

Abstract
All transglutaminases share the common enzymatic activity of transamidation, or the cross-linking of glutamine and lysine residues to form N epsilon (gamma-glutamyl) lysyl isopeptide bonds. The plasma proenzyme factor XIII is responsible for stabilizing the fibrin clot against physical and fibrinolytic disruption. Another member of the transglutaminase family, tissue transglutaminase or TG2 is abundantly expressed in cardiomyocytes, vascular cells and macrophages. The transglutaminases have a variety of functions independent of their transamidating activity. For example, TG2 binds and hydrolyzes GTP, thereby fostering signal transduction by several G protein coupled receptors. Accumulating evidence points to novel roles for factor XIII and TG2 in cardiovascular biology including: (a) modulating platelet activity, (b) regulating glucose control, (c) contributing to the development of hypertension, (d) influencing the progression of atherosclerosis, (e) regulating vascular permeability and angiogenesis (f) and contributing to myocardial signaling, contractile activity and ischemia/reperfusion injury. In this review, we summarize the cardiovascular biology of two members of the family of transglutaminases, Factor XIII and TG2.
Keywords: Angiogenesis, Atherosclerosis, Cross-Link, Factor XIII, G Protein, Heart Failure, Macrophage, Myocardial Infarction, Platelet, Transglutaminase, Review
2. INTRODUCTION
There are at least 9 members of the human transglutaminase family(1). Factor XIII is the plasma, platelet and macrophage proenzyme, traditionally viewed as a terminal component of the coagulation cascade. TG2 is another member of the transglutaminase family that is widely expressed in cardiovascular cells and in macrophages1. Recent studies have documented diverse roles for these transglutaminases in cardiovascular pathophysiology (Figure 1). Transglutaminases likely influence both the chronic as well as the acute manifestations of atherosclerosis (e.g., plaque rupture). Transglutaminases can modulate several cardiovascular risk factors, especially hypertension, by influencing angiotensin II-AT1 receptor signaling and by altering vascular structure. The transglutaminases have direct myocardial actions such as regulating myocardial growth, fibrosis, and wound healing after myocardial infarction. In this review, we will consider these and other aspects of the contributions of transglutaminases to cardiovascular health and disease.
Figure 1.
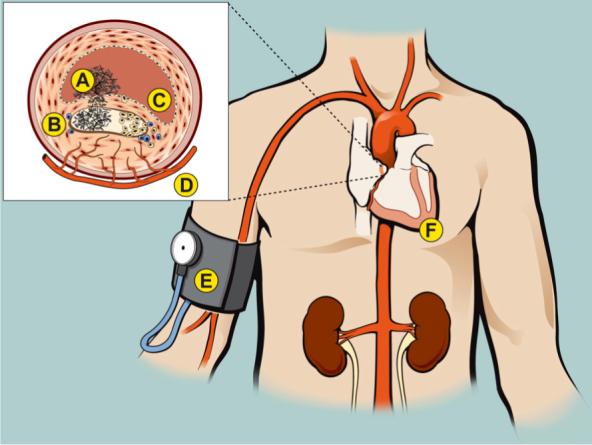
Effects of transglutaminases on vascular and cardiac diseases. The inset depicts a coronary artery with an atheroma that has undergone plaque rupture, with subsequent thrombus formation in the arterial lumen and within the lipid core. Stabilization of the thrombus. Factor XIIIa mediated cross-linking of fibrin and covalent attachment of plasmin inhibitor to fibrin renders the clot resistant to fibrinolysis. Factor XIIIa and tissue transglutaminase play roles in platelet activation, such as contributing to the formation of “coated” platelets. Coated platelets have surface-bound procoagulant proteins, resulting in enhanced ability to promote thrombin generation. Factor XIIIa and TG2 may also regulate platelet adhesion by increasing the ligand binding affinity of integrin receptors. A. Effects on the progression of atherosclerosis. Inhibitors of FXIIIa reduce the progression of atherosclerosis in hyperlipidemic mice. In contrast, mice that lack leukocyte expression of TG2 have more advanced atherosclerosis. Transglutaminase activity within the atheroma could lead to plaque progression by a variety of mechanisms including increasing the deposition of ECM proteins and activating TGF-β1. Tranglutaminase activity (especially TG2) also counteracts proteinases, for example, by attaching elafin to the ECM or by stabilizing the fibrous cap against MMP-mediated rupture. B. Endothelial barrier function. Transglutaminases stabilize