

Abstract
The influence of genetic polymorphism in inosine triphosphate pyrophosphatase (ITPA) on thiopurine-induced adverse events has not been investigated in the context of combination chemotherapy for acute lymphoblastic leukemia (ALL). This study investigated the effects of a common ITPA variant allele (rs41320251) on mercaptopurine metabolism and toxicity during treatment of children with ALL. Significantly higher concentrations of methyl mercaptopurine nucleotides were found in patients with the nonfunctional ITPA allele. Moreover, there was a significantly higher probability of severe febrile neutropenia in patients with a variant ITPA allele among patients whose dose of mercaptopurine had been adjusted for TPMT genotype. In a cohort of patients whose mercaptopurine dose was not adjusted for TPMT phenotype, the TPMT genotype had a greater effect than the ITPA genotype. In conclusion, genetic polymorphism of ITPA is a significant determinant of mercaptopurine metabolism and of severe febrile neutropenia, after combination chemotherapy for ALL in which mercaptopurine doses are individualized on the basis of TPMT genotype.
Approximately 80% of children with acute lymphoblastic leukemia (ALL) can be cured with combination chemotherapy.1,2 However, treatment-related toxicity can be life threatening and is the primary cause of interruption or discontinuation of chemotherapy, leading to an increase in relapse risk.1,3,4 Germline polymorphisms in genes encoding drug-metabolizing enzymes, drug transporters, and drug targets can significantly influence the pharmacokinetics and pharmacological effects of medications and can be significant determinants of the efficacy and toxicity of antileukemic therapy.5–7 Indeed, the influence of genetic polymorphism in thiopurine S-methyltransferase (TPMT) on the pharmacokinetics and toxicity of mercaptopurine is one of the clearest examples of a clinically important pharmacogenetic trait.8–10 Mercaptopurine, an analog of hypoxanthine, is widely used in leukemia therapy;1,2,11 mercaptopurine and its prodrug azathioprine are also used as immunosuppressive therapy in inflammatory bowel disease and other autoimmune disorders.12,13 Mercaptopurine requires intracellular activation to thiopurine nucleotides to exert its pharmacological effects. Intracellularly, mercaptopurine is converted into thioinosine monophosphate by hypoxanthine guanine phosphoribosyltransferase and is subsequently converted into thioguanosine monophosphate through a two-step process involving inosine monophosphate dehydrogenase and guanosine monophosphate synthetase.14 This process is in competition with methylation by TPMT, which is influenced by a common genetic polymorphism in the TPMT gene.9 TPMT converts mercaptopurine into inactive methyl mercaptopurine but also metabolizes thioinosine monophosphate into methyl thioinosine monophosphate, a molecule that can inhibit de novo purine synthesis.15 The molecular mechanisms of mercaptopurine’s antileukemic effects are related to interference with the activity of DNA-processing enzymes due to subtle structural changes in the DNA after incorporation of thioguanine nucleotides (TGNs).14,16 The importance of inhibition of de novo purine synthesis (mainly by methyl mercaptopurine nucleotides, MMPNs) is less well defined in the treatment of ALL.9
It is well established that nonfunctional variant alleles of the TPMT gene encode proteins that are rapidly degraded, resulting in low enzymatic activity. Inheritance of these variant alleles is associated with a marked increase in the concentration of TGNs and a significantly higher risk of hematopoietic toxicity after mercaptopurine treatment.10 However, it is less clear whether polymorphisms in genes encoding other enzymes involved in mercaptopurine metabolism (Figure 1) also influence its efficacy and toxicity. Indeed, some patients with wild-type TPMT alleles develop mercaptopurine-related adverse events, for reasons that are not fully understood.
Figure 1.

Schematic representation of the metabolism of mercaptopurine and the enzymes involved. GDP, guanosine diphosphate; GMP, guanosine monophosphate; GMPS, guanosine monophosphate synthase; GTP, guanosine triphosphate; HPRT, hypoxanthine phosphoribosyltransferase; IMPDH, inosine monophosphate dehdrogenase; IDP, inosine diphosphate; IMP, inosine monophosphate; ITP, inosine triphosphate; ITPA, inosine triphosphate pyrophosphatase; K, kinase; TPMT, thiopurine S-methyltransferase; XO, xanthine oxidase.
Among various possible candidate genes, inosine triphosphate pyrophosphatase (ITPA) has been associated, in some studies, with adverse events from azathioprine and mercaptopurine treatment of inflammatory bowel disease,17–19 whereas other studies have failed to show any significant association between ITPA polymorphism and adverse events from the use of these agents in treating inflammatory bowel disease;20–22 as such, this issue remains unresolved.23,24 ITPA is an enzyme that catalyzes the hydrolysis of inosine triphosphate (ITP) to inosine monophosphate (IMP).24 IMP is a central intermediate in purine metabolism and can be converted to ITP, to ATP through adenosine monophosphate, or to guanosine triphosphate through guanosine monophosphate. The putative role of ITPA is to protect cells from the accumulation of potentially harmful nucleotides, such as ITP or deoxyinosine triphosphate, that may be incorporated into the RNA and DNA.25 The single-nucleotide polymorphism (SNP) rs41320251 is a C>A transversion (minor allele frequency: 0.083 in Caucasians, 0.033 in Africans, and 0.11 in Asians26) located in exon 2 of the gene.25 It causes an amino acid change (P32T) that abolishes ITPA enzymatic activity in homozygous individuals and reduces the activity to 25% in heterozygous subjects;25,27,28 this pattern is consistent with impaired assembly of the dimeric structure of the enzyme resulting from the P32T amino acid change.29 is genetic polymorphism, leading to lower ITPA enzyme activity, gives rise to the physiological effect of abnormal accumulation of ITP in cells, which by itself is a clinically benign condition.25,30 Characterization of the ITPA haplotype structure has shown that the SNP rs41320251 is the most relevant polymorphism in determining low ITPA enzymatic activity.28
The influence of ITPA genetic polymorphism on mercaptopurine toxicity has not been defined in the context of combination chemotherapy for ALL. This study assessed the influence of nonfunctional variant alleles of TPMT and ITPA on mercaptopurine metabolism and toxicity in patients with ALL whose mercaptopurine dosages had been adjusted on the basis of TPMT genotype.
