

Abstract
Background
Long Dan Xie Gan Wan (LD), a Chinese herbal remedy formulation, is traditionally used to treat a range of conditions, including gall bladder diseases, hepatitis, hyperthyroidism, migraines but it is not used for the management or treatment of cancer. However some of its herbal constituents, specifically Radix bupleuri, Radix scutellariae and Rhizoma alismatis have been shown to inhibit the growth of cancer cells. Thus, the aim of the study was to investigate the impact of LD on cancer cells in vitro.
Methods
HL60 and HT29 cancer cell lines were exposed to water extracts of LD (1:10, 1:50, 1:100 and/or 1:1000 prepared from a 3 mg/30 ml stock) and for both cell lines growth, apoptotic induction, alterations in cell cycle characteristics and genotoxicity were investigated. The specificity of the action of LD on these cancer cell lines was also investigated by determining its effect on human peripheral blood lymphocytes. Preliminary chemical analysis was carried out to identify cytotoxic constituents of LD using HPLC and LCMS.
Results
LD was significantly cytotoxic to, and induced apoptosis in, both cell lines. Apoptotic induction appeared to be cell cycle independent at all concentrations of LD used (1:10, 1:50 and 1:100) for the HL60 cell lines and at 1:10 for the HT29 cell line. At 1:50 and 1:100 apoptotic induction by LD appeared to be cell cycle dependent. LD caused significant genotoxic damage to both cell lines compared to their respective controls. The specificity study showed that LD exerted a moderate cytotoxic action against non-proliferating and proliferating blood lymphocytes but not apoptosis. Chemical analysis showed that a number of fractions were found to exert a significant growth inhibitory effect. However, the molecular weights of compounds within these fractions did not correspond to those from the herbal constituents of LD.
Conclusion
It is possible that LD may have some chemotherapeutic potential. However, further studies are required to determine its cytotoxic constituents.
Background
Long Dan Xie Gan Wan (LD) is traditionally used for the treatment of a range of conditions, including gall stones, gall bladder diseases, hepatitis, herpes, shingles, cystitis, hyperthyroidism, migraines and jaundice. The traditional ingredients typically used to make LD are Radix Scutellariae (Huang Qui), Fructus Gardeniae (Zhi zi), Radix glycyrrhizae (Gan cao), Radix rehmanniae (Di huang), Radix Gentianae (Long dan), Radix angelicae sinensis (Dang gui), Semen Plantaginis (Che qian zi), Radix Bupleuri (Cai hu) and Rhizoma alismatis (Ze xie) and also Aristolochia manshuriensis (Mu Tong). However, LD remedies containing Aristolochia manshuriensis (Mu Tong) are no longer available as Aristolochia species contain the toxic and carcinogenic aristolochic acids [1] and this species has been replaced in many formulations of LD by Medulla tetrapanacis (Tong cao).
Long Dan Xie Gan Wan is not traditionally prescribed in the treatment of cancer, and, to the authors' knowledge, there is no research regarding the effects of LD in any biological context. However, there are herbs within LD that are prescribed for the treatment of cancer and are reported to inhibit the growth of cancer cells in vitro. These herbal constituents are Radix bupleuri (which is traditionally derived from the dried roots of Bupleurum chinense DC and B. scorzonerifolium Willd, however other species and variants of the Bupleurum genus are also used as Radix Bupleuri [2]), Radix scutellariae, also known as Scutellaria baicalensis, and Rhizoma alismatis [3-9].
As LD is a commonly used Chinese herbal remedy (CHR) that contains constituents reported to possess anti-cancer activity, the aim of this study was to investigate the impact of LD on cancer cell lines in vitro to ascertain if it possesses any potential chemotherapeutic activity. The cell lines used were the HL60- (human promyelotic leukaemia) cell line [10] and the HT29 (human colon adenocarcinoma) cell line. These cell lines are currently being used by the authors in the characterisation of CHRs said to possess anti-cancer activity.
Results and discussion
Growth Inhibition Study
The growth inhibition study showed the water extract of LD generally exerted a toxic effect at 1:10 (made from 3 mg/30 ml stock) on both cell lines after 48 and 72 hours of incubation (Figure 1A and 1B). The water extract of LD exerted its greatest toxic effect on the HL60 cell line (human promyelotic leukaemia cell lines) killing all HL60s after 72 hours of exposure. At 1:10 the water extract of LD also exerted a significant (p = 0.05) growth inhibitory effect on the HT29 cell line (human colon adenocarcinoma cell line) after 72 hours. The effect of the water extract of LD at 1:100 and 1:1000 (made from 3 mg/30 ml stock) was greatly reduced when compared the effects of LD at 1:10, only causing significant growth inhibition after 72 hours in the HL60 cell line.
Figure 1.

A and B: Incubation of water extracts of LD at 1:10, 1:100 and 1:1000 on the HL60 and HT29 cell lines for 24, 48 and 72 hours compared to normal (control) cell growth, ± standard deviation from the mean, *denotes statistical significance (p = 0.05).
Apoptosis Study
The Annexin V assay revealed apoptotic induction in both HL60 and HT29 cell lines exposed to a 1:10 (made from 3 mg/30 ml stock) concentration of LD. Figure 2A shows control values for an unchallenged HL60 population, with 85.5% of cells viable and 6.4% of the cell population undergoing apoptosis. After a 6 hour exposure to LD the population of actively apoptosing cells had risen to 14.7% (Figure 2B), after 12 hours to 26.4% (Figure 2C), and after 16 hours to 39.4% (Figure 2D). In the HT29 control (Figure 2E) the apoptotic population was 4.5%. There was a slight increase in apoptosing cells after 12 and 16 hours (Figures 2F and 2G), however it was not until 24 hours after initial exposure (Figure 2H) that a distinct apoptotic population was observed, accounting for 18.2% of the cell population.
Figure 2.
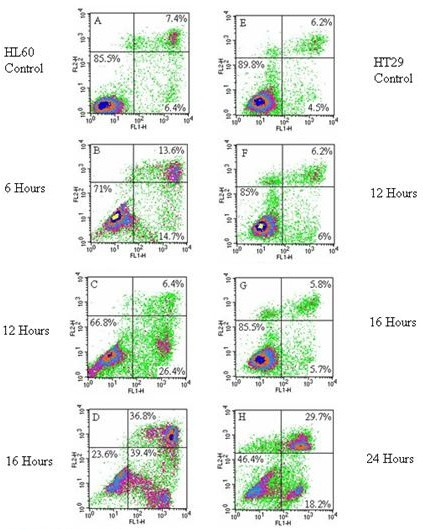
A-H: Annexin V staining of HL60 and HT29 cell lines exposed to the water extract of LD at 1:10. The x-axis shows Annexin V-FITC binding and the y-axis staining of the vital dye propidium iodide. Cells in the lower left quadrant are viable, cells in the lower right are apoptotic and those in the upper right are non-viable dead/late stage apoptotic cells. 2A: HL60 control, 2B: HL60 cells exposed to LD (1:10) for 6 hrs, 2C: HL60 cells exposed to LD (1:10) for 12 hours, 2D: HL60 cells exposed to LD (1:10) for 16 hours, 2E: HT29 cells control, 2F: HT29 cells exposed to LD (1:10) for 12 hours, 2G: HT29 cells exposed to LD (1:10) for 16 hours, 2H: HT29 cells exposed to LD (1:10) for 24 hours.