the endothelial barrier, reducing endothelial permeability. Reduced EC barrier function could contribute to a variety of clinical conditions including plaque progression, myocardial edema and pleural effusions. C. Effects of transglutaminases on angiogenesis. The atheroma often has a rich neovascular supply. Factor XIIIa enhances angiogenesis by a variety of mechanisms such as enhancing VEGF receptor interaction with integrins and by down-regulating thrombospondin-1, an inhibitor of angiogenesis. In different animal models, TG2 has been reported to either enhance or inhibit angiogenesis. The effect of transglutaminases on angiogenesis could influence a variety of cardiovascular events including plaque progression and stability, coronary collateral formation and would healing after myocardial infarction. D. Effects of transglutaminases on blood pressure. Factor XIIIa induces the formation of dimers of the AT1 receptor that are hyper-activated. AT1 dimers occur more commonly in hypertensive than in normotensive patients. TG2 activity promotes the inward remodeling of arterioles in response to flow reduction. Increased expression of TG2 increases vascular tone, which could promote hypertension. E. Effects of tranglutaminases on the myocardium. Factor XIII knockout mice have poor myocardial healing and are prone to rupture after an MI. Transgenic mice that overexpress TG2 have LVH, myocardial fibrosis and reduced LVEF. The function of TG2 as a G protein (Gh), coupling α-adrenergic receptors to phospholipase C is dimished in ischemic and dilated cardiomypathies.
3. ROLES OF FACTOR XIIIA IN HEMOSTASIS AND THROMBOSIS
The traditional role of plasma Factor XIII is to prevent hemorrhage by stabilizing the incipient hemostatic thrombus against premature physical and enzymatic disruption. Human Factor XIII deficiency is characterized by a life-long severe bleeding disorder, with intracranial hemorrhage (2) and recurrent abortions (3) being a significant risk. Factor XIII subunit A−/− mice have a similar phenotype with impaired clot formation and reduced clot stability (4), and also suffer from spontaneous miscarriages due to uterine bleeding (5).
The clot stabilizing activity of factor XIIIa may impede both endogenous and therapeutic fibrinolytic activity. For example, Factor XIIIa cross-linking has been demonstrated to play a role in the resistance of experimental pulmonary emboli to fibrinolysis (6). Several inhibitors of factor XIIIa have been designed to enhance thrombolytic activity, including L-722,151, an agent that enhances fibrinolysis when administered early after experimental thrombus induction (7,8). However, since Factor XIIIa catalytic activity has short half of ∼ 19 minutes within the clot (9), it is uncertain whether factor XIIIa inhibitors will be clinically useful to enhance thrombolysis.
Factor XIIIa stabilizes fibrin by cross-linking the gamma-chains (10) and alpha-chains (6,11) of fibrin to each other and by attaching plasmin inhibitor to the fibrin alpha-chain (12). Thrombin-activable fibrinolysis inhibitor (TAFI, or plasma procarboxypeptidase B) is also cross-linked to fibrin by factor XIIIa and TG2, providing another anti-fibrinolytic effect (13). Other mechanisms of altering clot stability likely exist. Furthermore, emerging evidence supports the concept that FXIIIa activity can promote thrombus formation. For example, when the N-terminus of fibronectin is crosslinked by factor XIIIa to the C-terminus of the fibrin alpha-chain (13), the cross-linked product enhances platelet thrombus formation ex vivo in a parallel flow chamber to a greater extent than surfaces coated with fibrin alone or fibronectin alone (15). Elevated factor XIIIa activity has been detected in selected patients with a prothrombotic tendency. For example, factor XIIIa activity is elevated in patients with antiphospholipid syndrome in whom it may contribute both to thrombosis and to accelerated atherosclerosis (16).
