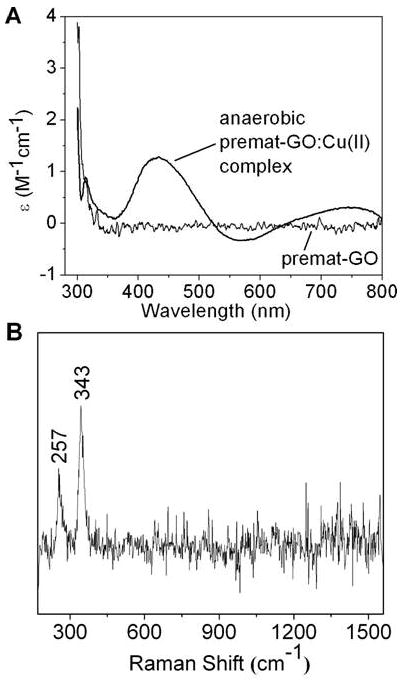
Copper–thiolate protein complex. (A) CD spectrum of the anaerobic Cu(II)–premat-GO complex (thick line) and copper-free premat-GO (thin line). (B) Resonance Raman spectrum of the anaerobic Cu(II)–premat-GO complex.
Decay of the Anaerobic Stoichiometric Cu(II)-Premat- GO Complex
Following maximal formation of the Cu(II) thiolate-protein complex (k) 0.022 (0.002 min-1) (Figure 5A,C), the 406 nm band decayed (k) 0.003 (0.0003 min-1) (Figure 5B,C), converting to a “bleached” optical species with weak absorption bands at approximately 430 and 625 nm. SDS-PAGE samples taken during the reaction indicated that while the cross-link was not present in the yellow complex (406 nm), it was formed as this complex decayed. The observed rate of decay of the 406 nm species, contemporaneous with cross-link formation, is comparable with the rate of formation of the oxidized enzyme (k) 0.0023 min-1) formed by exposure of the 406 nm species to air (28).
Figure 5.
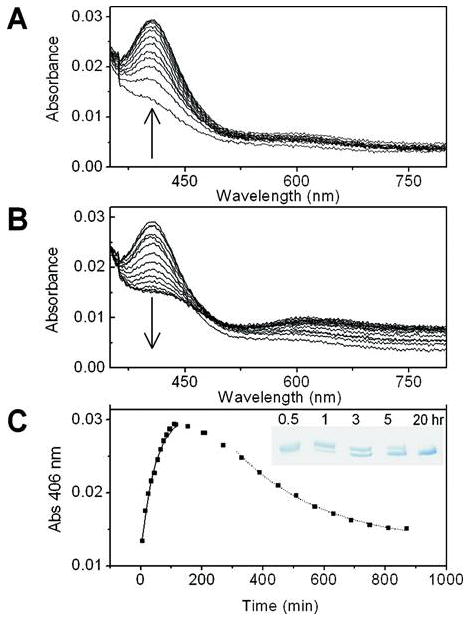
Anaerobic reaction of premat-GO (10 μm) in 50 mM Pipes (pH 6.8) with Cu(II) (10 μM) over time. Under these conditions, a peak at 406 nm appears (A) followed by its decay (B). The time course for the 406 nm absorption band is shown in panel C, where the inset shows a 8% Tris-HCl SDS–PAGE analysis of formation of the C228–Y272 bond over the time course of 0.5–20 h.
Anaerobic C228-Y272 Cofactor Formation in Premat- GO with Excess Cu(II)
Upon addition of a single anaerobic aliquot of Cu(II) (3.5-fold excess) to premat-GO (Figure 6A), a peak at 406 nm assigned as the transient copper thiolate species appeared immediately, followed by its rapid decay (k406decay = 0.245 ± 0.022 min-1) to form a species with an absorption band at ∼435 nm. The broad absorption band centered around 625 nm is assigned as an aqueous Cu(II) band and did not diminish over time. Anaerobic C228-Y272 cross-link formation, as shown by SDS-PAGE, occurred within the time of decay of the 406 nm species and shows that the cross-link forms in the absence of O2.
Figure 6.
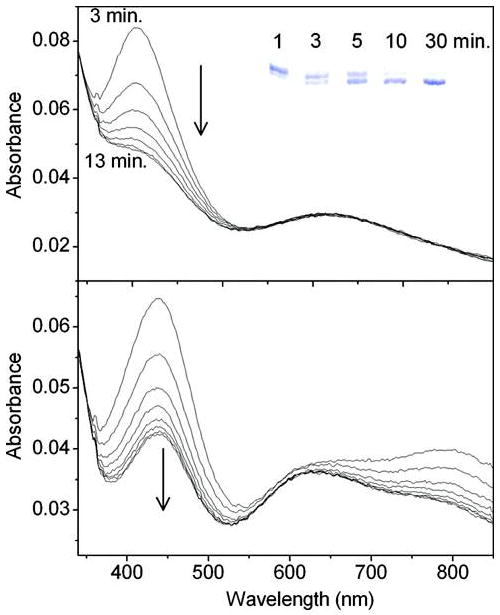
Formation of oxidized galactose oxidase following the addition of dioxygen-saturated buffer to the bleached protein form. (A) Cu(II) (190 μm) was added in a single aliquot to premat-GO (53 μm) in 50 mM Pipes buffer (pH 6.8) with spectra recorded at 3, 5, 7, 9, 11, 12, and 13 min. The inset shows the formation of the cross-link as analyzed by 7% Tris-actate SDS–PAGE, sampled at 1, 3, 5, 10, and 30 min. (B) An equal volume of oxygenated buffer was added to the bleached protein form with spectra recorded at 1, 3, 5, 7, 9, 11, 13, and 14 min.