Propidium iodide (PI) staining of the HL60 cell line exposed to a 1:10 water extract of LD for 4 and 8 hours revealed apoptotic induction, and after 24 hours in the HT29 cell line. For the HL60 4 hour control, 1.3% of cells within the population were apoptotic (Figure 3A). HL60 cells exposed to LD for 4 hours had 30.2% of cells apoptosing (Figure 3B). After 8 hours the apoptotic population had risen to 56.5% (Figure 3D). There was no evidence of apoptotic induction in the HT29 cell line after 4 and 8 hours of exposure (Figures 3F and 3H respectively). The HT29 24 hour control contained an apoptotic population of 2.12% (Figure 3I) and for HT29 cells exposed to LD for 24 hours, 68.91% of cells were apoptotic (Figure 3J).
Figure 3.
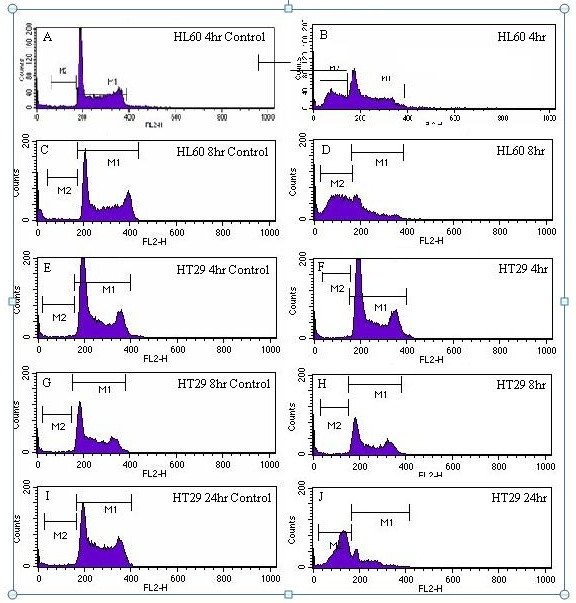
A - J: Cell cycle histograms of HL60 and HT29 cell lines exposed to LD at 1:10. The x-axis shows relative DNA content, and the y-axis cell number Cells in region M1 are viable, and those in the Sub-G1 region, M2, are apoptotic. 3A: HL60 4 hour control. 3B: HL60 cells exposed to LD for 4 hours. 3C: HL60 8 hour control. 3D: HL60 cells exposed to LD for 8 hours. 3E: HT29 4 hour control. 3F: HT29 cells exposed to LD for 4 Hours. 3G: HT29 8 hour control. 3H: HT29 cells exposed to LD for 8 hours. 3I: HT29 24 hour control. 3J: HT29 cells exposed to LD for 24 hours.
The results from the TUNEL assay indicate apoptotic induction after 4 hours of exposure to the water extract of LD in the HL60 cell line, and after 24 hours in the HT29 cell line, with no evidence of apoptotic induction in the HT29 cell line after 4 hours of exposure (Figure 4D). The HL60 and HT29 controls (Figures 4A, C and 4E) show normal populations of cells spread between channels 200 and 400 on the x-axis. The distinct cell population above that of the viable cells (as seen in the controls) for the 4 hour-exposed HL60 and 24 hour exposed HT29 cell lines (Figures 4B and 4F, respectively) are indicative of apoptosis as these regions contain cells with an increased proportion of double-strand breaks compared to controls.
Figure 4.
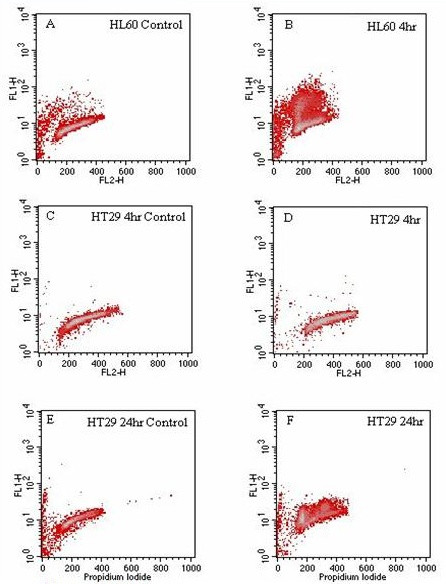
A-F: TUNEL analysis of the HL60 and HT29 cell lines exposed to LD at 1:10. The x-axis shows relative DNA content and the y-axis the number of double strand breaks within cells. 4A: the HL60 control and 4B: effect of a 4 hour incubation of HL60s with LD. 4C: the HT29 4 hour control and 4D: the effect of a 4 hour exposure to LD. 4E: the HT29 24 hour control and 4F: the effect of a 24 hour exposure to LD.
Alterations in cell cycle characteristics by LD
When analysed using Cyclred, cell cycle histograms of HL60 cells exposed to LD at 1:50 and 1:100 (made from 3 mg/30 ml stock) for 24, 48 and 72 hours did not generate a consistent set of time and dose-related data. However, visual inspection of the cell cycle histograms generated showed dose and time dependent apoptotic induction. It can be seen clearly that significantly more cells are apoptosing when exposed to LD at 1:50 after 24, 48 and 72 hours (Figures 5A, B and 5C respectively) than cells exposed to LD at the lower concentration of 1:100, for 24, 48 and 72 hours (Figures 5D, E and 5F, respectively) for which there were low levels of apoptosing cells at all time points. In addition, for HL60 cells exposed to LD at 1:50, at all time points there were consistent levels of apoptosing cells and over time a decrease in the relative numbers of viable cells. Furthermore, the levels of apoptosing cells (during exposure to LD at 1:50) were greater than that of control HL60 cells (Figures 3A and 3C) and the levels for the controls were similar to those for HL60 cells exposed to LD at 1:100. Although the controls were not set up for the same time periods as those used to investigate the effect of LD (1:50 and 1:100) Figure 1A clearly shows exponential growth of control HL60 cells over a 72 hr period thus indicating little to no apoptosis of control HL60 cells.
Figure 5.