RESULTS
Patients and TPMT/ITPA genotyping
For patients enrolled in the Total 13B protocol, TPMT and ITPA genotypes were determined in 244 of the 246 children included; in 2 patients ITPA genotype was not evaluable because the SNP call rate was <95% and failed replicate analysis by TaqMan assay. Among the 244 patients with both TPMT and ITPA genotypes, 11 (4.5%) were found to have one variant allele of the TPMT gene and one wild-type ITPA allele, 33 (13.5%) had one variant ITPA allele and one wild-type TPMT allele, and 1 patient (0.41%) had one variant allele for both TPMT and ITPA; all of the other 199 patients (81.6%) were wild type for both TPMT and ITPA. Because only 1 patient had a variant allele for both TPMT and ITPA, we performed parallel analyses in which data from this patient were excluded or included. The significant findings did not differ between these two analyses. Results presented in this article are those attained excluding this patient; those obtained when this patient was included are reported in the Supplementary Data S1 online.
For patients enrolled in the Total 12 protocol, TPMT and ITPA genotypes were determined in 101 of the 188 patients: the other patients were not studied, either because DNA was not collected or a sufficient quantity was no longer available. Among the 101 patients studied, 12 (11.9%) were found to have one variant allele of the TPMT gene and one wild-type ITPA allele, 19 (18.8%) had one variant ITPA allele and one wild-type TPMT allele, and no patient had one variant allele for both TPMT and ITPA; all of the other 70 patients (69.3%) were wild type for both TPMT and ITPA. The demographic characteristics of the patients enrolled are reported in Table 1.
Table 1.
Demographic and clinical characteristics of patients with acute lymphoblastic leukemia enrolled in each treatment protocol
| Protocol | Total 12 | Total 13B |
|---|---|---|
| Total number of patients | 101 | 244 |
| Age in years (median and range) | 4.3 (0.60–18.7) | 5.9 (0.08–18.8) |
| Gender (n (%)) | ||
| Female | 46 (45.5) | 101 (41.4) |
| Male | 55 (54.4) | 143 (58.6) |
| Ethnic group (n (%)) | ||
| White | 91 (90.1) | 187 (76.6) |
| Black | 9 (8.9) | 45 (18.4) |
| Other | 1 (0.99) | 19 (7.8) |
| Treatment arm/risk groupa (n(%)) | ||
| Standard/high | — | 128 (52.5) |
| Low | — | 116 (47.5) |
Risk group classification was done only for patients in Total 13B protocol according to criteria previously described in detail.31
The genotype distributions were in Hardy–Weinberg equilibrium in both cohorts of patients.
Effect of TPMT/ITPA genotype on rate of de novo purine synthesis (Total 13B)
De novo purine synthesis was measured in ALL cells from 196 patients; 47 patients were not evaluable because of insufficient ALL cell yields from the bone marrow aspirate. No significant difference was found in the rate of de novo purine synthesis at diagnosis between the patients with wild-type ITPA (median 90.8 fmol/nmol/h, range 0–5,018, n = 171) and variant ITPA (median 52.8 fmol/nmol/h, range 0–1,441, n = 25) or between those with wild-type TPMT (median 89.6 fmol/nmol/h, range 0–5,018, n = 186) and variant TPMT (median 33.8 fmol/nmol/h, range 0.82–819.1, n = 10); no significant effect was found even when analyzed by the multilocus genotypes. The lack of an effect of TPMT and ITPA genotype was confirmed by multivariate analysis after adjusting for age, race, sex, and treatment arm.
Effect of TPMT/ITPA genotypes on mercaptopurine metabolites in bone marrow leukemia cells after initial therapy with mercaptopurine (Total 13B)
These studies were performed in the subgroup of patients treated with mercaptopurine alone: estimations were performed in 56 patients; TGN was measurable in only 49 patients because for 7 patients the assay failed for technical reasons. Neither ITPA nor TPMT genotypes were significantly related to TGN levels in bone marrow ALL cells from patients treated with an initial single dose of intravenous high-dose mercaptopurine, even after adjusting for the potential confounders: age, race, and sex (Figure 2). MMPN concentrations were significantly higher in patients with a variant ITPA allele and lower in those with a variant TPMT allele, both as per univariate analysis (ITPA variant vs. ITPA wild type, P = 0.0038; TPMT variant vs. TPMT wild type, P = 0.026) and in the pairwise analysis for the multilocus genotype (TPMT wild type/ITPA variant vs. TPMT wild type/ITPA wild type, P = 0.0056; TPMT variant/ITPA wild type vs. TPMT wild type/ITPA wild type, P = 0.030, Figure 3). Multivariate analysis adjusted for age, race, sex, and treatment arm confirmed, after initial treatment with mercaptopurine, the significant effect of the ITPA genotype (P = 0.00030), but not the TPMT genotype (P = 0.060), on MMPN concentrations. Boxplots showing mercaptopurine metabolite concentrations independently for ITPA and TPMT genotypes are available in Supplementary Data S1 online (Figures 1S and 2S, respectively).
Figure 2.
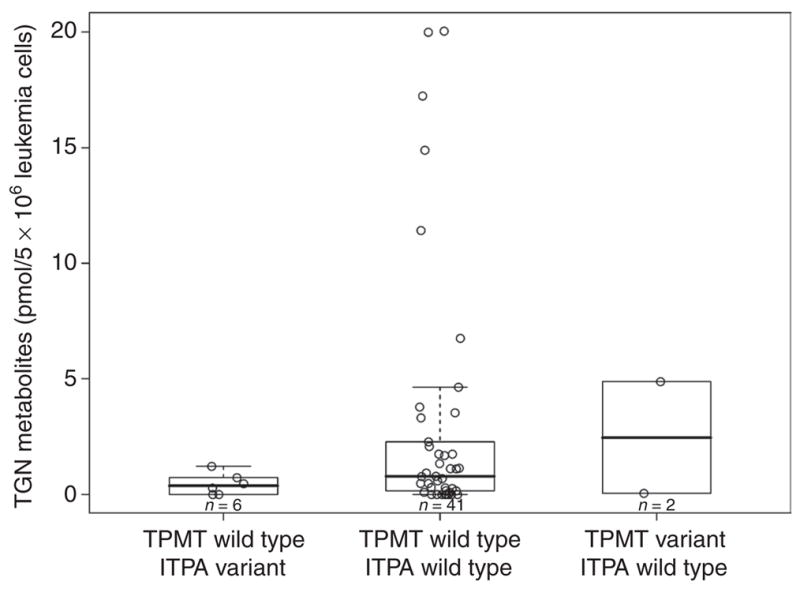
TPMT/ITPA multilocus genotype and concentrations of mercaptopurine thioguanine nucleotide metabolite (TGN) measured in bone marrow leukemia cells after initial treatment with mercaptopurine alone; concentrations did not differ significantly by TPMT/ITPA genotype. ITPA, inosine triphosphate pyrophosphatase; TPMT, thiopurine S-methyltransferase.