To determine whether excess Cu(II) could accept electrons in place of O2 during anaerobic C228-Y272 cross-link formation, EPR quantification was performed. After addition of 3.5 equiv of Cu(II), only 0.9 equiv was reduced, whereas SDS-PAGE showed all protein molecules had formed the thioether bond. The conclusion drawn is that 1 equiv of Cu(II) is reduced during bleaching of the 406 nm complex and associated anaerobic formation of the thioether bond. It is reasonable to conclude, on the basis of the spectral data and copper-soaked crystal structures (see below), that this single equivalent of copper is located at the active site.
Reaction of the Bleached Anaerobic Complex with Oxygen
Addition of an equal volume of oxygen-saturated buffer to the anaerobic 3.5-fold excess of Cu(II)-premat-GO complex immediately generated a spectrum resembling that of oxidized mature GO with characteristic peaks at ∼445 and 810 nm (Figure 6B). Unexpectedly, the newly formed Y272· decayed within minutes (k = 0.24 ± 0.21 min-1). Addition of a 5-fold excess of anaerobic potassium ferricyanide to the anaerobic bleached form generated only a minimal amount of the Y272· species as determined by absorption at 445 and 800 nm. In an anaerobic control experiment, potassium ferricyanide treatment fully generated Y272· in mature GO. Control experiments, using mature oxidized GO, showed that tyrosyl radical decay was not accelerated by the presence of the experimental Tris buffer used here (45).
Cu(II) Preferentially Supports Formation of the Thioether Bond under Anaerobic Conditions
To investigate whether Cu(I) supports anaerobic thioether bond formation, premat- GO was incubated with a stoichiometric amount of Cu(I) in a glovebox. The sample was divided, and one aliquot was exposed to air while the second was maintained under anaerobic conditions. After 5 min, the anaerobic sample had not been processed while 50% of the C228-Y272 cofactor had formed aerobically (Figure 7B). Premat-GO with a stoichiometric amount of Cu(I) was incubated under anaerobic conditions, and aliquots were removed for analysis at longer time points (Figure 7C). Compared with the rate of processing with Cu(II) [k = 0.03 min-1 (Figure 2B)], the rate of cross-link formation with Cu(I) was much slower (k = 0.0047 min-1) and may indeed reflect slow conversion of Cu(I) to Cu(II). These data indicate that Cu(II) is the preferred species for mediating formation of the thioether bond during the anaerobic processing of premat-GO. Our results indicate that formation of the thioether bond, though not final formation of the Y272 radical state, can proceed in the absence of oxygen. Whittaker and Whittaker (28) have previously shown that an EDTA-Cu(II) complex forms as indicated by a weak 750 nm band when CuSO4 is added to unprocessed GO and that biogenesis of the thioether bond is prevented by EDTA. We have also demonstrated that EDTA inhibits the formation of the thioether bond under anaerobic conditions (Figure 7A).
Figure 7.
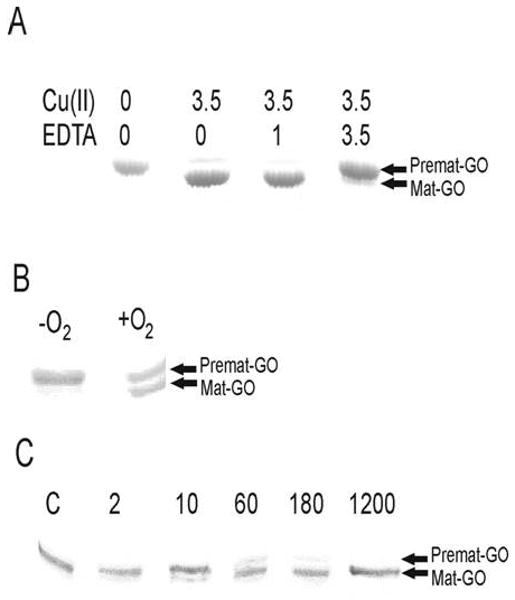
Effect of EDTA and Cu(I) on premat-GO processing analyzed by SDS–PAGE. (A) Migration of protein treated with no EDTA (0), an equimolar amount of EDTA (1), or a 3.5-fold molar excess of EDTA (3.5) prior to addition of a 3.5-fold molar excess of Cu(II). (B) Samples following a 5 min exposure to a stoichiometric amount of Cu(I) under anaerobic (−O2) or aerobic (+O2) conditions. (C) Samples of premat-GO incubated with a stoichiometric amount of Cu(I) anaerobically were removed for SDS–PAGE analysis at 2, 10, 60, 180, and 1200 min.
Crystal Structures of Premat-GO and Cu(II)-Soaked Premat-GO Crystals ReVeal a Potential Processing Intermediate
Premat-GO was crystallized using previously established copper-free conditions (46). The structure of premat-GO was determined to 1.9 Å resolution (Table 1) and was found to largely resemble the mature structure (PDB entry 1GOG) with the exception of the active site region (overall CR rms deviation of 0.42 Å; PDB entry 2VZ1). The thioether bond is not present, and C228 and Y272 are in conformations more similar to those found in the pro-GO structure (42), though Y272 is not well ordered and has been modeled in two conformations (Figure 8A). Similarly, W290 is not found in its mature stacking position, and residues 290-293 have not been modeled.
Table 1.