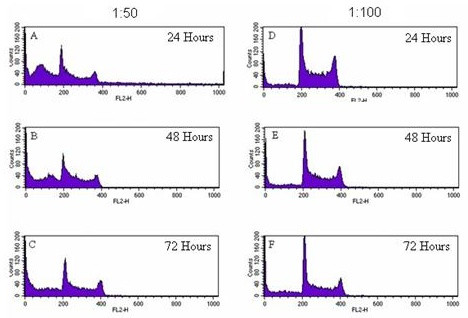
A-F: Cell cycle histograms of HL60 cells exposed to LD at 1:50 and 1:100. The peak in channel 200 shows cells in G1, and the peak in 400 cells in G2 of the cell cycle. The Sub-G1 region is apoptotic cells. 5A-5C show the effects of a LD at 1:50 on cell cycle characteristics after 24, 48 and 72 hours, respectively. 5D-5F show the effects of a 1:100 extract after 24, 48 and 72 hours, respectively.
Cell cycle analysis of HT29 cells exposed to 1:50 and 1:100 concentrations of LD for 24, 48 and 72 hours did generate some consistent results (represented as bar charts) (Figures 6A, B and 6C). Results showed a relative decrease in cells in G1 phase of the cycle compared to the control, with a concomitant increase in cells in S phase at all time points, and a small increase in the relative number of cells in G2 phase of the cell cycle.
Figure 6.

A-C: The effect of LD at 1:50 and 1:100 on the cell cycle characteristics of HT29 cells compared to controls. 6A: the effect after 24 hours, 6B: the effect after 48 hours and 6C: the effect after 72 hours.
TUNEL data examining the cell cycle stage in which HL60 and HT29 cells were apoptosing after exposure to LD at 1:10 for 4 and 24 hours respectively (see Figures 4B and 4F) revealed apoptosis occurring in all cell cycle stages (Figures 7A, B, C and 7D).
Figure 7.
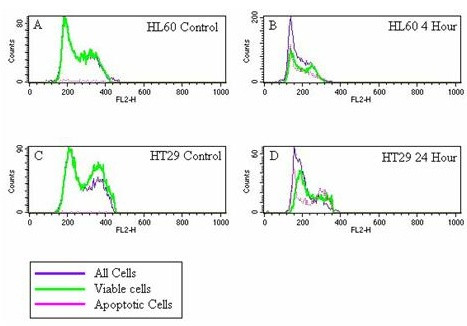
A-D: Overlaid cell cycle histograms generated using gated TUNEL assay data. The x axis shows the relative DNA content of individual cells, the y axis shows cell number. The first peaks, above channel 200 represent cells in G1, the second peak represents cells in G2 phase of the cell cycle. The purple line represents all cells analyzed, the green line represents those cells that are still viable and the pink line represents cells with an increased number of double strand breaks, which is indicative of apoptosis. 7A: HL60 control, 7B: HL60 cells exposed to LD for 4 hours, 7C: HT29 control and 7D: HT29 cells exposed to LD for 24 hours.
Comet Assay
HL60 and HT29 cells exposed to LD for 4 hours at 1:10, 1:50 and 1:100 (made from 3 mg/30 ml stock) showed statistically significant increases in tail length when compared to their respective controls (Figures 8A and 8B). For the HL60 cell line, tail lengths increased significantly from 18.97 μm in the control to 46.29 μm, 33.84 μm and 32.23 μm after exposure to LD for 4 hours at 1:10, 1:50 and 1:100, respectively, and for the HT29 cell line tail lengths increased significantly from a control length of 13.06 μm to 17.93 μm, 20.58 μm and 21.08 μm at 1:10, 1:50 and 1:100, respectively. At all concentrations the observed genotoxic effect on the HL60 cell line was greater than that observed on the HT29 cell line.
Figure 8.

A & A: Average tail lengths (μm) of 8A: HL60 and 8B: HT29 cell lines exposed to aqueous extracts of LD for 4 hours at 1:10, 1:50 and 1:100 (compared to controls) ± standard deviation from the mean (n = 100). * denotes statistical significance (p = 0.01).
The effect of LD extract on unstimulated (non-proliferating) and stimulated (proliferating) primary human blood lymphocytes
For non-stimulated primary blood lymphocytes (PBLs) exposed to 1:10 LD (made from 3 mg/30 ml stock) there was a decrease in the number of viable cells in G0 as time passed, however viable cells were still present after 72 hours of exposure (Figures 9A, B and 9C)). There was no evidence of cell cycle progression in PHA-stimulated PBLs exposed to LD, however viable cells were still present after 72 hours of exposure (Figures 9D, E and 9F).
Figure 9.
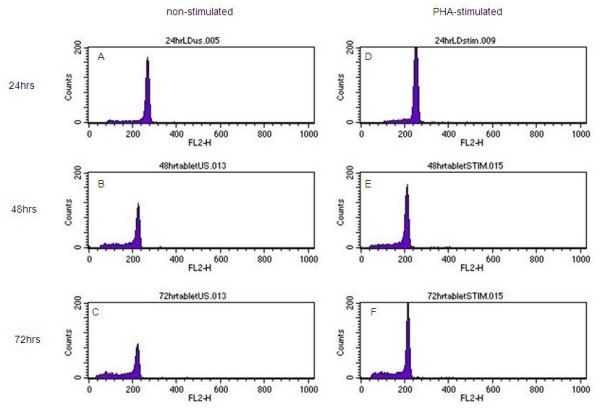
A-F: Histograms showing PI stained non-stimulated and PHA-stimulated PBLs exposed to LD at 1:10 for 24, 48 and 72 hours. The x axis of each histogram shows relative DNA content, with cells in G0/G1 over channel 200. The sub-G1 region represents apoptotic cells. The y axis represents cell number. 9A: non stimulated PBLs exposed to LD (1:10) for 24 hours, 9B: non stimulated PBLs exposed to LD (1:10) for 48 hours, 9C: non stimulated PBLs exposed to LD (1:10) for 72 hrs, 9D: PHA stimulated PBLS exposed to LD (1:10) for 24 hours, 9E: PHA stimulated PBLS exposed to LD (1:10) for 48 hours, 9F: PHA stimulated PBLS exposed to LD (1:10) for 72 hours.
Identification of bioactive fractions and chemical analysis of LD using HPLC and LC-MS
The HPLC trace for LD revealed a total of 24 peaks, each possibly relating to a single compound (Figure 10A). Analysis of the cytotoxic effect of the first 10 fractions on the HL60 cell line after 48 hours of exposure revealed fractions 1 (p = 0.01), 2, 4 and 7 (p = 0.05) exerted a statistically significant cytotoxic effect (Figure 10B). A single compound was detected in fraction 2 by LC-MS, while 2 compounds were found in fraction 7 (Table 1). For the LD repeat a total of 25 peaks were recorded at 253 nm (Figure 11A). Fractions 2, 3, 4 and 7 exerted a toxic effect on the HL60 cell line after 48 hours (p = 0.05) (Figure 11B). A single compound was detected by LC-MS in fraction 2, 2 compounds were found in fraction 4 and a single compound in fraction 7 (Table 2).