Figure 3.

TPMT/ITPA multilocus genotype and concentrations of mercaptopurine metabolite methylmercaptopurine nucleotide (MMPN) measured in bone marrow leukemia cells after initial treatment with mercaptopurine alone; concentrations were higher in patients with a multilocus genotype TPMT wild type/ITPA variant (P = 0.0056, Wilcoxon test) and lower in patients with a multilocus genotype TPMT variant/ITPA wild type (P = 0.030, Wilcoxon test) as compared to patients with a wild-type genotype for both genes. Boxes include data between the 25th and 75th percentiles, and whiskers indicate the minimal and maximal values excluding the outliers. ITPA, inosine triphosphate pyrophosphatase; TPMT, thiopurine S-methyltransferase.
Effect of TPMT/ITPA genotypes on mercaptopurine metabolites in erythrocytes during continuation therapy that includes mercaptopurine (Total 13B)
Concentrations of thiopurine metabolites were measured in erythrocytes obtained during continuation therapy with chronic oral mercaptopurine (75 mg/m2 daily). For TGN, 534 estimations were made in 113 patients (median of five measurements per patient, range 1–9); the median concentration measured for each patient was 267.5 pmol/8 × 108 erythrocytes (range 58.4–1,023.0 pmol/8 × 108) and, as a measure of intrapatient variability, the median interquartile range across patients was 82.2 (range 0.10–499.80 pmol/8 × 108). For MMPN, 278 measurements were made in 107 patients (median of two measurements per patient, range 1–8); the median concentration measured for each patient was 10,440.0 pmol/8 × 108 erythrocytes (range 54.5–37,780.0 pmol/8 × 108) and, as a measure of intrapatient variability, the median interquartile range across patients was 2,776.0 (range 21.0–13,730.0 pmol/8 × 108). TGN concentrations (Figure 4) were higher in patients with the variant TPMT genotype (TPMT variant vs. TPMT wild-type, P = 0.0077); no effect of ITPA genotype on TGN was found (ITPA variant vs. ITPA wild type, P = 0.95). Pairwise analysis of the multilocus genotype confirmed the effect of TPMT only on TGN concentrations. MMPN concentrations (Figure 5) were higher in patients with a variant ITPA allele and lower in those with a variant TPMT allele, both as per univariate analysis (ITPA variant vs. ITPA wild type, P = 0.0057; TPMT variant vs. TPMT wild type, P = 0.032) and in the pairwise analysis for the multilocus genotype (TPMT wild type/ITPA variant vs. TPMT wild type/ITPA wild type, P = 0.0086; TPMT variant/ITPA wild type vs. TPMT wild type/ITPA wild type, P = 0.048). No significant effect of age, race, or sex on any of these correlations was found. Boxplots showing mercaptopurine metabolite concentrations independently for ITPA and TPMT genotypes are available in Supplementary Data S1 online (Figure 3S and 4S, respectively).
Figure 4.

Multilocus TPMT/ITPA genotype and median concentrations of mercaptopurine metabolite thioguanine nucleotide (TGN) measured during chronic treatment with mercaptopurine in accordance with the St Jude Total 13B protocol. Mercaptopurine doses were adjusted for TPMT genotype so as to avoid toxic TGN concentrations. Red-blood-cell TGN concentrations differed by TPMT genotype, resulting in higher concentrations (P = 0.0095, pairwise Wilcoxon test) in patients with a multilocus genotype of TPMT variant/ITPA wild type than in patients with a wild-type genotype for both genes. ITPA, inosine triphosphate pyrophosphatase; TPMT, thiopurine S-methyltransferase.
Figure 5.

Multilocus TPMT/ITPA genotype and median concentrations of mercaptopurine metabolite methylmercaptopurine nucleotide (MMPN) measured during chronic treatment with mercaptopurine in accordance with the St Jude Total 13B protocol. Mercaptopurine doses were adjusted for TPMT genotype so as to avoid toxic TGN concentrations. Red-blood-cell MMPN concentrations differed among patients according to TPMT/ITPA genotypes, showing higher concentrations in patients with a multilocus genotype TPMT wild type/ITPA variant (P = 0.0086, pairwise Wilcoxon test) and lower concentrations in patients with a multilocus genotype of TPMT variant/ITPA wild type (P = 0.048, pairwise Wilcoxon test) as compared to patients who were wild type for both loci. Boxes include data between the 25th and 75th percentiles, and whiskers indicate the minimal and maximal values excluding the outliers. ITPA, inosine triphosphate pyrophosphatase; TPMT, thiopurine S-methyltransferase.
TPMT and ITPA genotypes and mercaptopurine dose adjustment during continuation therapy (Total 13B)
The dose of mercaptopurine was adjusted for patients enrolled in the Total 13B protocol, in accordance with patient tolerance and TPMT genotype. Among the 205 patients who completed treatment, 8 (3.9%) had a variant TPMT genotype. Among the 197 patients with wild-type TPMT, 6 (3.0%) were on a mercaptopurine dose that had been reduced by 30% or more by completion of therapy. Among the 8 patients with variant TPMT, 2 (25.0%) were taking a dose reduced by 30% or more; the probability of being prescribed a lower dose of mercaptopurine was significantly higher for those with a TPMT variant allele than for those with a wild-type allele (odds ratio (OR) = 10.6; 95% confidence interval (CI) = 1.8–63.9; P = 0.0099). This was confirmed in a multivariate analysis adjusted for age, race, sex, and treatment arm. None of the patients with a variant ITPA genotype was found to be on a reduced dose of mercaptopurine at the end of the treatment, and there was no statistical evidence for a relationship between ITPA genotype and mercaptopurine dose reduction during continuation therapy.