It is possible that factor XIII contributes to the observation that levels of total plasma fibrinogen levels correlate with risk of myocardial infarction and stroke (17). Interestingly, factor XIII binds to a variant of the fibrinogen gamma chain known as gamma prime. This interaction occurs by binding the factor XIII B chain to the gamma prime site (18). The gamma prime isoform comprises approximately 8% of the total gamma-chain population in plasma and is derived by alternative spicing of the carboxyl terminus of the gamma chain in which the common sequence (AGDV) is replaced by a new C-terminus (VRPEHPAETEYDSLYPEDDL). The gamma prime extended C-terminus may be prothrombotic by serving as a binding site for factor XIII (19,20). Alternatively, the gamma variant may also have a role in suppressing thrombin generation by binding to thrombin, a role that was historically referred to as “antithrombin-I” (20).
The vascular roles of factor XIIIa-mediated fibrin cross linking are not limited to clot stability. Migration of VSMC into factor XIIIa-crosslinked fibrin gels is increased 2-fold compared to non-crosslinked gels (21). Thus, factor XIIIa may influence the response to vascular injury, where it may function as it does in other forms of wound healing (22). Plaque healing that occurs after rupture has been cited as a mechanism for rapid plaque growth (23). Accordingly, the extent of thrombus cross-linking by factor XIIIa could affect plaque progression.
4. TRANSGLUTAMINASES AND PLATELET FUNCTION
Platelets contain FXIII A subunit dimers (24) as well as TG2 (25) in their cytoplasm. Megakaryocytes have a 140-fold increased expression of factor Xlll A subunit relative to stem cell precursors (26). As a result, platelets are a major source of Factor XIII, containing up to 50% of the total factor XIIIa activity in blood (27). Despite its abundance, the role of platelet Factor XIII is not thoroughly understood. Platelet factor XIII may be activated by distinct mechanisms, without the need for the proteolytic cleavage of an N-terminal peptide as occurs with plasma factor XIII (28). Platelets not only contain significant quantities of Factor XIII in their cytoplasm, but factor XIII binds to the surface of activated platelets, at a site that is sensitive to plasmin proteolysis (29).
Recently, platelet Factor XIIIa has been reported to be involved in “coated” platelet (formerly known as collagen and thrombin activated, or COAT-platelet) formation (30). Coated platelets have high surface content of α-granule proteins (such as fibrinogen, VWF, fibronectin, factor V and thrombospondin) as well as exposed phosphatidyl serine (31). There is evidence that transglutaminase activity is involved in coated platelet formation, since transglutaminase inhibitors block their production (30). The α-granule proteins that are retained on the surface of coated-platelets have been serotonylated by transglutaminase-mediated cross-linking activity (32). The serotonin-conjugated proteins bind to serotonin binding sites on fibrinogen and thrombospondin (32). Although Factor XIIIa was considered the prime candidate for the transglutaminase involved in coated platelet formation, factor XIIIA−/− mice were found to form coat platelets as well as controls, suggesting the possibility that platelet-derived tissue transglutaminase was involved (33).
Platelet transglutaminase activity also contributes to the secretion of alpha-granule contents during aggregation. Upon platelet activation, intracytoplasmic Ca++ levels rise, triggering transglutaminase activity. Serotonin is conjugated to small GTPase proteins including RhoA and Rab4, which then become constitutively active (34). The interaction of the small GTPases with their downstream effectors results in cytoskeletal rearrangement and secretion of alpha-granule contents (34).
With platelet stimulation, cytoplasmic factor XIII cross links platelet cytoskeletal proteins, including filamin, vinculin, actin, and myosin as well as the heat shock protein hsp27 (24,35). The function of these intracytoplasmic cross-linked proteins in activated platelets is currently unknown. Platelet factor XIII, along with calpain, also regulates platelet membrane microvesiculation and modulates the conversion of platelets from a proadhesive to a procoagulant phenotype (36).