Figure 10.

A: HPLC readout (253 nm) of LD eluted at 3 ml/minute with the corresponding fraction number labelled on the y axis. 10B: the growth inhibitory effect of each fraction on the HL60 cell line after 48 hours of exposure. The pink line represents control cell growth, the blue line represents the number of cells/ml after 48 hours of exposure to CHR ± standard deviation form the mean, * and ** denote statistical significance p = 0.05 and p = 0.01 respectively.
Table 1.
Molecular weights of compounds within active fractions
| Fraction 2 DMSO:Water (50:50) | |
| Fraction 2 MeOH:Chloroform (50:50) | 362.1 |
| Fraction 7 DMSO:Water (50:50) | 278.1 |
| Fraction 7 MeOH:Chloroform (50:50) | 222.9 |
The molecular weights of compounds isolated from bioactive fractions by LC-MS. Each fraction was evaporated and taken up in DMSO:Water (50:50) and MeOH:Chloroform (50:50) for LC-MS analysis.
Figure 11.

A: HPLC readout (253 nm) of LD eluted at 3 ml/minute with the corresponding fraction number labelled on the y axis. (Repeat analysis). 11B: the growth inhibitory effect of each fraction on the HL60 cell line after 48 hours of exposure. The pink line represents control cell growth, the blue line represents the number of cells/ml after 48 hours of exposure to CHR ± standard deviation form the mean, * and ** denote statistical significance p = 0.05 and p = 0.01 respectively. (Repeat experiment)
Table 2.
Molecular weights of compounds within active fractions
| Fraction 2 DMSO:Water (50:50) | ||
| Fraction 2 MeOH:Chloroform (50:50) | 418.3 | |
| Fraction 4 DMSO:Water (50:50) | ||
| Fraction 4 MeOH:Chloroform (50:50) | 259.2 | 362.1 |
The results of this investigation show that water extracts of LD exert a growth inhibitory effect and apoptosis on both the HL60 and HT29 cancer cell lines and that there is consistency between the growth inhibitory and apoptotic effects of LD on these cell lines with growth inhibition and apoptosis being greater for HL60 than for HT29. The difference between HL60 and HT29 concerning the extent of apoptosis suffered by these two cell lines may be explained by previous studies that have shown HL60 cells to readily apoptose in response to a variety of stimuli, while HT29 cells are generally resistant to apoptosis [11].
There is a paucity of literature on the cytotoxic and apoptotic action of LD on cancer cells (in vivo and in vitro) however studies on the mechanism of action of some of its constituents provide some material that can be used to begin to attempt to elucidate the mechanism of action of this formulation.
Radix bupleuri, Radix scutellariae, Rhizoma alismatis and Fructus gardenia and their chemical constituents (saikosaponin D - a triterpene derivative, flavonoids including baicalin and baicalein, triterpenes and acetylated glycoside geniposide respectively) are reported to demonstrate cytotoxic and/or apoptotic activity against cancer cells in vitro, and/or in vivo [3-9,12-18] thus, at first inspection, it is possible that the action of LD observed in this present study may be related to the actions of these herbal constituents and their chemical constituents.
The results of the cell cycle analysis reflect those of the growth inhibition and apoptosis studies as LD was generally more toxic to the HL60 cell line than to the HT29 cell line. For HL60 these results suggest that the dose-related growth inhibitory action of LD at 1:100, as seen in the growth inhibition study, was related to apoptotic induction and not cell cycle arrest. Furthermore, overlaid histograms of TUNEL data showed HL60 cells were dying irrespective of their position in the cell cycle when exposed to LD at 1:10 for 4 hours, thus suggesting the cytotoxic action of LD at 1:10 is also cell cycle-independent. For HT29 there was a clear change in cell cycle characteristics, with a dose-related decrease in cells in G1 at all time points, an increase in cells in S phase at all time points, and a small change in the number of cells in G2 compared to the control. Thus, suggesting that the growth inhibitory action of LD at 1:50 and 1:100 in this cell line may be related to transient growth arrest during S-phase of the cell cycle. However, in contrast, overlaid histograms of TUNEL data revealed that at 1:10, LD induced apoptosis in a cell cycle-independent fashion. Thus, these results may suggest that at high concentrations LD triggers apoptosis through the induction of large amounts of cellular damage and at lower concentrations, less damage is induced by LD and S phase cell cycle arrest is triggered in the HT29 cell line. As S phase arrest can be triggered by compounds that directly damage DNA or inhibit DNA synthesis [19] the results of the cell cycle analysis suggest that water extracts of LD may possess one or more of these types of compounds, and that these may be responsible for the ability of LD to transiently arrest S phase progression in HT29 cells at 1:50 and 1:100.
Geniposide (more specifically its acetylated derivative) a constituent of Fructus gardenia and saikosaponin D, a constituent of Radix bupleuri, have been shown to cause cell cycle arrest at the G0 and/or G1 phase(s) of the cell cycle of cancer cells in vitro via activation of pro-apoptotic genes including p53 [12,18]. However, the results of the present study do not provide evidence of LD inducing cell cycle arrest at G1 in the HT29 cell line. This difference in results may be due to the HT29 cell line being p53 negative [20] and as a lack of functional p53 can prevent the prolonged arrest of cells with DNA damage at the G1 checkpoint, this could also account for the lack of G1 arrest observed in this study. However, it may be that the compounds are not present in the water extract of LD used in the present study as the preparation of both geniposide and saikosaponin D used in the studies cited differed considerably from that of the LD water extract used in the present study: the preparation of geniposide involved reflux in ether and extractions using methanol and butanol [21]. The saikosaponin D used by Hsu et al [12] was a pure product dissolved in DMSO. The absence of chemical constituents in the LD water extract may also explain the differences between this extract and that of the Bupleurum scorzonerifolium Willd (from which Radix bupleuri is traditionally derived [2]) used by Chen et al [7]. In the present study only slight and transient G2/M arrest was seen after 24 hours of exposure to the water extract of LD. In the study by Cheng et al [7] three preparations were used an acetone, a methanol and a water extract but it was only the acetone extract that induced cell cycle arrest at the G2/M checkpoint in A549 lung cancer cells.