Effect of TPMT/ITPA genotypes on the incidence of toxicity
During continuation therapy in the Total 13B protocol, in which mercaptopurine doses were adjusted on the basis of TPMT genotype and mercaptopurine metabolite concentrations, children with an ITPA variant allele experienced a higher incidence of grade 3/4 febrile neutropenia as compared with children with a homozygous wild-type ITPA genotype (OR = 3.0; 95% CI = 1.2–7.3; P = 0.018, Table 2). There was no significant difference in the incidence of grade 3/4 febrile neutropenia in children with the TPMT variant allele as compared to those with a homozygous wild-type TPMT genotype (OR = 1.4; 95% CI = 0.3–6.9; P = 0.71). Cumulative incidence curves for grade 3/4 febrile neutropenia during continuation therapy, in relation to ITPA genotype, are shown in Figure 6.
Table 2.
Logistic regression model for the incidence of grade 3/4 febrile neutropenia in patients with ALL during continuation therapy that includes mercaptopurine according to the St Jude Total 13B protocol and ITPA genotype
| OR (95% CI) | P value | |
|---|---|---|
| ITPA variant vs. ITPA wild type | 2.98 (1.21–7.35) | 0.018 |
| Age | ||
| <1 Year vs. 1–10 years | 0.57 (0.06–5.49) | 0.063 |
| More than 10 vs. 1–10 years | 1.05 (0.39–2.84) | 0.92 |
| Race | ||
| Black vs. white | 1.20 (0.41–3.50) | 0.74 |
| Other vs. white | 0.52 (0.18–1.51) | 0.23 |
| Sex | ||
| Female vs. male | 1.23 (0.56–2.73) | 0.61 |
| Treatment arm | ||
| High/standard risk vs. low risk | 0.86 (0.34–2.17) | 0.74 |
ALL, acute lymphoblastic leukemia; CI, confidence interval; ITPA, inosine triphosphate pyrophosphatase; OR, odds ratio.
Figure 6.
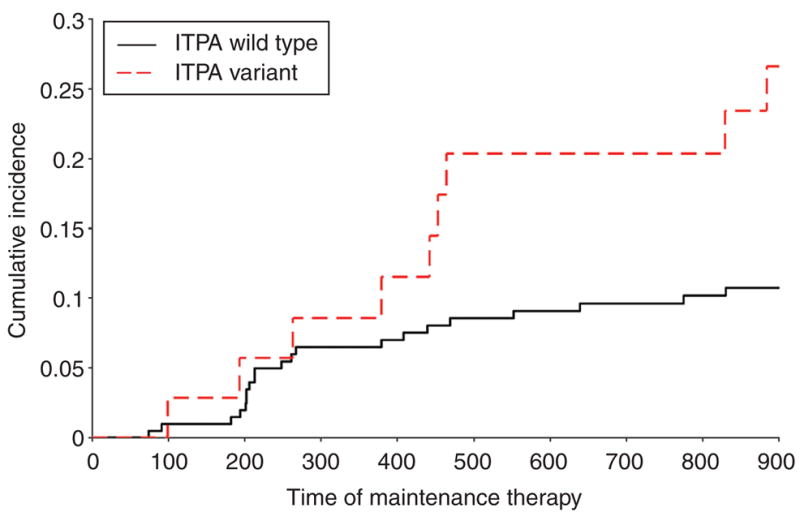
ITPA genotype and cumulative incidence curves for the risk of grade 3/4 febrile neutropenia in patients with acute lymphoblastic leukemia during chronic continuation therapy that included mercaptopurine in accordance with the St Jude Total 13B protocol. At day 900 of maintenance therapy, the estimate of incidence of these adverse events was 10.7% ± 2.2 in patients with wild-type ITPA and 26.6% ± 7.8 in patients with variant ITPA. ITPA, inosine triphosphate pyrophosphatase; TPMT, thiopurine S-methyltransferase.
A significantly higher proportion of children with grade 4 febrile neutropenia have an ITPA variant allele when compared with children with no febrile neutropenia or children with grade 1–2 febrile neutropenia (excluding grade 3) (OR = 5.2; 95% CI = 1.3–20.9; P = 0.021).
There was no significant effect of age, ethnicity, or sex on any correlation of toxicity with the TPMT or ITPA genotypes described. None of the other toxicities that were evaluated (e.g., gastrointestinal toxicity and infection) was significantly associated with the ITPA or TPMT genotype in patients enrolled in the Total 13B protocol.
During continuation therapy in the Total 12 protocol, in which mercaptopurine doses were not adjusted on the basis of TPMT genotype, children with a TPMT variant allele experienced a higher incidence of grade 3/4 infection as compared with children with a homozygous wild-type TPMT genotype (OR = 4.1; 95% CI = 1.2–14.0; P = 0.026). There was no significant difference in the incidence of grade 3/4 infection in children with the ITPA variant allele as compared to those with a homozygous wild-type genotype (OR = 0.90; 95% CI = 0.34–2.4; P = 0.84). Age, ethnicity, and sex had no significant effect on the incidence of grade 3/grade 4 infection. Gastrointestinal toxicity, which was the other evaluated toxicity, was not significantly associated with the ITPA or TPMT genotype in patients enrolled in the Total 12 protocol.
Effect of TPMT/ITPA genotypes on the efficacy of treatment in patients treated according to Total 13B protocol
With aggressive supportive care of treatment-related toxicities, no significant effect of the TPMT or ITPA genotypes was found on the long-term efficacy of treatment, measured as event-free survival at 5, 8, or 10 years after diagnosis.