Platelet transglutaminases may mediate platelet adhesion to other cells or to the extracellular matrix. For example, factor XIIIa supports the adhesion of platelets to endothelial cells. Factor XIIIa binds to endothelial cells via the alphavbeta3 integrin and to platelets via GPIIb/IIIa (37-40). Conceivably, intrinsic platelet transglutaminase activity serves to counterbalance platelet-derived matrix metalloproteinase activity. MMPs have been shown to cleave GPVI (41,42), GPIbalpha (43), P-selectin (44,45). Cleavage of these receptors would have an anti-adhesive effect, which could be reversed by transglutaminase activity. On the other hand, intrinsic platelet MMP activity would likely oppose platelet transglutaminase effects. Using tumor cell models, it has been demonstrated that membrane-type MMPs degrade TG2, suppressing cell adhesion to and migration on fibronectin (46). MT1-MMP also activates MMP-2, which proteolyzes domain II of TG2, destroying its catalytic and adhesive activities (47).
The interactions of TG2 with integrins have broad implications in cardiovascular biology, extending beyond the scope of platelet function. Nearly all TG2 molecules on the cell surface have a direct 1:1 non-covalent interaction with integrins, primarily β1 and β3 integrin subunits (48,49). Cell surface TG2 serves as a link between integrins and fibronectin by acting as an integrin-associated co-receptor that promotes cell adhesion, spreading and migration. This adhesive function of cell surface TG is independent from its enzymatic activities (48,49). The TG2-integrin complex provides a binding site for fibronectin and facilitates cell adhesion and migration (49-51). Recently the importance of TG2 in regulating signals from the extracellular matrix has been reinforced by the discovery that it is a ligand for GPR56 (52). GPR56 is a member of a newly described family of G protein coupled receptors. In melanoma cells, GPR56 is markedly down-regulated in the metastatic variants (52). Furthermore, overexpresssion of GPR56 suppresses tumor growth and metastasis whereas inhibition of GPR56 enhances tumor progression (52). The implications of these findings for cardiovascular biology have not yet been explored.
5. EXPRESSION OF TRANSGLUTAMINASES IN VASCULAR CELLS AND CARDIOMYOCYTES
Factor XIII is expressed primarily by bone-marrow derived cells including platelets, monocytes and macrophages (53). In contrast, TG2 is widely expressed by cardiac and vascular cells including endothelial and smooth muscle cells which constitutively express TG2 at high levels (54). In the human fetus, TG2 is expressed in human cardiac myoblasts and vascular endothelial smooth muscle cells at 5−8 weeks post fertilization (55). The cardiac expression tended to decrease with further fetal development, whereas expression in vascular smooth muscle remained intense up to 14 weeks (55). Cardiomyocytes from adult hearts also express TG2, especially those derived from the subendocardial and subepicardial regions (54). Tissue transglutaminase expression in autopsied human hearts has been correlated with areas of “mucinous degeneration”, an intracytoplasmic basophilic lesion that occurs primarily in the elderly (56).
Chondrocytes, which under rare circumstances can exist within the arterial wall (57,58), express both Factor XIII A chains and tissue transglutaminase (59). The expression of transglutaminases in arterial cells including chondrocytes suggests that TG2 or FXIII could have roles in arterial calcification similar to those that have been demonstrated for transglutaminases in bone mineralization (60,61).
6. ROLE OF TRANSGLUTAMINASES IN INFLAMMATION
TG2 is expressed by a wide array of cells involved in immunity and inflammation including lymphocytes, neutrophils and monocytes (62). Although tissue transglutaminase and factor XIII are both expressed by monocytes and macrophages, TG2 is markedly upregulated while factor XIII is downregulated in the process of monocyte to macrophage maturation (63). The expression of Factor XIII in macrophages has been described as a marker of the alternative pathway of activation (i.e. via Th2 cytokines rather than Th1 cytokines) (64). Macrophages activated by the alternative pathway are involved in tissue repair and fibrosis, whereas those activated “classically” promote tissue degradation by the release of matrix metalloproteinases (64).