Regarding the possible role of other chemical constituents, Lee et al [15] reported that baicalein, a constituent of Radix scutellariae, induced apoptosis in human lung carcinoma CH27 cells through arrest in S phase of the cell cycle. Thus, although commercially available baicalein was used by Lee et al [15] one could, based solely on cell culture experiments, speculate that this compound may potentially have contributed to the action of LD on the cell cycle of the HT29 cell line in the present study.
The results of the comet assay suggest that LD induced DNA damage in both cell lines through the induction of double strand breaks, single strand breaks or alkali label sites. The ability of LD or its constituents to induce DNA damage in cancer cell lines in vitro has not previously been examined. However, the ability of LD to induce DNA damage is in-keeping with the previous results of this investigation, in that LD has been found to elicit a cytotoxic action and induce apoptosis in the HL60 and HT29 cell lines, and induce cell cycle arrest in the HT29 cell line; all these actions can all occur as a result of the induction of DNA damage. Furthermore, the results of this investigation suggest that LD exerts a greater genotoxic action on the HL60 cell line than the HT29 cell line, which is also consistent with previous results obtained in this investigation.
To further elucidate the mechanism of action of LD and to characterize its chemotherapeutic potential its effect on non cancer cells was investigated. The results of this part of the study suggest that at 1:10 LD may exert a moderate cytotoxic action against non-proliferating PBLs and that LD may have induced some form of cytotoxic damage that prevented those cells from progressing through the G0/G1 checkpoint (as discussed above). However, as LD was found to significantly inhibit the growth of the HL60 and HT29 cell lines, at 1:10, these results suggest that LD may exert a more significant cytotoxic action against cancer cells than non-cancer cells.
The preliminary chemical analysis showed that for LD a number of fractions were found to exert a significant growth inhibitory effect at the 5% level (p = 0.05), however none of the fractions tested exerted a significant effect at the 1% level (p = 0.01) which may suggest that LD contains a number of moderately cytotoxic compounds that interact to elicit a significant cytotoxic effect in vitro. However, LC-MS analysis of the cytotoxic fractions revealed that the molecular weights of the compounds within these fractions do not correspond to the molecular weights of the chemical constituents discussed above. Thus, the results of this analysis suggest that previously unreported compounds are responsible for the in vitro inhibitory action of LD, on the growth of the HL60 and HT29 cancer cells lines, observed in this current study.
Experimental
Preparation of LD
LD was obtained from a local TCM practitioner (London, UK). This formulation of LD, which was in capsule form, contains: Radix gentianiae (Long Dan), Radix bupleuri (Cai hu), Rhizoma alismatis (Ze xie), Semen plantaginis (Che qian zi), Radix rehmanniae (Di huang) (the approximate relative quantity of each of these herbal constituents in this formulation of LD is 14.29%) Fructus gardeniae (Zhi zi), Radix scutellariae (Huang qin) Radix angelicae sinensis (Dang gui) Radix glycyrrhizae (Gan cao), Medulla tetrapanacis (Tong cao) (the approximate relative quantity of each of these herbal constituents in this formulation of LD is 7.1%). So as to adhere as closely as possible to the traditional method of preparation and ingestion of this CHR, aqueous extracts were prepared as follows: LD was obtained in tablet form and was therefore not boiled, instead 3 g of the contents of each pill were crushed then incubated in water (30 ml) at 37°C for 1 hour, after which time the water extract was centrifuged and sterile filtered using a 0.45 μm sterile filter (Nalgene, Hereford, UK). This water extract of 3 g in 30 ml was the stock solution used to prepare the concentrations (1:10, 1:50, 1:100 and 1:1000) of the LD water extract used in the experiments described below.
Cell Culture
HL60 (human promyelotic leukaemia) and HT29 (human colon adenocarcinoma) cancer cell lines were obtained from The European Collection of Cell Cultures (ECACC, Sailsbury, UK). The HL60 cell line was grown in RPMI 1640 medium (Gibco, Paisley, UK) containing 10% foetal bovine serum (FBS) (Gibco, Paisley, UK). The HT29 cell line was grown in minimum essential medium (MEM) (Gibco, Paisley, UK) containing 10% FBS, 2 mM L-glutamine and 2 mM NEAAs. Cells were incubated at 37°C in an atmosphere containing 5% CO2.
Growth Inhibition Study
For the growth inhibition study, the growth inhibitory actions of water extracts of LD were investigated at final concentrations of 1:10, 1:100 and 1:1000, prepared using the stock solution described above, on each of the cell lines over a period of 72 hours. Growth curves were carried out in 24-multiwell plates; the seeding concentration was 1 × 105cells/ml. Three identical plates were made up for each of the experiments at 0 hrs, and then one of these was counted and discarded after 24, 48 and 72 hours. Cell viability was measured by determining whole cell numbers using a haemocytometer, and viable cells were excluded using trypan blue (Gibco, Paisley, UK).
Apoptosis study
Apoptosis studies were carried out to ascertain the mode of cell death. The Annexin V assay, propidium iodide staining and the TUNEL assay were all used to investigate not only the impact of LD on apoptosis but to determine at what stage this process is induced by this CHR.
The Annexin V assay
The Annexin V assay provides information about one of the early morphological characteristics unique to apoptotic cell death, which is the loss of membrane asymmetry before loss of membrane integrity. In the Annexin V assay, the translocation of phosphatidylserine (PS) residues from the internal to the outer face of the plasma membrane before loss of membrane integrity is used as a marker of apoptotic induction [22]. Annexin V conjugated with the fluorochrome fluorescin isothiocyanate (FITC) and the vital dye propidium iodide (PI) were used to distinguish between viable, early apoptotic and late stage apoptotic/dead cells.
The Annexin V assay was performed using the BD Pharmingen™ Annexin V-FITC Apoptosis Detection Kit (Becton Dickenson, Oxford, UK) and 5 μl of 50 μg/ml of propidium iodide (Sigma, Poole, UK) were used.
HL60 and HT29 cell lines (1 × 105cell/ml) were exposed to LD (1:10) for 6,12 16 and 24 hours (for HT29 only) then analysed using a BD FacsCalibur Flow Cytometer running CellQuest Pro (Becton Dickenson, Oxford, UK). The fluorochrome FITC was analyzed using the FL1 detector, while PI was detected using FL2.
PI staining for sub-G1 peaks
Although propidium iodide (PI) staining was used as part of the Annexin V assay to distinguish cell populations, separate PI staining provides evidence of the presence of one of the later characteristics of apoptosis which is fragmentation of the genome by identifying reductions in DNA content.