DISCUSSION
This study has documented that inheritance of a nonfunctional variant allele for either TPMT or ITPA is associated with significant modification in the metabolism of mercaptopurine during treatment of ALL. Although the importance of the TPMT genetic polymorphism is very well known and characterized,8–10,32 this is the first report showing a significant effect of the ITPA genetic polymorphism in the context of mercaptopurine therapy individualized on the basis of TPMT genotype. Here we document significantly higher concentrations of the methylated nucleotide metabolites of mercaptopurine in leukemia cells and erythrocytes of patients who have inherited a nonfunctional ITPA allele. In contrast, inheritance of a variant ITPA allele was not associated with differences in TGN concentrations in either leukemia cells or erythrocytes. Although ITPA is known to be involved in mercaptopurine metabolism (Figure 1), the mechanism by which ITPA variant alleles influence the accumulation of methylated thionucleotides has not been fully elucidated.
It is known that individuals with reduced activity of ITPA have physiologically higher concentrations of the endogenous nucleotide, ITP.25 The methylated nucleotide metabolites of mercaptopurine can be produced by direct methylation of thioITP by TPMT;15 alternatively, methylthioITP can be produced by conversion of methylthioIMP to methylthioITP, as is known to occur with the physiological nucleotides.33 Interestingly, it has been recently shown that methylthioITP has less affinity for ITPA than does thioITP, and this could account for the greater accumulation of methylthioITP in patients with ITPA deficiency (i.e., ITPA heterozygotes).24 The assay we used to measure these nucleotide concentrations does not discriminate among the mono-, di-, and triphosphate nucleotides,33 and therefore it is not known whether one or all of these nucleotides were affected by the reduction in ITPA activity; however, because ITPA primarily cleaves thioITP to thioIMP, patients with reduced ITPA activity (i.e., patients with the ITPA variant) should have an increased concentration of primarily the triphosphate nucleotide (thioITP), leading to an accumulation of methylthioITP, documented in this study as an increase in MMPN.
The level of de novo purine synthesis can influence mercaptopurine pharmacology, but our studies revealed no relationship between de novo purine synthesis in ALL cells and ITPA genotype.
These studies reveal that the cumulative incidence of febrile neutropenia in patients receiving a chemotherapy protocol that includes mercaptopurine individualized for TPMT is significantly greater among those who have inherited an ITPA variant allele. The association was particularly evident during the second half of continuation therapy (Figure 6), when the treatment was predominantly chronic (i.e., daily mercaptopurine and weekly methotrexate treatment31), and remained significant when the analysis was limited to only life-threatening events (i.e., grade 4 fever and neutropenia). Febrile neutropenia in cancer patients is a serious complication of cytotoxic chemotherapy, and it generally leads to hospitalization for evaluation and treatment. Even when properly treated it can be associated with significant morbidity, mortality, and costs.34–37 Moreover, febrile neutropenia leads to treatment delays and reductions in chemotherapy dosage, which may negatively affect the long-term outcome of the treatment for malignancy.34–37 In the current analysis, ITPA genotype was shown to have a significant influence on the risk of fever and neutropenia (and the morbidity and costs associated with its treatment), but fortunately this did not influence event-free survival in the patients. We postulate that, although the ITPA polymorphism significantly influences the risk of toxicity (febrile neutropenia), this did not influence the efficacy of the treatment because we immediately and aggressively treated febrile neutropenia with antibiotics, assuming it to be a result of infection until proven otherwise. By avoiding toxicity-related deaths associated with the higher frequency of fever and neutropenia, there was no adverse effect of ITPA polymorphism on the efficacy of ALL therapy. Given the higher risk of this toxicity, patients with variant ITPA and wild-type TPMT should be closely monitored for the risk of febrile neutropenia during treatment with mercaptopurine.
The higher frequency of febrile neutropenia observed in patients with a variant ITPA allele may have been caused by the higher concentration of methylated thiopurine nucleotides that we documented in patients with a variant nonfunctional ITPA allele; these metabolites are known to have cytotoxic properties, and their accumulation may contribute to a more persistent neutropenia, increasing the likelihood of febrile neutropenia events.9,38
It is also noteworthy that in a prior cohort of St Jude patients with ALL whose mercaptopurine doses were not adjusted on the basis of TPMT genotype or TGN concentrations (St Jude Protocol Total 12, 1988–1991)39 we documented a higher probability of grade 3/4 infections in patients who had inherited TPMT deficiency, but that this was not significantly associated with ITPA genotype (Figure 7). That is, if mercaptopurine doses are not individualized on the basis of TPMT genotype, then TPMT will be the predominant determinant of severe hematopoietic toxicity; whereas, if doses are adjusted for TPMT, then ITPA has a significant influence on the risk of febrile neutropenia.
Figure 7.

TPMT genotype, ITPA genotype, and the odds ratio of severe toxicity during continuation therapy of children with acute lymphoblastic leukemia for whom the dose of mercaptopurine was not adjusted on the basis of TPMT genotype (St Jude Total 12) and in those for whom the dose was adjusted on the basis of TPMT genotype (St Jude Total 13B). Toxicity measured prospectively according to the Total 12 protocol (i.e., no mercaptopurine dose adjustment on the basis of TPMT) was grade 3/4 infection, whereas the comparable toxicity measured according to the Total 13B (i.e., mercaptopurine dose adjusted for TPMT) was grade 3/4 febrile neutropenia. Odds ratios are from a weighted logistic regression model and are adjusted for treatment arm and patient’s age, race, and sex. ITPA, inosine triphosphate pyrophosphatase; TPMT, thiopurine S-methyltransferase. v, variant allele; wt, wild-type allele.
All previous studies that evaluated the role of ITPA polymorphism in the toxicity of thiopurine have been carried out in patients with inflammatory bowel disease, with contradictory results.17–24 Most of these studies involved patients on doses of mercaptopurine that were not systematically adjusted on the basis of TPMT genotype, and our findings indicate that this is probably the reason for the inconsistent results relating to the effect of ITPA in these earlier studies.
In summary, we have shown that genetic polymorphism of ITPA (rs41320251) is a significant determinant of mercaptopurine metabolism and of severe, life-threatening febrile neutropenia during treatment in patients with ALL who are treated with combination chemotherapy involving mercaptopurine doses individualized on the basis of TPMT genotype and the concentration of TGNs. This illustrates the evolution of pharmacogenetics in clinical practice; as treatment is individualized for one genetic determinant of drug response, the importance of other genetic polymorphisms emerges.