Crystallographic Data Collection and Refinement Statistics for Premat-GO and Copper Soaks
premat-GO, copper-free (PDB entry  ) ) |
2 mM Cu(II) soak, 3 min | 10 mM Cu(II) soak, 3 min | 2 mM Cu(II) soak, 24 h (PDB entry  ) ) |
|
|---|---|---|---|---|
| space group | C2 | C2 | C2 | C2 |
| unit cell (Å) | a = 96.9 Å | a = 97.0 Å | a = 97.0 Å | a = 97.0 Å |
| b = 88.4 Å | b = 88.5 Å | b = 88.5 Å | b = 88.5 Å | |
| c = 85.3 Å | c = 85.4 Å | c = 85.4 Å | c = 85.4 Å | |
| β = 117.2° | β = 117.2° | β = 117.2° | β = 117.2° | |
| resolution (Å) | 30–1.9 | 30–2.0 | 30–2.0 | 30–1.9 |
| no. of observed reflections | 158746 | 146102 | 126858 | 124536 |
| no. of unique reflections | 49382 | 42764 | 37423 | 47220 |
| overall completeness (%) (final shell) | 98.7 (93.8) | 99.6 (97.5) | 86.5 (72.1) | 94.3 (88.8) |
| overall mean I/σ(I) | 11.4 | 8.7 | 11.0 | 11.7 |
| overall Rsym (%)a (final shell) | 4.0 (12.6) | 5.9 (17.1) | 4.3 (18.9) | 3.6 (11.4) |
| Wilson B-factor (Å2) | 11.7 | 18.8 | 18.1 | 16.7 |
| Rcryst (%)b (highest-resolution shell) | 17.8 (20.2) | 16.8 (19.5) | ||
| Rfree (%)c (highest-resolution shell) | 22.4 (26.7) | 20.1 (25.1) | ||
| no. of atoms | 5391 | 5231 | ||
| average overall B-factor (Å2) | 18.6 | 18.7 | ||
| rmsd for bond lengths (Å) | 0.012 | 0.008 | ||
| rmsd for bond angles (deg) | 1.4 | 1.2 |
Rsym = Σhkl[ΣI(|Ihkl,I − 〈Ihkl〉|)]/Σhkl,IIhkl,I, where Ihkl,I is the intensity of an individual reflection and 〈Ihkl〉 is the mean intensity of that reflection.
Rcryst = Σhkl(‖Fobs,hkl| − |Fcalc,hkl‖)/|Fobs,hkl|, where |Fobs,hkl| and |Fcalc,hkl| are the observed and calculated structure factor amplitudes, respectively, for reflections used during refinement (working set).
Rfree is equivalent to Rcryst but is calculated with reflections omitted from the refinement process (test set).
Figure 8.
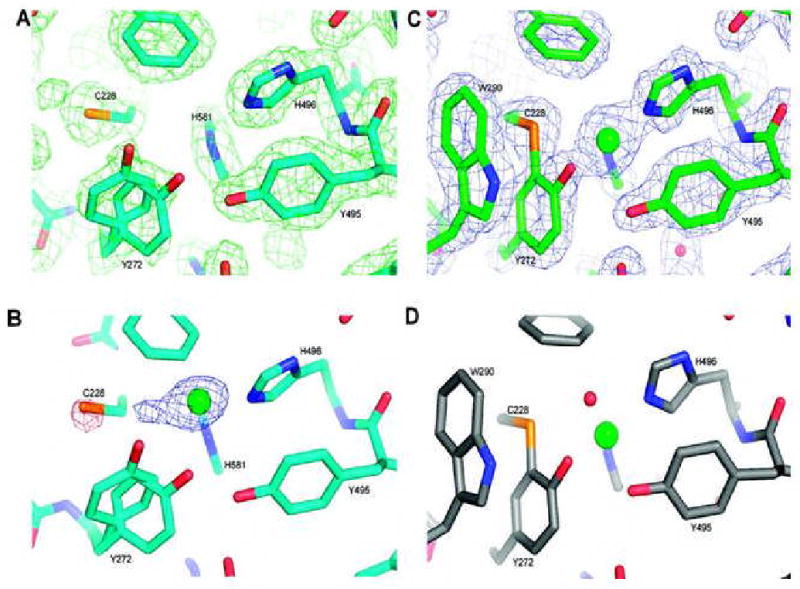
Crystal structures of the active site of GO processing forms. (A) Premat-GO prepared and crystallized under copper-free conditions, showing the final 2Fo – Fc map contoured at 1.0σ. (B) After a 3 min Cu(II) anaerobic soak, the Fo(2 mM copper soak) – Fo(premat copper free) map contoured at 4σ indicates coordination of copper to the active site histidines. A negative peak on the C228 sulfur and a positive peak adjacent to the copper are consistent with rotation of C228 to coordinate to the incoming copper. The structure shown is the premat-GO with a modeled copper ion (green). Note that W290 is not well ordered and is not shown. (C) After a 24 h Cu(II) anaerobic soak, the final Fo – Fc map contoured at 1.0σ shows the thioether bond clearly formed and with W290 now in a stacking interaction with the C228–Y272 feature. (D) Fully processed mature GO (PDB entry 1GOG) for comparison with panel C.
To trap intermediates during anaerobic thioether bond formation, crystals were soaked in the crystallization buffer also containing copper acetate and diffraction data were collected to 2.0 Å. An Fo(2 mM Cu(II) soak) - Fo(premat) map from a crystal that had been soaked for 3 min in a solution containing 2 mM copper acetate contained a strong positive peak in the active site region, identified as a copper ion, and two smaller peaks, one positive peak adjacent to the copper peak and one negative peak of comparable size centered on the cysteine sulfur (Figure 8B). The maps strongly suggest that copper binds to the two active site histidines (H581 and H496) and that the C228 side chain rotates around χ1 to coordinate to the copper. The copper occupancy appeared to be low (approximately 20%), however, so to increase the amount of copper at the active site, the soak was repeated with 10 mM copper acetate. The resulting maps indicated there was more disorder than with the lower-concentration soak. Several regions of the protein, similar to those that differed between pro-GO and the mature protein (42), are disordered (Y189-I201, D216-F227, G244-D258, and W290-F295), which may indicate the transitory nature of the sulfur-liganded copper species. These two copper soaks suggest that the approximate Cu-S distance is 2.2 Å, with the three copper ligands in an almost T-shaped arrangement. It appears that there is no fourth ligand, equatorial or axial, though Y495 is present approximately 3.2 Å from the copper, and therefore not directly coordinating. The possibility of a fourth equatorial ligand, such as water, cannot be excluded, but this ligand may be unidentifiable due to the mixture of species in the crystals. The electron density also suggests that a greater proportion of Y272 is positioned as in the pro- GO crystal structure with the plane of the ring rotated approximately 90° from its position in the mature GO. In this position, Y272 sterically hinders W290 from stacking over the tyrosyl side chain as in the mature GO. Because of the low occupancy of the copper site, and large regions of disorder, these structures were not fully refined.