The factor XIII A subunit can be synthesized in the liver (65) and in the bone marrow (53). The liver cells that synthesize Factor XIII A chains include hepatocytes, Kupffer cells and connective tissue histocytes (66). However, the quantity of factor XIII A subunits contributed by the liver appears insignificant relative to that produced by cells derived from the bone marrow (27). The Factor XIII B subunit, which functions primarily as a carrier and stabilizing protein for the A chain, is synthesized in the liver (67).
Increased TG2 activity is a common feature of several inflammatory diseases. TG2 is involved in enhanced inflammation by participating in an inflammatory loop with the “master switch” for inflammation, NF-kappaB, reducing free I-KkappaBalpha, leading to the translocation of free NF-kappaB into the nucleus (68). Since the TG2 promoter contains a NF-kappaB binding motif (69), TG2 is induced by NF-kappaB, completing the cycle. In addition to NF-kappaB, several inflammatory cytokines including IFN-gamma (70) and TGF-beta1(71) are known to upregulate TG2. The upregulation of TG2 by TGF-beta1 leads to another positive feedback loop. TG2 activates TGF-beta by cross linking the latent TGF-beta binding protein to the extracellular matrix (72). TGF-beta expression down regulates inflammatory and autoimmune responses (73). TG2−/− mice fail to activate TGF-beta, have delayed clearance of apoptotic cells and have evidence of tissue inflammation and autoimmunity (74).
Factor XIIIa and TG2 effect post-translational modifications of phospholipase A2, an enzyme that releases arachadonic acid from cell membrane glycerophospholipids, leading to the synthesis of inflammatory eicosanoids (75). Cordella-Miele and colleagues demonstrated that transglutaminases can modify secretory isoforms of PLA2 in two distinct ways. First, TG2 and Factor XIIIa mediated intramolecular cross-links that promoted noncovalent dimerization of the enzyme (76). Second, transglutaminases incorporated polyamines into sPLA2 (77). Both modifications significantly augmented the activity of sPLA2. Peptide inhibitors of TG2 or peptides with dual inhibition of TG2 and PLA2 significantly inhibited the inflammation associated with allergic conjunctivitis induced by ragweed in a guinea pig model (78) and lung inflammation induced by lipopolysaccharide (79). Both factor XIII and tissue TG have roles in regulating macrophage adhesion, migration and phagocytosis (50,74,80). TG2 additionally promotes the transendothelial migration of CD8+ but not CD4+ T cells (62). By mechanisms that are currently unknown, TG2 mediates the expression of the gp91phos subunit of NADPH oxidase expression in neutrophils, a gene product that is essential for oxidative killing by generation of superoxide anions (81). TG2 also contributes to neutrophil migration into areas of inflammation. TG2 −/− mice have reduced neutrophil nitroblue tetrazolium reduction capacity, decreased superoxide anion generation and decreased gp91phos expression, but increased neutrophil phagocytic activity compared to wild type mice (81).
TG2 has been implicated in several autoimmune diseases (82). A variety of TG2 substrates have been identified as autoantigens in autoimmune diseases. Furthermore, antibodies to TG2 have been identified in celiac disease (83), dermatitis herpetiformis (84), type I diabetes (85) and lupus (86). The mechanism by which TG2 might contribute to autoimmunity is unclear. In the case of celiac disease, TG2 is thought to generate neoepitopes from gliadin through deamidation of some of the glutamine residues that constitute 40% of the amino acids residues of this molecule (82). TG2 also cross links itself to gliadin, a process that appears to foster the generation of anti-gliadin and anti-TG2 antibodies (82).