METHODS
Patients
We studied children enrolled as patients in two single-institution clinical protocols for the treatment of newly diagnosed ALL. All the patients and/or their parents provided informed consent for the institutional review board–approved protocols.
The study was initially focused on patients receiving treatment according to the Total 13B protocol to assess the associations between different clinical and pharmacological phenotypes related to mercaptopurine treatment. For patients enrolled in the Total 12 protocol, only toxicity was analyzed for its relation to TPMT and ITPA genotypes.
For both the Total 12 and Total 13B protocols, the therapy schedule has been previously described in detail (refs. 31,39; see Supplementary Data S1 online). Of the 188 patients enrolled in the Total 12 protocol, 101 had genomic DNA samples available for inclusion in this analysis. Of the 247 patients who were enrolled in the St Jude Total 13B protocol, 246 had genomic DNA samples available for inclusion in this analysis.3
Genotyping
The major nonfunctional variant alleles of TPMT (TPMT*2, TPMT*3A, and TPMT*3C, defined by SNPs rs1142345, rs1800460, and rs1800462) were determined using methods that we have previously described in detail;32,40 the major nonfunctional variant allele of ITPA (SNP rs41320251) was genotyped on the basis of a specific probe (SNP_A-1646349) in the Affymetrix chip array (Mapping50K_Hind240) and confirmed using a TaqMan assay (Prometheus Labs, San Diego, CA). All genotyping was done on germline DNA extracted from patients’ blood samples.
Rate of de novo purine synthesis in leukemic blasts
In patients enrolled in the Total 13B protocol, the rate of de novo purine synthesis in treatment-naive bone marrow ALL cells was determined by quantifying unlabeled and radio-labeled purine bases (adenine and guanine) after acid hydrolysis of a 2-h ex vivo incubation of lymphoblasts with 14C-formate using methods that we have previously described in detail.41
Measurement of mercaptopurine metabolites in leukemia cells
In patients enrolled in the Total 13B protocol, concentrations of mercaptopurine metabolites (TGN and MMPN) were measured in bone marrow ALL cells using methods that we have previously described in detail.33 These analyses were performed on leukemia cells obtained after initial therapy with mercaptopurine in a subgroup of patients whose initial therapy consisted of a single intravenous infusion of mercaptopurine (1,000 mg/m2 infused IV over 6 h); the other patients received initial therapy with a combination of mercaptopurine and methotrexate and were therefore not included in the analysis of mercaptopurine metabolism in ALL cells.31
Measurement of mercaptopurine metabolites in erythrocytes
The concentrations of the two principal metabolites (TGN and MMPN) were measured in patients’ erythrocytes using previously described methods.42,43 Samples were collected during continuation therapy from patients who were compliant with their mercaptopurine therapy and who had taken a stable dose for at least 12 of the prior 14 days; the median value of multiple measurements relating to each patient was used in the analyses.
Mercaptopurine dose adjustment during continuation chemotherapy
For patients enrolled in the Total 12 protocol, the dose of mercaptopurine was 75 mg/m2, and it was not adjusted prospectively for TPMT genotype. Rather, the mercaptopurine dose was modified only if patients developed dose-limiting toxicity attributable to mercaptopurine.10 For those enrolled in the Total 13B protocol, the standard dose of mercaptopurine was 75 mg/m2, but the dose was adjusted prospectively according to each patient’s TPMT genotype and tolerance to therapy (see Supplementary Data S1 online).
Toxicity
Of the 246 patients enrolled in this study, 240 were evaluable for the toxicity analysis: 5 patients with Down syndrome and 1 patient with cystic fibrosis were excluded because their underlying conditions could influence toxicity. During continuation therapy, adverse events were documented and graded prospectively using the National Cancer Institute Common Toxicity Criteria version 1.0, as previously described.3 For the analyses, the estimation of toxicity was dichotomized as “present” (grades 3–4) or “absent” (grades 0–2). The toxicities considered in this study were febrile neutropenia, gastrointestinal toxicity, and infection in those on the Total 13B protocol; and gastrointestinal toxicity and infection for those on the Total 12 protocol. Febrile neutropenia events were not recorded independently of infection for the Total 12 protocol patients. For patients enrolled in the Total 13B protocol, a separate analysis was carried out to compare the incidence of grade 4 febrile neutropenia with the incidence of nil febrile neutropenia and of grades 1 and 2 febrile neutropenia, separately from grade 3 events.
Efficacy of treatment
Efficacy of treatment was defined as event-free survival and overall survival at 5, 8, and 10 years after the diagnosis of leukemia.
Statistical analysis
The associations between the genotypes and the pharmacological or metabolic phenotypes were evaluated using Wilcoxon test for two-group comparisons and pairwise comparisons among multiple groups defined by the multilocus genotypes.
Logistic regression was used to assess the association between mercaptopurine dose reductions at the end of treatment and TPMT and ITPA genotypes.
A weighted logistic regression model that takes time at risk of each patient into account3 was used to test the association between genotype and incidence of the toxicity events.
Event-free survival rates were estimated using the Kaplan–Meier method and were compared between genotypes using the log rank test (see Supplementary Data S1 online).
To test for potential confounders, all associations between the considered phenotypes and the genotypes were confirmed by multivariate analyses using linear or generalized linear models (see Supplementary Data S1 online).
Statistical analyses were performed using SAS version 9.1.3 Service Pack 4 (SAS institute, Cary, NC) and R version 2.6.1 (http://www.r-project.org).
Supplementary Material
Acknowledgments
We thank the patients and their parents for their participation in this study and our clinical staff for providing protocol-based patient care. We are grateful to our research nurses, Sheri Ring, Lisa Walters, Terri Kuehner, Margaret Edwards, and Paula Condy. We also thank Yaqin Chu, May Chung, Margaret Needham, and Emily Melton for their outstanding technical assistance; and Nancy Kornegay and Mark Wilkinson for their computer and database expertise. Funding sources include grants from the National Institutes of Health (R37 CA36401 to WEE, MVR, C-HP; R01 CA78224 to WEE, MVR, C-HP; RO1 CA51001 to MVR, C-HP; U01 GM61393 to MVR, WEE; Cancer Center Support Grant CA21765), and the American Lebanese Syrian Associated Charities. The funding agencies had no role in the design of the study; the collection, analysis, and interpretation of the data; the writing of the manuscript; or in the decision to submit the manuscript.