Several anaerobic soaks with 2 mM copper acetate were carried out and crystals flash-frozen at longer intervals. Disorder in crystals soaked for less than 24 h precluded diffraction data analysis. After 24 h, however, the Cu(II)-soaked crystals diffracted to 1.9 Å with an R-factor of 16.8% and an Rfree of 20.1% (Table 1; PDB entry 2VZ3). The structure was found to be largely identical to that of mature GO (Cα rms deviation of 0.36 Å) except for a few significant active site differences. The maps clearly showed that the thioether bond has formed with rotation of Y272 and movement of W290 to stack over the C228-Y272 feature (Figure 8C). Copper coordination was similar to that of mature GO (Figure 8D) provided by the three equatorial ligands, H496, H581, and Y272, and Y495 was observed as the axial ligand with a distance of 2.9 Å (22, 23). Interestingly, a fourth equatorial ligand, water or acetate in the original crystal structure (22, 23), was not observed, although an acetate molecule was observed close to the copper ion hydrogen bonding to Nη1 and Nη2 of R330 and the hydroxyl group of Y272. An acetate molecule has not previously been observed in this position.
The structures described here show that C228-Y272 processing can go to completion, anaerobically in crystals. The short copper soak revealed a copper geometry not previously observed in galactose oxidase and appears to represent a Cu(I) thiolate-liganded form of the protein lacking the cross-link (Figure 8B). The 24 h soak is suggested to represent the bleached species that has a fully formed thioether bond (Figure 8C).
Discussion
Our previous studies have shown that processing of galactose oxidase requires four post-translational events: cleavage of the secretion signal, cleavage of the N-terminal pro sequence, formation of the C228-Y272 thioether bond, and, finally, one-electron oxidation generating the tyrosyl radical Y272· (27, 42). The latter three processing steps have been shown to proceed in vitro without the involvement of additional processing enzymes. To separate the oxidative processing steps from the pro peptide cleavage step, we have studied in vitro processing of premat-GO, a recombinant form of GO without the pro sequence (29) which was expressed and isolated under copper-depleted conditions.
First, we examined aerobic processing of premat-GO following addition of 3.5 equiv of Cu(II). Mature active enzyme was generated with a specific activity of 560 μmol min-1 mg-1, corresponding to 70% of the specific activity of the wild-type enzyme produced under copper-supplemented conditions. Polarographic studies showed that ∼1.3 mol of dioxygen was reduced per mole of premat-GO. These results may be compared with those of Whittaker and Whittaker (28), showing that essentially fully active enzyme was produced when their premat-GO was incubated aerobically with substoichiometric amounts of Cu(I) and that 1.8 mol of dioxygen was reduced per mole of protein. The difference in the polarography data when Cu(II) as opposed to Cu(I) is used suggests that with Cu(II), one electron from the protein reduces Cu(II) to Cu(I).
Our aerobic processing data further show that the rate constant for thioether formation (0.3 ± 0.05 min-1) is comparable to the rate constant for Y272 radical formation (0.21 ± 0.03 min-1), suggesting that thioether bond formation is rate-limiting in the oxidative steps of processing. Rapid aerobic processing with substoichiometric amounts of Cu(I), which is ∼4700-fold faster than with Cu(II) (28), suggested that the physiologically relevant form is Cu(I), which is the form of copper present intracellularly. It is clear that aerobic processing can be initiated by either Cu(I) or Cu(II) depending upon which valence state of copper is available and can proceed rapidly with Cu(II) in excess. To separate the chemical steps involved in the two oxidative steps of premat-GO processing (thioether bond formation and activation to the Y272 radical), anaerobic reactions were performed. Monitoring formation of the C228-Y272 thioether bond by SDS-PAGE showed that anaerobic addition of 0.8 equiv of Cu(II) to premat-GO resulted in slow formation of the bond. The possibility that this was due to the presence of contaminating dioxygen was precluded by the observation of similar rates of thioether bond formation with and without the dioxygen scavenging system of glucose and glucose oxidase. Spectrophotometric studies showed that anaerobic addition of 0.8 equiv of Cu(II) to premat-GO leads to formation of a species with a λmax of 406 nm. This species was previously shown to appear transiently following aerobic addition of Cu(II) to the pro sequence form of GO (27) and has been observed when Cu(II) is added to a premat-GO under anaerobic conditions (28). The rate constant for formation of the 406 nm species measured under anaerobic conditions (0.036 ± 0.001 min-1) is similar to that previously reported (0.019 min-1) (28). Whereas Whittaker and Whittaker (28) find that the 406 nm species is stable anaerobically, we observe decay to a bleached species with a rate constant of 0.003 min-1. During this bleaching, the thioether bond forms to ∼80% completion, which represents full processing by the added Cu(II) and evidence that only 1 equiv of Cu(II) is required for bond formation. This conclusion is supported by the EPR copper quantification experiments which showed that in the presence of a 3.5-fold molar excess of Cu(II), only 1 molar equiv of Cu(II) was reduced.
The 406 nm species has been characterized by CD and resonance Raman spectroscopies which together indicate cysteine-s- and histidine-copper coordination. Whittaker and Whittaker (28) have previously assigned the 406 nm absorption to a Cu(II) thiolate ligand-to-metal charge transfer based on resonance Raman evidence. A similar optical absorption feature has been observed in designed four-helix bundle proteins bearing a copper center (47, 48) and in designed variants of blue copper proteins (49, 50).