HL60 and HT29 cell lines at a starting concentration of 1 × 105cell/ml were made up and exposed to water extracts of LD (1:10 - prepared as described above) for 4, 8 and 24 hours (for HT29 cells only). Following exposure, cells were permeabilised using ice-cold 70% ethanol. Cells were then washed twice in ice cold PBS and re-suspended in PBS (1 ml). To remove double stranded RNA, 1 unit of DNAse free RNAse A (Promega, Hertfordshire, UK) was added to the cell suspension, and incubated for 30 minutes at 37°C. Finally, PI (100 μl of 50 μg/ml) was added to the cell suspension, and cells were stored on ice and protected from light until analysis.
Flow cytometry was used to analyse samples. For apoptotic analysis 10,000 of all events detected by the flow cytometer were counted and saved. In order to generate accurate histograms gating was performed to separate single cells that had passed through the flow cell from two (doublets) or more cells that had passed through the flow cell at the same time (which are scored as a single event by the flow cytometer).
TUNEL assay
The TUNEL assay was performed using the Promega DeadEnd™ Fluoremetric TUNEL system (Promega, Southampton, UK). This assay is similar to PI staining in that it looks for evidence of genomic fragmentation however as a marker it is more specific as it provides evidence of 3' hydroxyl-termini DNA strand breaks within cells.
Flasks were seeded with HL60 and HT29 cells at a starting concentration of 1 × 105 cells/ml. After exposure to water extracts of LD (1:10 - prepared as described above)for 4, 8 and 24 hours (for HT29 cell only)
Cell cycle analysis using PI staining and TUNEL
To address the issue of whether any inhibitory effects of LD on HL60 and HT29 cells occurred as a result of cell cycle arrest, the effect of LD on cell cycle characteristics was investigated by determining how it affected cell cycle checkpoints that, via a series of sensors, can detect cellular damage and ultimately inhibit cell cycle progression, to allow for repair, or induce apoptosis if the damage is irreparable [23]. These cell cycle check points are G1 (during which cells prepare for replication), S (during which DNA is replicated and a complete copy of each chromosome is made) and G2/M (M is the last phase of the cell cycle during which new chromosomes are equally segregated between two daughter cells before division).
To examine the role of cell cycle arrest as a consequence of any growth inhibitory actions due to LD HL60 and HT29 cell lines were seeded at a starting concentration of 105 cells/ml and water extracts of LD added at final concentrations of 1:50 and 1:100 (prepared using the stock solution described above) for 24, 48 and 72 hours. Following incubation, cells were permeabilized using ice-cold 70% ethanol, then washed twice in ice cold PBS and re-suspended in PBS (1 ml). To remove double stranded RNA, 1 unit of DNAse free RNAse A was added to the cell suspension, and incubated for 30 minutes at 37°C. Finally, PI (100 μl) was added to the cell suspension, and cells were stored on ice and protected from light until analysis. For cell cycle analysis 15,000 of all events were saved and the percentage of cells in each stage of the cell cycle calculated using the freeware Cyclred [24].
TUNEL data for HL60 and HT29 cells exposed to water extracts of LD (1:10 - prepared as described above)for 4 hrs and 24 hrs respectively were collected (as described above) to analyse the cell cycle stage in which apoptosis was induced for each of these cell lines. Analysis of the cell cycle stage in which apoptosis was being induced are expressed as overlaid histogram plots showing all cells, viable cells and apoptotic cells, with data gated from TUNEL density plots and FL2-Area versus FL2-Width plots.
Determination of genotoxicity using the Comet assay
To look for evidence of DNA damage (genotoxicity) which can trigger apoptosis, the comet assay was used. The pH>13 version of the experiment was used, which looks for evidence of single strand breaks, alkali labile sites, DNA-DNA/DNA-protein cross linking and single strand breaks (SSB) associated with incomplete excision repair sites [25]. Media (1.5 ml) containing 1 × 105 cells/ml was placed in 24 well multi-well plates. HL60 and HT29 cell lines were exposed to water extracts of LD at 1:10, 1:50 and 1:100 (prepared as described above); a control was also set up. Cells were incubated at 37°C in an atmosphere containing 5% CO2 for 4 hours. After the incubation, the cells were centrifuged at 800 rpm for 5 minutes and re-suspended in low melting point (LMP) agarose (Promega, Southampton, UK) (0.5%) heated to 37-42°C and pipetted on to slides. The slides were placed in lysis solution (2.5 M NaCl, 100 mM EDTA, 10 mM Tris and 1%v/v Triton® X-100 (added immediately before use)) for 1 hour at 4°C. The slides were then removed, washed in PBS and placed in electrophoresis solution (0.3 M NaOH, 1 mM EDTA, pH>13) for 30 minutes at 4°C, then subject to electrophoresis for 30 minutes at 4°C at 25 V/300 mA. After electrophoresis, slides were washed in neutralisation buffer (0.4 M Tris, pH 7.5 with 10 M HCl) 3 times for 5 minutes each. Slides were then dried and ethidium bromide (20 μg/ml) (Sigma, Dorset, UK) was pipetted on to the gel. Slides were analyzed using a Zeiss Axioskop microscope (Carl Zeiss LTD, Hertfordshire, UK) connected to a Nikon DN100 digital camera (Nikon, Kingston upon Thames, UK). Tail length was measured live at 1000× magnification using the Eclipse Net image analysis package (Nikon, Kingston upon Thames, UK) The comet assay was carried out in duplicate and for each run 50 tails scored, yielding a total of 100 counts per experiment.
The effect of LD extract on unstimulated (non-proliferating) and stimulated (proliferating) primary human blood lymphocytes
To determine if the cytotoxic effect of LD was cancer cell specific its effect on non cancer cells peripheral human blood lymphocytes, PBLs (both dividing (stimulated) and non-dividing (non-stimulated) was investigated.
Isolation of PBLs
Primary human blood lymphocytes (PBLs) were isolated from 20 ml heparinized venous whole blood using Ficoll-Paque Plus. Whole blood (15 ml) was diluted 1:1 with PBS then layered on top of 20 ml Ficoll in a 50 ml falcon tube and centrifuged for 30 minutes at 800 rpm. Lymphocytes were removed from the interface between the plasma and Ficoll layers.
Culture and stimulation of PBLs
Isolated PBLs were cultured in RPMI 1640 media containing 10% FBS supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C. Cells were incubated at 37°C in an atmosphere containing 5% CO2. The effects of LD on non-proliferating and proliferating PBLs were examined. In order to stimulate isolated PBLs to proliferate in vitro, phytohaemagglutinin (PHA) was used. Phytohaemagglutinin is a plant lectin isolated from the red kidney bean (Phaseolus vulgaris), and is a potent mitogen [26]. To stimulate PBLs, 10 mM PHA (Sigma, Dorset, UK) was added to appropriate flasks. Flasks were seeded at 1 × 105 cells/ml and incubated with water extracts of LD at 1:10 (prepared as described above) for 24, 48 and 72 hours in the presence and absence of PHA. These exposure times were used so as to allow the PHA-stimulated PBLs to progress through the cell cycle in the presence of LD. Controls, for each time point, were also set up. Experiments were carried out in duplicate.