Footnotes
CONFLICT OF INTEREST
WE Evans is a co-inventor on a patent awarded for the molecular diagnosis of the major TPMT variant alleles. The other authors declared no conflict of interest.
SUPPLEMENTARY MATERIAL is linked to the online version of the paper at http://www.nature.com/cpt
References
- 1.Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354:166–178. doi: 10.1056/NEJMra052603. [DOI] [PubMed] [Google Scholar]
- 2.Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008;371:1030–1043. doi: 10.1016/S0140-6736(08)60457-2. [DOI] [PubMed] [Google Scholar]
- 3.Kishi S, et al. Ancestry and pharmacogenetics of antileukemic drug toxicity. Blood. 2007;109:4151–4157. doi: 10.1182/blood-2006-10-054528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rivera GK, et al. Unexpectedly severe toxicity from intensive early treatment of childhood lymphoblastic leukemia. J Clin Oncol. 1985;3:201–206. doi: 10.1200/JCO.1985.3.2.201. [DOI] [PubMed] [Google Scholar]
- 5.Evans WE, Relling MV. Moving towards individualized medicine with pharmacogenomics. Nature. 2004;429:464–468. doi: 10.1038/nature02626. [DOI] [PubMed] [Google Scholar]
- 6.Evans WE, McLeod HL. Pharmacogenomics—drug disposition, drug targets, and side effects. N Engl J Med. 2003;348:538–549. doi: 10.1056/NEJMra020526. [DOI] [PubMed] [Google Scholar]
- 7.Evans WE, Relling MV. Pharmacogenomics: translating functional genomics into rational therapeutics. Science. 1999;286:487–491. doi: 10.1126/science.286.5439.487. [DOI] [PubMed] [Google Scholar]
- 8.Krynetski EY, Evans WE. Pharmacogenetics of cancer therapy: getting personal. Am J Hum Genet. 1998;63:11–16. doi: 10.1086/301941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krynetski E, Evans WE. Drug methylation in cancer therapy: lessons from the TPMT polymorphism. Oncogene. 2003;22:7403–7413. doi: 10.1038/sj.onc.1206944. [DOI] [PubMed] [Google Scholar]
- 10.Relling MV, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst. 1999;91:2001–2008. doi: 10.1093/jnci/91.23.2001. [DOI] [PubMed] [Google Scholar]
- 11.McLeod HL, Krynetski EY, Relling MV, Evans WE. Genetic polymorphism of thiopurine methyltransferase and its clinical relevance for childhood acute lymphoblastic leukemia. Leukemia. 2000;14:567–572. doi: 10.1038/sj.leu.2401723. [DOI] [PubMed] [Google Scholar]
- 12.Schwab M, et al. Azathioprine therapy and adverse drug reactions in patients with inflammatory bowel disease: impact of thiopurine S-methyltransferase polymorphism. Pharmacogenetics. 2002;12:429–436. doi: 10.1097/00008571-200208000-00003. [DOI] [PubMed] [Google Scholar]
- 13.Teml A, Schaeffeler E, Herrlinger KR, Klotz U, Schwab M. Thiopurine treatment in inflammatory bowel disease: clinical pharmacology and implication of pharmacogenetically guided dosing. Clin Pharmacokinet. 2007;46:187–208. doi: 10.2165/00003088-200746030-00001. [DOI] [PubMed] [Google Scholar]
- 14.Elion GB. The purine path to chemotherapy. Science. 1989;244:41–47. doi: 10.1126/science.2649979. [DOI] [PubMed] [Google Scholar]
- 15.Krynetski EY, Krynetskaia NF, Yanishevski Y, Evans WE. Methylation of mercaptopurine, thioguanine, and their nucleotide metabolites by heterologously expressed human thiopurine S-methyltransferase. Mol Pharmacol. 1995;47:1141–1147. [PubMed] [Google Scholar]
- 16.Somerville L, et al. Structure and dynamics of thioguanine-modified duplex DNA. J Biol Chem. 2003;278:1005–1011. doi: 10.1074/jbc.M204243200. [DOI] [PubMed] [Google Scholar]
- 17.Marinaki AM, et al. Adverse drug reactions to azathioprine therapy are associated with polymorphism in the gene encoding inosine triphosphate pyrophosphatase (ITPase) Pharmacogenetics. 2004;14:181–187. doi: 10.1097/00008571-200403000-00006. [DOI] [PubMed] [Google Scholar]
- 18.von Ahsen N, et al. Association of inosine triphosphatase 94C>A and thiopurine S-methyltransferase deficiency with adverse events and study drop-outs under azathioprine therapy in a prospective Crohn disease study. Clin Chem. 2005;51:2282–2288. doi: 10.1373/clinchem.2005.057158. [DOI] [PubMed] [Google Scholar]
- 19.Zelinkova Z, et al. Inosine triphosphate pyrophosphatase and thiopurine S-methyltransferase genotypes relationship to azathioprine-induced myelosuppression. Clin Gastroenterol Hepatol. 2006;4:44–49. doi: 10.1016/j.cgh.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 20.Gearry RB, Roberts RL, Barclay ML, Kennedy MA. Lack of association between the ITPA 94C>A polymorphism and adverse effects from azathioprine. Pharmacogenetics. 2004;14:779–781. doi: 10.1097/00008571-200411000-00010. [DOI] [PubMed] [Google Scholar]
- 21.van Dieren JM, van Vuuren AJ, Kusters JG, Nieuwenhuis EE, Kuipers EJ, van der Woude CJ. ITPA genotyping is not predictive for the development of side effects in AZA treated inflammatory bowel disease patients. Gut. 2005;54:1664. [PMC free article] [PubMed] [Google Scholar]
- 22.Hindorf U, et al. Pharmacogenetics during standardised initiation of thiopurine treatment in inflammatory bowel disease. Gut. 2006;55:1423–1431. doi: 10.1136/gut.2005.074930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Dieren JM, Hansen BE, Kuipers EJ, Nieuwenhuis EE, Van der Woude CJ. Meta-analysis: inosine triphosphate pyrophosphatase polymorphisms and thiopurine toxicity in the treatment of inflammatory bowel disease. Aliment Pharmacol Ther. 2007;26:643–652. doi: 10.1111/j.1365-2036.2007.03412.x. [DOI] [PubMed] [Google Scholar]