X-ray crystallographic studies with premat-GO crystals incubated anaerobically with Cu(II) for 3 min reveal that the copper ion is coordinated to H581, H496, and C228. This tricoordinate geometry of the Cu(II) could favor reduction to Cu(I). C228 has undergone movement from its premat- GO position to coordinate to copper. Y272 does not occupy its mature enzyme position, but the plane of the ring is rotated by some 90°. The C228 S atom appears to be overlaying the π-system of Y272 which is appropriately positioned for a cross-linking reaction.
The rate of anaerobic thioether bond formation with Cu(II) matches the rate of decay of the 406 nm species. The resulting bleached species is probably that characterized by X-ray crystallography when premat-GO crystals were incubated anaerobically with Cu(II) for 24 h. In this structure, the thioether bond is formed and there is no coordination of copper to C228 (PDB entry 2VZ3). Taken together, these observations lead to the conclusion that under anaerobic conditions, the 406 nm species represents a competent processing intermediate species preceding thioether bond formation. There is evidence of thioether formation between Cu(II)-coordinated cysteine and added tyrosine under anaerobic conditions in a model system (51).
The results reported here allow proposal of a chemical mechanism for thioether bond formation in premat-GO following addition of Cu(II) under both anaerobic and aerobic conditions (Scheme 1). Overall, C228-Y272 bond formation requires removal of two electrons and two protons. A proton is lost in generating C228 thiolate which then coordinates to the Cu(II) (species B) under the pH 6.8 conditions of the reaction. A group with a pKa of 7.3 is associated with higher reaction rates in the deprotonated form and is proposed to be C228 by Whittaker and Whittaker (28). An electron is then transferred from thiolate to Cu(II), forming a thiyl radical and Cu(I) (species C). The thiyl radical is proposed to attack the aromatic ring of Y272 to give species D where the unpaired electron is delocalized on the Y272 ring. All the steps described above could proceed under both aerobic and anaerobic conditions, and given the similar rates of decay of the 406 nm species, the rate-limiting step in conversion of the carbonium ion intermediate to give the thioether- bonded species is probably the same under both anaerobic and aerobic conditions. Subsequently, under aerobic conditions, two electrons from species D reduce dioxygen, forming H2O2 with reoxidation of Cu(I) to Cu(II) (species E). The identity of the second electron acceptor participating in the anaerobic conversion of species D to species F is unclear. Potential candidates include C515 and C518, but these can be ruled out as the cystine bond between these residues is intact in the 24 h crystal structure. We note that species D would be reactive, probably with a low redox potential capable of losing an electron and yielding the carbonium ion (species F). Loss of a proton would give species G.
scheme 1.
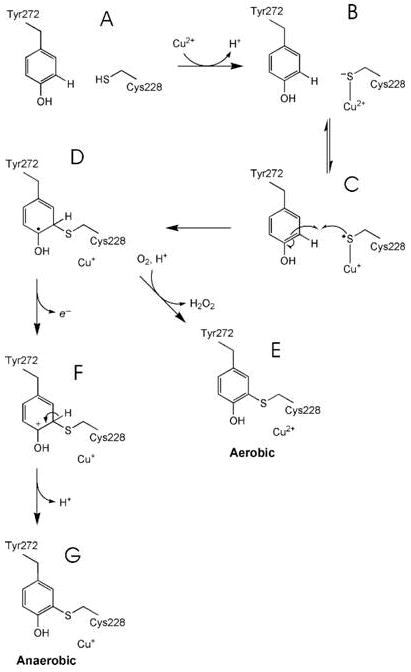
In our earlier paper describing the structure of the pro sequence precursor form of GO (42), possible thioether bond formation mechanisms, based on established chemistry, were discussed. The first of these postulated mechanisms involved initial coordination of Y272 to Cu(II) and subsequent formation of the Y272 radical. However, there is no evidence of coordination of Y272 to Cu(II) in the crystal structure of premat- GO soaked for 3 min with Cu(II); rather, this new structure points strongly to initial coordination of Cu(II) to C228 and favors the mechanism shown in Scheme 1. The second mechanism postulated from the pro-GO crystal structure involved early oxidation of C228 based on our observation of electron density which could be attributed to sulfenate modification. However, similar extra electron density at C228 was not observed in the structure of premat-GO reported here. Moreover, anaerobic processing argues against a cysteine sulfenate intermediate. Most probably, the electron density attributed to sulfenate in the pro sequence precursor form is an oxidative artifact resulting from X-ray exposure.
The observed instability of mature active GO following incubation of the bleached 406 nm species with dioxygen is attributed to some minor difference in the active site, which could also account for the inability of ferricyanide to convert the bleached 406 nm species to mature active GO under the in vitro conditions.
Is the anaerobic processing pathway physiologically significant? In eukaryotic cells, free copper is highly toxic, so its uptake and intracellular trafficking are carefully controlled by transporter proteins and intracellular chaperones (52). Copper homeostasis in eukaryotes has recently been reviewed (53). The substrate for the copper transporter (Ctr) proteins is Cu(I) which is generated from Cu(II) by plasma membrane reductases. Intracellularly, the Cu(I) is acquired by a high-affinity chaperone of the Atx type that translocates the copper to the ATPase (ATP7A-type) transporter located in the trans-Golgi network. This transporter delivers copper to proteins in the secretory pathway which require copper for activity (53). Little is known about the copper transfer processes in the trans-Golgi network, although, as pointed out by Davis and O'Halloran (54), copper trafficking proteins bind Cu(I) exclusively whereas the recipient copper enzymes often accommodate both Cu(II) and Cu(I) oxidation states. It seems likely that GO would be competent to accept copper in the trans-Golgi compartment as it has an open active site architecture near the surface of the protein (22). There is apparently no information about the redox potential or oxygen levels in the trans- Golgi network, but it seems unlikely that conditions are appropriate for final oxidation of GO to the radical form. The redox potential within the cytoplasm is in the range of -190 to -250 mV (55), but the secretory pathway is more oxidizing as disulfide bonds must be formed within the ER where the redox potential is some -120 to -180 mV (56, 57). Although the trans-Golgi network may be more oxidizing than the ER, it is unlikely to achieve a redox potential of 400 mV required for oxidation of the Cu(II)-Y272 species to a radical, even in the presence of cellular oxygen. Indeed, biologically the production of an active GO would likely be detrimental to the cell due to the potential for GO-mediated oxidation of various sugars on glycosylated proteins in transit through the trans-Golgi network. It seems more likely that formation of a thioether bond occurs in the trans-Golgi network, but that the enzyme is not oxidized to the active radical form until it is secreted into the extracellular milieu where the environment is more oxidizing and a wide range of oxidants would be available.