Evidence of apoptotic induction and alterations in cell cycle characteristics were investigated using PI staining and FACS analysis, as described above. In order to analyse the cell cycle-related and apoptosis-inducing effects of LD on proliferating and non-proliferating PBLs using PI staining, singlets had to be gated and used for analysis. Data are represented as histograms showing cell number (x-axis) versus relative PI fluorescence intensity (y-axis).
Identification of bioactive fractions and chemical analysis of LD using HPLC and LC-MS
LD was split into fractions using a LichroCART® 250-10, LIChrospher® 100 RP-18e (10 μm) HPLC column (Merck, Darmstadt, Germany). LD 500 ul was injected into the column and the flow rate set at 3 ml/minute. Fractions (3 ml) were collected at 1 minute intervals. Water was used as the mobile phase. The HPLC prep column was attached to a Perkin-Elmer series 410 LC pump and a Perkin-Elmer LC-235 Diode array detector. Absorbance was measured at 253 nm. Individual fractions were sterile filtered using a 0.45 μm sterile filter. HL60 cells at a starting concentration of 1 × 105/ml were exposed to LD fractions for 48 hours then cell numbers compared to control cell growth using the trypan blue exclusion assay.
LC-MS analysis was carried out on those fractions that exerted the strongest statistically significant (p = 0.01) growth inhibitory effects using atmospheric pressure chemical ionization (APCI) and sample detection using a Finnigan LCQ MS detector. Samples (1.5 ml) were initially found to be too dilute, and along with a distilled water control were evaporated under vacuum at 55°C. These were then taken up in DMSO: Water 50:50 (200 μl) and sonicated before analysis. Any remaining residue was taken up in MeOH: Chloroform 50:50 (200 μl) and also sonicated before analysis.
Data presentation and statistical analysis
Data for the growth inhibition study are expressed as mean ± standard deviation from the mean. The mean values of each concentration at each time point were compared to control values using ANOVA. Annexin V data were collected in the form of dot plots and then represented as density plots. Cells in the bottom left quadrant of the density plots are viable (Annexin V-FITC and PI negative), cells in the lower right quadrant are apoptotic (Annexin V-FITC positive and PI negative), and cells in the upper right quadrant are late stage apoptotic/necrotic (Annexin V-FITC and PI positive). PI staining data are presented as histograms with the x axis showing relative DNA content and the y axis showing cell number. TUNEL data are represented as density plots showing DNA content on the x axis (using FL2) and the relative number of DNA strand breaks on the y-axis (using FL2). Cell cycle analysis data are represented as histograms showing cell number (x-axis) versus relative PI fluorescence intensity (y-axis). Data from the comet assay are expressed as mean ± SD. The tail lengths of DNA from the exposed cells were compared to those of the controls using the t-test. HPLC data are represented as graphs showing relative growth inhibition compared to control cell growth (± standard deviation). The mean growth inhibitory effect of each fraction was compared to control cell growth using ANOVA.
Conclusion
To the authors' knowledge, this is the first investigation to suggest that LD may elicit a cytotoxic action against cancer cell lines in vitro. The results of this investigation suggest that water extracts of LD are toxic to cancer cells in vitro, triggering apoptosis possibly as a result of inducing some form of genotoxic damage. However, preliminary chemical analysis of this CHR indicates that this cytotoxic activity is due to the actions of constituents other than those discussed above. One main reason for this is linked to the differences between the preparations used in the cytotoxicity studies reported in this paper [4,6,8,9,12-18] and that used in the current study i.e. a water extract of LD. However, the possibility that interactions between the constituents in this water extract may have influenced the results observed cannot be ruled out [27].
In conclusion, based on the findings of this investigation, the water extract of LD appears to have some chemotherapeutic potential However, further studies are required to determine what constituents within this CHR are responsible for its cytotoxic action.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SW participated in the design of the study; carried out the cell growth and apoptosis studies; the cell cycle analysis; and comet assay; and participated in and collected the HPLC and LC-MS data; performed the statistical analysis; interpreted the data; and made a significant contribution to drafting the initial manuscript. LJ conceived of the study and participated in its design and coordination. JB conceived of the study, participated in its design and coordination and supervised the HPLC analysis. EIO conceived of the study and participated in its design and coordination and the interpretation of data; made a significant contribution to drafting and critically revising the manuscript; and gave the final approval of the version to be published. All authors read and approved the manuscript
Acknowledgments
Acknowledgements
Shaun Willimot was the recipient of a Biomedical and Pharmaceutical Sciences Research Group, Kingston University, funded Ph.D. LC-MS analysis was conducted by Dr Mirza at the Institute of Cancer Research (ICR, Surrey, UK)
Contributor Information
Shaun Willimott, Email: swillimott@yahoo.co.uk.
James Barker, Email: j.barker@kingston.ac.uk.
Lucy A Jones, Email: l.jones@kingston.ac.uk.
Elizabeth I Opara, Email: e.opara@kingston.ac.uk.