- 24.Bierau J, Lindhout M, Bakker JA. Pharmacogenetic significance of inosine triphosphatase. Pharmacogenomics. 2007;8:1221–1228. doi: 10.2217/14622416.8.9.1221. [DOI] [PubMed] [Google Scholar]
- 25.Sumi S, et al. Genetic basis of inosine triphosphate pyrophosphohydrolase deficiency. Hum Genet. 2002;111:360–367. doi: 10.1007/s00439-002-0798-z. [DOI] [PubMed] [Google Scholar]
- 26.Frazer KA, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maeda T, et al. Genetic basis of inosine triphosphate pyrophosphohydrolase deficiency in the Japanese population. Mol Genet Metab. 2005;85:271–279. doi: 10.1016/j.ymgme.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 28.von Ahsen N, Oellerich M, Armstrong VW. Characterization of the inosine triphosphatase (ITPA) gene: haplotype structure, haplotype–phenotype correlation and promoter function. Ther Drug Monit. 2008;30:16–22. doi: 10.1097/FTD.0b013e318161a21a. [DOI] [PubMed] [Google Scholar]
- 29.Stenmark P, et al. Crystal structure of human inosine triphosphatase. Substrate binding and implication of the inosine triphosphatase deficiency mutation P32T. J Biol Chem. 2007;282:3182–3187. doi: 10.1074/jbc.M609838200. [DOI] [PubMed] [Google Scholar]
- 30.Vanderheiden BS. Genetic studies of human erythrocyte inosine triphosphatase. Biochem Genet. 1969;3:289–297. doi: 10.1007/BF00521144. [DOI] [PubMed] [Google Scholar]
- 31.Pui CH, et al. Improved outcome for children with acute lymphoblastic leukemia: results of Total Therapy Study XIIIB at St Jude Children’s Research Hospital. Blood. 2004;104:2690–2696. doi: 10.1182/blood-2004-04-1616. [DOI] [PubMed] [Google Scholar]
- 32.Tai HL, et al. Thiopurine S-methyltransferase deficiency: two nucleotide transitions define the most prevalent mutant allele associated with loss of catalytic activity in Caucasians. Am J Hum Genet. 1996;58:694–702. [PMC free article] [PubMed] [Google Scholar]
- 33.Dervieux T, Chu Y, Su Y, Pui CH, Evans WE, Relling MV. HPLC determination of thiopurine nucleosides and nucleotides in vivo in lymphoblasts following mercaptopurine therapy. Clin Chem. 2002;48:61–68. [PubMed] [Google Scholar]
- 34.Kuderer NM, Dale DC, Crawford J, Cosler LE, Lyman GH. Mortality, morbidity, and cost associated with febrile neutropenia in adult cancer patients. Cancer. 2006;106:2258–2266. doi: 10.1002/cncr.21847. [DOI] [PubMed] [Google Scholar]
- 35.Santolaya ME, et al. Admission clinical and laboratory factors associated with death in children with cancer during a febrile neutropenic episode. Pediatr Infect Dis J. 2007;26:794–798. doi: 10.1097/INF.0b013e318124aa44. [DOI] [PubMed] [Google Scholar]
- 36.Kuderer NM, Dale DC, Crawford J, Lyman GH. Impact of primary prophylaxis with granulocyte colony-stimulating factor on febrile neutropenia and mortality in adult cancer patients receiving chemotherapy: a systematic review. J Clin Oncol. 2007;25:3158–3167. doi: 10.1200/JCO.2006.08.8823. [DOI] [PubMed] [Google Scholar]
- 37.Klaassen RJ, Goodman TR, Pham B, Doyle JJ. “Low-risk” prediction rule for pediatric oncology patients presenting with fever and neutropenia. J Clin Oncol. 2000;18:1012–1019. doi: 10.1200/JCO.2000.18.5.1012. [DOI] [PubMed] [Google Scholar]
- 38.Gerson SL, Talbot GH, Hurwitz S, Strom BL, Lusk EJ, Cassileth PA. Prolonged granulocytopenia: the major risk factor for invasive pulmonary aspergillosis in patients with acute leukemia. Ann Intern Med. 1984;100:345–351. doi: 10.7326/0003-4819-100-3-345. [DOI] [PubMed] [Google Scholar]
- 39.Evans WE, Relling MV, Rodman JH, Crom WR, Boyett JM, Pui CH. Conventional compared with individualized chemotherapy for childhood acute lymphoblastic leukemia. N Engl J Med. 1998;338:499–505. doi: 10.1056/NEJM199802193380803. [DOI] [PubMed] [Google Scholar]
- 40.Yates CR, et al. Molecular diagnosis of thiopurine S-methyltransferase deficiency: genetic basis for azathioprine and mercaptopurine intolerance. Ann Intern Med. 1997;126:608–614. doi: 10.7326/0003-4819-126-8-199704150-00003. [DOI] [PubMed] [Google Scholar]
- 41.Dervieux T, et al. De novo purine synthesis inhibition and antileukemic effects of mercaptopurine alone or in combination with methotrexate in vivo. Blood. 2002;100:1240–1247. doi: 10.1182/blood-2002-02-0495. [DOI] [PubMed] [Google Scholar]
- 42.Erdmann GR, France LA, Bostrom BC, Canafax DM. A reversed phase high performance liquid chromatography approach in determining total red blood cell concentrations of 6-thioguanine, 6-mercaptopurine, methylthioguanine, and methylmercaptopurine in a patient receiving thiopurine therapy. Biomed Chromatogr. 1990;4:47–51. doi: 10.1002/bmc.1130040202. [DOI] [PubMed] [Google Scholar]
- 43.Lennard L. Assay of 6-thioinosinic acid and 6-thioguanine nucleotides, active metabolites of 6-mercaptopurine, in human red blood cells. J Chromatogr. 1987;423:169–178. doi: 10.1016/0378-4347(87)80340-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.