Some of our observations on anaerobic formation of the C228-Y272 cofactor seem likely to be physiologically relevant for galactose oxidase maturation. The anaerobic processing is also of chemical interest and may provide a precedent for cross-link formation in proteins found in anaerobic environments.
Acknowledgments
We thank David J. Lowe and Shirley Fairhurst (John Innes Centre) for the anaerobic facilities, EPR spectroscopy, and helpful discussions; Brad Elmore and Doreen Brown for experimental assistance; Joe Jaeger and Carrie Wilmot for helpful discussions; Helen Dawkes and the staff of Daresbury SRS for assistance with data collection; and Richard J. Bushby for helpful discussions on the mechanistic scheme. Thanks to Mark Reinholz for excellent technical support.
Footnotes
This work was supported by the Biotechnology and Biological Sciences Research Council (24/B11662) and NIH (GM 27659). S.J.F. was supported by an EPSRC studentship.
PDB entries 2VZ1 and 2VZ3.
Abbreviations: EDTA, ethylenediaminetetraacetic acid; Pipes, piperazine-1,4-bis(2-ethanesulfonic acid); Tris, tris(hydroxymethyl)- aminomethane.
References
- 1.Okeley NM, van der Donk WA. Novel cofactors via post-translational modifications of enzyme active sites. Chem Biol. 2000;7:R159–R171. doi: 10.1016/s1074-5521(00)00140-x. [DOI] [PubMed] [Google Scholar]
- 2.Stubbe JA. Protein radical involvement in biological catalysis. Annu Rev Biochem. 1989;58:257–285. doi: 10.1146/annurev.bi.58.070189.001353. [DOI] [PubMed] [Google Scholar]
- 3.Stubbe JA, van der Donk WA. Protein radicals in enzyme catalysis. Chem ReV. 1998;98:705–762. doi: 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]
- 4.Frey PA. Radical mechanisms of enzymatic catalysis. Annu Rev Biochem. 2001;70:121–148. doi: 10.1146/annurev.biochem.70.1.121. [DOI] [PubMed] [Google Scholar]
- 5.Banerjee R. Introduction: Radical enzymology. Chem Rev. 2003;103:2081–2081. [Google Scholar]
- 6.Janes SM, Mu D, Wemmer D, Smith AJ, Kaur S, Maltby D, Burlingame AL, Klinman JP. A new redox cofactor in eukaryotic enzymes: 6-Hydroxydopa at the active site of bovine serum amine oxidase. Science. 1990;248:981–987. doi: 10.1126/science.2111581. [DOI] [PubMed] [Google Scholar]
- 7.Ruggiero CE, Smith JA, Tanizawa K, Dooley DM. Mechanistic studies of topa quinone biogenesis in phenylethylamine oxidase. Biochemistry. 1997;36:1953–1959. doi: 10.1021/bi9628836. [DOI] [PubMed] [Google Scholar]
- 8.Kim M, Okajima T, Kishishita S, Yoshimura M, Kawamori A, Tanizawa K, Yamaguchi H. X-ray snapshots of quinone cofactor biogenesis in bacterial copper amine oxidase. Nat Struct Biol. 2002;9:591–596. doi: 10.1038/nsb824. [DOI] [PubMed] [Google Scholar]
- 9.Samuels NM, Klinman JP. Investigation of Cu(I)- dependent 2,4,5-trihydroxyphenylalanine quinone biogenesis in Hansenula polymorpha amine oxidase. J Biol Chem. 2006;281:21114–21118. doi: 10.1074/jbc.M601501200. [DOI] [PubMed] [Google Scholar]
- 10.Wang SX, Mure M, Medzihradszky KF, Burlingame AL, Brown DE, Dooley DM, Smith AJ, Kagan HM, Klinman JP. A crosslinked cofactor in lysyl oxidase: Redox function for amino acid side chains. Science. 1996;273:1078–1084. doi: 10.1126/science.273.5278.1078. [DOI] [PubMed] [Google Scholar]
- 11.Yamada Y, Fujiwara T, Sato T, Igarashi N, Tanaka N. The 2.0 Å crystal structure of catalase-peroxidase from Haloarcula marismortui. Nat Struct Biol. 2002;9:691–695. doi: 10.1038/nsb834. [DOI] [PubMed] [Google Scholar]
- 12.Ghiladi RA, Knudsen GM, Medzihradszky KF, de Montellano PRO. The Met-Tyr-Trp cross-link in Mycobacterium tuberculosis catalase-peroxidase (KatG): Autocatalytic formation and effect on enzyme catalysis and spectroscopic properties. J Biol Chem. 2005;280:22651–22663. doi: 10.1074/jbc.M502486200. [DOI] [PubMed] [Google Scholar]
- 13.Smulevich G, Jakopitsch C, Droghetti E, Obinger C. Probing the structure and bifunctionality of catalase-peroxidase (KatG) J Inorg Biochem. 2006;100:568–585. doi: 10.1016/j.jinorgbio.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 14.Ostermeier C, Harrenga A, Ermler U, Michel H. Structure at 2.7 angstrom resolution of the Paracoccus denitrificans two-subunit cytochrome c oxidase complexed with an antibody FV fragment. Proc Natl Acad Sci USA. 1997;94:10547–10553. doi: 10.1073/pnas.94.20.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshikawa S, Shinzawa-Itoh K, Nakashima R, Yaono R, Yamashita E, Inoue N, Yao M, Fei MJ, Libeu CP, Mizushima T, Yamaguchi H, Tomizaki T, Tsukihara T. Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science. 1998;280:1723–1729. doi: 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]