References
- Cosyns JP, Goebbels RM, Liberton V, Schmeiser HH, Bieler CA, Bernard AM. Chinese herbs nephropathy-associated slimming regimen induces tumours in the forestomach but no interstitial nephropathy in rats [abstract] Arch toxicol. 1998;72:738–43. doi: 10.1007/s002040050568. [DOI] [PubMed] [Google Scholar]
- Yang ZY, Zhi C, Huo KK, Hui X, Tian ZP, Pan SL. ITS sequence analysis used for molecular differentiation of the Bupleurum species from northwestern China. Phyomedicine. 2007;14:416–422. doi: 10.1016/j.phymed.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Kok LD, Wong CK, Leung KN, Tsang SF, Fung KP, Choy YM. Activation of the anti-tumor effector cells by Radix bupleuri. Immunopharmacology. 1995;30:79–87. doi: 10.1016/0162-3109(95)00010-Q. [DOI] [PubMed] [Google Scholar]
- Ikemoto S, Sugimura K, Yoshida N, Yasumoto R, Wada S, Yamamoto K, Kishimoto T. Antitumour effects of Scutellariae radix and its components baicalein, baicalin, and wogonin on bladder cancer cell lines. Urology. 2000;55:951–955. doi: 10.1016/S0090-4295(00)00467-2. [DOI] [PubMed] [Google Scholar]
- Cheng YL, Chang WL, Lee SC, Liu YG, Lin HC, Chen CJ, Yen CY, Yu DS, Lin SZ, Harn HJ. Acetone extracts of Bupleurum scorzonerifolium inhibits proliferation of A549 human lung cancer cells via inducing apoptosis and suppressing telomerase activity. Life Sci. 2003;73:2383–2394. doi: 10.1016/S0024-3205(03)00648-9. [DOI] [PubMed] [Google Scholar]
- Sonoda M, Nishiyama T, Matsukawa Y, Moriyasu M. Cytotoxic activities of flavonoids from two Scutellaria plants in Chinese medicine. J Ethnopharmacol. 2004;91:65–68. doi: 10.1016/j.jep.2003.11.014. [DOI] [PubMed] [Google Scholar]
- Chen YL, Lee SC, Lin SZ, Chang WL, Chen YL, Tsui NM, Liu YC, Tzao C, Yu DS, Ham HJ. Antiproliferative activity of Bupleurum scrozonerifolium in A549 human lung cancer cells in vitro and in vivo. Cancer Lett. 2005;222:183–193. doi: 10.1016/j.canlet.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Huang YT, Huang DM, Chueh SC, Teng CM, Guh JH. Alisol B acetate, a triterpene from Alismatis rhizome, induces Bax nuclear translocation and apoptosis in human hormone-resistant prostate cancer PC-3 cells. Cancer Lett. 2006;231:270–278. doi: 10.1016/j.canlet.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Baumann S, Fas SC, Giaisi M, Müller WW, Merling A, Gülow K, Edler L, Krammer PH, Li-Weber M. Wogonin preferentially kills malignant lymphocytes and suppresses T-cell tumor growth by inducing PLCγ1- and Ca2+-dependent apoptosis. Blood. 2008;111:2354–2363. doi: 10.1182/blood-2007-06-096198. [DOI] [PubMed] [Google Scholar]
- Willimot S, Barker J, Jones L, Opara EI. Apoptotic effect of Oldenlandia diffusa on the leukaemic cell line HL60 and human lymphocytes. J Ethnopharmacol. 2007;114:290–299. doi: 10.1016/j.jep.2007.08.030. [DOI] [PubMed] [Google Scholar]
- Shao RG, Shimizu T, Pommier Y. Brefeldin is a potent inducer of apoptosis in human cancer cells independently of p53. Exp Cell Res. 1996;227:190–196. doi: 10.1006/excr.1996.0266. [DOI] [PubMed] [Google Scholar]
- Hsu Yl, Kuo PL, Lin CC. The proliferative inhibition and apoptotic mechanism of saikosaponin D in humam non-small cell lung cancer A549 cells. Life Sci. 2004;75:1231–1242. doi: 10.1016/j.lfs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Lee HZ, Leung HW, Lai MY, Wu CH. Baicalein induced cell cycle arrest and apoptosis in human lung squamous carcinoma CH27 cells. Anticancer Res. 2005;25:959–964. [PubMed] [Google Scholar]
- Chan FL, Choi HL, Chen Zy, Chan PSF, Huang Y. Induction of apoptosis in prostate cancer cell lines by a flavonoid, baicalin. Cancer Lett. 2000;160:219–228. doi: 10.1016/S0304-3835(00)00591-7. [DOI] [PubMed] [Google Scholar]
- Lee S, Kho Y, Min B, Na M, Kang S, Maeng H, Bae K. Cytotoxic triterpenoids from Alismatis Rhizoma. Arch Pharm Res. 2001;24:524–6. doi: 10.1007/BF02975158. [DOI] [PubMed] [Google Scholar]
- Chang YC, Tseng TH, Lee MJ, Hsu JD, Wang CY. Induction of apoptosis by penta-acetyl geniposide in rat C6 glioma cells. Chem-Biol Interact. 2002;141:243–257. doi: 10.1016/S0009-2797(02)00073-X. [DOI] [PubMed] [Google Scholar]
- Peng CH, Tseng TH, Liu JY, Hsieh YH, Huang CN, Hsu SP, Wang CJ. Penta-acetyl geniposide-induced C6 glioma cell apoptosis was associated with the activation of protein kinase C-delta. Chem-Biol Interact. 2004;147:287–296. doi: 10.1016/j.cbi.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Peng CH, Tseng TH, Huang CN, Hsu SP, Wang CJ. Apoptosis induced by penta-acetyl geniposide in C6 glioma cells is associated with JNK activation and Fas ligand induction. Toxicol Appl Pharm. 2005;202:172–179. doi: 10.1016/j.taap.2004.06.016. [DOI] [PubMed] [Google Scholar]
- American Cancer Society http://www.cancer.org/docroot/ETO/content/ETO_1_4X_What_Are_The_Different_Types_Of_Chemotherapy_Drugs.asp?sitearea=ETO
- Rodrigues NR, Rowan A, Smith ME, Kerr IB, Bodmer WF, Gannon JV, Lane DP. p53 mutations in colorectal cancer. Proc Natl Acad Sci USA. 1990;87:7555–7559. doi: 10.1073/pnas.87.19.7555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CJ, Tseng TH, Lin JK. Penta-acetyl geniposide: isolation, identification and primary effect on C6 glioma cells in vitro. Anticancer Res. 1992;12:911–916. [PubMed] [Google Scholar]
- Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescin labelled annexin V. Journal Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-I. [DOI] [PubMed] [Google Scholar]
- Lukas J, Lukas C, Bartek J. Mammalian cell cycle checkpoints: signalling pathways and their organization in space and time. DNA Repair. 2004;3:997–1007. doi: 10.1016/j.dnarep.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Vanhuyse M, Kluza J, Tardy C, Otero G, Cuevas C, Bailly C, Lansiaux A. Lamellarin D: a novel pro-apoptotic agent from marine origin insensitive to P-glycoprotein-mediated drug efflux. Cancer Lett. 2005;221:165–175. doi: 10.1016/j.canlet.2004.09.022. [DOI] [PubMed] [Google Scholar]
- Tice RR, Agurell E, Anderson D, Burlinson B, Hartmann A, Kobayashi H, Miyamae Y, Rojas E, Ryu J-C, Sasaki YF. Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicology testing. Environ Mol Mutagen. 2000;35:206–221. doi: 10.1002/(SICI)1098-2280(2000)35:3<206::AID-EM8>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Serke S, Serke M, Brudler O. Lymphocyte activation by phytohaemagglutinin and pokeweed mitogen. Journal Immunol Methods. 1987;99:167–172. doi: 10.1016/0022-1759(87)90122-0. [DOI] [PubMed] [Google Scholar]
- Yuan R, Lin Y. Traditional Chinese medicine: an approach to scientific proof and clinical validation. Pharmacol Therapeut. 2000;86:191–198. doi: 10.1016/S0163-7258(00)00039-5. [DOI] [PubMed] [Google Scholar]