- 16.Proshlyakov DA, Pressler MA, DeMaso C, Leykam JF, DeWitt DL, Babcock GT. Oxygen activation and reduction in respiration: Involvement of redox-active tyrosine 244. Science. 2000;290:1588–1591. doi: 10.1126/science.290.5496.1588. [DOI] [PubMed] [Google Scholar]
- 17.Schnell R, Sandalova T, Hellman U, Lindqvist Y, Schneider G. Siroheme- and Fe-4-S-4-dependent NirA from Mycobacterium tuberculosis is a sulfite reductase with a covalent Cys-Tyr bond in the active site. J Biol Chem. 2005;280:27319–27328. doi: 10.1074/jbc.M502560200. [DOI] [PubMed] [Google Scholar]
- 18.McCoy JG, Bailey LJ, Bitto E, Bingman CA, Aceti DJ, Fox BG, Phillips GN. Structure and mechanism of mouse cysteine dioxygenase. Proc Natl Acad Sci USA. 2006;103:3084–3089. doi: 10.1073/pnas.0509262103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simmons CR, Liu Q, Huang QQ, Hao Q, Begley TP, Karplus PA, Stipanuk MH. Crystal structure of mammalian cysteine dioxygenase: A novel mononuclear iron center for cysteine thiol oxidation. J Biol Chem. 2006;281:18723–18733. doi: 10.1074/jbc.M601555200. [DOI] [PubMed] [Google Scholar]
- 20.Whittaker MM, Kersten PJ, Cullen D, Whittaker JW. Identification of catalytic residues in glyoxal oxidase by targeted mutagenesis. J Biol Chem. 1999;274:36226–36232. doi: 10.1074/jbc.274.51.36226. [DOI] [PubMed] [Google Scholar]
- 21.Whittaker MM, Kersten PJ, Nakamura N, SandersLoehr J, Schweizer ES, Whittaker JW. Glyoxal oxidase from Phanerochaete chrysosporium is a new radical-copper oxidase. J Biol Chem. 1996;271:681–687. doi: 10.1074/jbc.271.2.681. [DOI] [PubMed] [Google Scholar]
- 22.Ito N, Phillips SEV, Stevens C, Ogel ZB, McPherson MJ, Keen JN, Yadav KDS, Knowles PF. Novel thioether bond revealed by a 1.7 Å crystal structure of galactose oxidase. Nature. 1991;350:87–90. doi: 10.1038/350087a0. [DOI] [PubMed] [Google Scholar]
- 23.Ito N, Phillips SEV, Yadav KDS, Knowles PF. Crystal structure of a free radical enzyme, galactose oxidase. J Mol Biol. 1994;238:794–814. doi: 10.1006/jmbi.1994.1335. [DOI] [PubMed] [Google Scholar]
- 24.Johnson JM, Halsall HB, Heineman WR. Redox activation of galactose oxidase: Thin layer electrochemical study. Biochemistry. 1985;24:1579–1585. doi: 10.1021/bi00328a001. [DOI] [PubMed] [Google Scholar]
- 25.Rogers MS, Dooley DM. Post-translationally modified tyrosines from galactose oxidase and cytochrome C oxidase. Adv Protein Chem. 2001;58:387–436. doi: 10.1016/s0065-3233(01)58009-2. [DOI] [PubMed] [Google Scholar]
- 26.Rokhsana D, Dooley DM, Szilagyi RK. Systematic development of computational models for the catalytic site in galactose oxidase: Impact of outer-sphere residues on the geometric and electronic structures. J Biol Inorg Chem. 2008;13:371–383. doi: 10.1007/s00775-007-0325-8. [DOI] [PubMed] [Google Scholar]
- 27.Rogers MS, Baron AJ, McPherson MJ, Knowles PF, Dooley DM. Galactose oxidase pro-sequence cleavage and cofactor assembly are self-processing reactions. J Am Chem Soc. 2000;122:990–991. [Google Scholar]
- 28.Whittaker MM, Whittaker JW. Cu(I)-dependent biogenesis of the galactose oxidase redox cofactor. J Biol Chem. 2003;278:22090–22101. doi: 10.1074/jbc.M300112200. [DOI] [PubMed] [Google Scholar]
- 29.Deacon SE, Mahmoud K, Spooner RK, Firbank SJ, Knowles PF, Phillips SEV, McPherson MJ. Enhanced fructose oxidase activity in a galactose oxidase variant. ChemBioChem. 2004;5:972–979. doi: 10.1002/cbic.200300810. [DOI] [PubMed] [Google Scholar]
- 30.McPherson MJ, Ogel ZB, Stevens C, Yadav KDS, Keen JN, Knowles PF. Galactose oxidase of Dactylium dendroides: Gene cloning and sequence analysis. J Biol Chem. 1992;267:8146–8152. [PubMed] [Google Scholar]
- 31.Kosman DJ, Ettinger MJ, Weiner RE, Massaro EJ. Molecular properties of copper enzyme galactose oxidase. Arch Biochem Biophys. 1974;165:456–467. doi: 10.1016/0003-9861(74)90271-9. [DOI] [PubMed] [Google Scholar]
- 32.McPherson MJ, Parsons MR, Spooner RK, Wilmot CW. Galactose oxidase. In: Messerschmidt A, Huber R, Poulos T, Wieghardt K, editors. Handbook of Metalloproteins. John Wiley & Sons, Ltd.; Chichester, U.K.: 2001. pp. 1272–1283. [Google Scholar]
- 33.Jones RW, Garland PB. Sites and specificity of reaction of bipyridylium compounds with anaerobic respiratory enzymes of Escherichia coli: Effects of permeability barriers imposed by cytoplasmic membrane. Biochem J. 1977;164:199–211. doi: 10.1042/bj1640199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Englander SW, Calhoun DB, Englander JJ. Biochemistry without Oxygen. Anal Biochem. 1987;161:300–306. doi: 10.1016/0003-2697(87)90454-4. [DOI] [PubMed] [Google Scholar]
- 35.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;267:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 36.Bailey S. The CCP4 Suite: Programs for protein crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 37.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 38.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 39.Baron AJ, Stevens C, Wilmot C, Seneviratne KD, Blakeley V, Dooley DM, Phillips SEV, Knowles PF, McPherson MJ. Structure and mechanism of galactose oxidase: The free radical site. J Biol Chem. 1994;269:25095–25105. [PubMed] [Google Scholar]
- 40.McPherson MJ, Stevens C, Baron AJ, Ogel ZB, Seneviratne K, Wilmot C, Ito N, Brocklebank I, Phillips SEV, Knowles PF. Galactose oxidase: Molecular analysis and mutagenesis studies. Biochem Soc Trans. 1993;21:752–756. doi: 10.1042/bst0210752. [DOI] [PubMed] [Google Scholar]
- 41.Dominy JE, Jr, Hwang J, Guo S, Hirschberger LL, Zhang S, Stipanuk MH. Synthesis of amino acid cofactor in cysteine dioxygenase is regulated by substrate and represents a novel post-translational regulation of activity. J Biol Chem. 2008;283:12188–12201. doi: 10.1074/jbc.M800044200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Firbank SJ, Rogers MS, Wilmot CM, Dooley DM, Halcrow MA, Knowles PF, McPherson MJ, Phillips SEV. Crystal structure of the precursor of galactose oxidase: An unusual self-processing enzyme. Proc Natl Acad Sci USA. 2001;98:12932–12937. doi: 10.1073/pnas.231463798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whittaker MM, Whittaker JW. The active site of galactose oxidase. J Biol Chem. 1988;263:6074–6080. [PubMed] [Google Scholar]
- 44.Rogers MS, Knowles PF, Baron AJ, McPherson MJ, Dooley DM. Characterization of the active site of galactose oxidase and its active site mutational variants Y495F/H/K and W290H by circular dichroism spectroscopy. Inorg Chim Acta. 1998;276:175–181. [Google Scholar]
- 45.Whittaker MM, Ballou DP, Whittaker JW. Kinetic isotope effects as probes of the mechanism of galactose oxidase. Biochemistry. 1998;37:8426–8436. doi: 10.1021/bi980328t. [DOI] [PubMed] [Google Scholar]
- 46.Vinecombe E. PhD Thesis. University of Leeds; Leeds, U.K.: 1999. X-ray crystallographic studies on copper containing oxidases. [Google Scholar]
- 47.Schnepf R, Haehnel W, Wieghardt K, Hildebrandt P. Spectroscopic identification of different types of copper centers generated in synthetic four-helix bundle proteins. J Am Chem Soc. 2004;126:14389–14399. doi: 10.1021/ja0484294. [DOI] [PubMed] [Google Scholar]
- 48.Schnepf R, Horth P, Bill E, Wieghardt K, Hildebrandt P, Haehnel W. De novo design and characterization of copper centers in synthetic four-helix-bundle proteins. J Am Chem Soc. 2001;123:2186–2195. doi: 10.1021/ja001880k. [DOI] [PubMed] [Google Scholar]
- 49.Hellinga HW. Construction of a blue copper analogue through iterative rational protein design cycles demonstrates principles of molecular recognition in metal center formation. J Am Chem Soc. 1998;120:10055–10066. [Google Scholar]
- 50.van Amsterdam IMC, Ubbink M, van den Bosch M, Rotsaert F, Sanders-Loehr J, Canters GW. A new type 2 copper cysteinate azurin: Involvement of an engineered exposed cysteine in copper binding through internal rearrangement. J Biol Chem. 2002;277:44121–44130. doi: 10.1074/jbc.M202977200. [DOI] [PubMed] [Google Scholar]
- 51.Lee Y, Lee DH, Narducci Sarjeant AA, Karlin KD. Thiol-copper(I) and disulfide-dicopper(I) complex O2-reactivity leading to sulfonate-copper(II) complex or the formation of a cross-linked thioether-phenol product with phenol addition. J Inorg Biochem. 2007;101:1845–1858. doi: 10.1016/j.jinorgbio.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 52.Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O'Halloran TV. Undetectable intracellular free copper: The requirement of a copper chaperone for superoxide dismutase. Science. 1999;284:805–808. doi: 10.1126/science.284.5415.805. [DOI] [PubMed] [Google Scholar]
- 53.Balamurugan K, Schaffner W. Copper homeostasis in eukaryotes: Teetering on a tightrope. Biochim Biophys Acta. 2006;1763:737–746. doi: 10.1016/j.bbamcr.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 54.Davis AV, O'Halloran TV. A place for thioether chemistry in cellular copper ion recognition and trafficking. Nat Chem Biol. 2008;4:148–151. doi: 10.1038/nchembio0308-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radical Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 56.Sevier CS, Kaiser CA. Formation and transfer of disulphide bonds in living cells. Nat Rev Mol Cell Biol. 2002;3:836–847. doi: 10.1038/nrm954. [DOI] [PubMed] [Google Scholar]
- 57.Hwang C, Sinskey AJ, Lodish HF. Oxidised redox state of glutathione in the endoplasmic reticulum. Science. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]