7. INTERACTIONS BETWEEN DIABETES AND TISSUE TRANSGLUTAMINASE
Tissue transglutaminase knockout mice were originally reported to have a normal phenotype (87,88). However, subsequent studies have demonstrated that TG2−/− mice have several subtle phenotypes including impaired glucose stimulated insulin secretion. TG2 has a role in the calcium-dependent, glucose-induced insulin release from beta-cells in the pancreas, as well as intracellular processing of the insulin receptor (89). However, the diabetic phenotype was partly ameliorated by increased peripheral sensitivity to insulin. This enhanced sensitivity was correlated with increased phosphorylation of the endogenous insulin receptor substrate, IRS-2 (89). The mild impairment of glucose metabolism in the TG2−/− mice is similar to that observed in type 2 diabetes and MODY (Maturity-Onset Diabetes of the Young). An analysis of TG2 in MODY patients revealed a missense mutation (N333S) in the active site of the enzyme. This same mutation was observed in a patient's father who was also diabetic (89).
8. HYPERTENSION AND TRANSGLUTAMINASES
Recently, it has been suggested that factor XIIIa can contribute to the development or to the exacerbation of elevated blood pressure (90). Immunoblotting of monocyte membranes from normotensive subjects revealed that the AT1 receptor migrated predominately as a monomer. In patients with hypertension, however, the receptor was a dimer that was stable to denaturing and reducing conditions (90). Patients with factor XIII deficiency failed to form AT1 receptor dimers (90). This dimeric form of the AT1 receptor is hyperactive, with increased coupling to Gq/11, generation of inositol phosphate, and mobilization of intracellular calcium. Monocytes from hypertensive patients had higher FXIIIa activity than those from normotensive subjects (90). Treatment of hypertensive subjects with ACE inhibitors for three months increased the abundance of the monomeric form of AT1 receptors, decreased factor Xllla levels (90), and reduced monocyte adhesion (90). In contrast to FXIIIa, TG2 was not effective in forming dimers of AT1 (90).
9. TRANSGLUTAMINASES AND ARTERIAL REMODELING
Although tissue transglutaminase does not cross-link the AT1 receptor, this enzyme can contribute to hypertension by other mechanisms. Tissue transglutaminase in VSMC can enhance vasoconstriction by stimulating RhoA/ROCK-2 kinase binding and ROCK-2 autophosphorylation (91). Persistent arteriolar vasoconstriction or reduced shear stress can lead to “inward remodeling” in which there is a sustained reduction in vascular diameter (92). This inward remodeling involves the rearrangement or structural reorganization of a constant amount of vascular tissue with a reduced lumen diameter (93). Tissue transglutaminase activity regulates the inward remodeling that occurs after blood flow reduction in the mesenteric arteries of the rat. Inhibitors of TG2 block inward remodeling that was induced by endothelin-1. Enhanced expression of endogenous TG2 or the application of exogenous TG2 increases inward remodeling in several animal models (94). In a porcine model of renal artery stenosis, simvastatin inhibited the inward remodeling of the intrarenal microvasculature, partly by decreasing the upregulation of TG2 expression in the ischemic kidney (95). In TG2 null mice, inward remodeling of mesenteric arteries is delayed in response to low flow states compared to wild type mice. However, factor XIII that is present in adventitial macrophages is able to provide a redundant source of transglutaminase that promotes inward remodeling, albeit at a slower rate than TG2 (96). Eliminating monocytes/macrophages reduces the contribution of factor XIII to inward remodeling in TG2 null mice (96).
Increased expression of TG2 is associated with increased vascular tone, while inhibition of TG2 activity causes vasodilation. Interestingly, the activity of TG2 appears to be regulated, in part, by the application of pressure to the arterial wall (94). Both TG2 and FXIII transglutaminase activities are also regulated by nitric oxide. Of the 18 free cysteine residues in TG2, 15 can be nitrosylated and denitrosylated in a calcium-dependent manner, resulting in enzymatic inhibition or activation, respectively (97). Factor XIIIa is similarly inhibited by NO (98). Thus, a healthy endothelium that generates a robust supply of NO, will inhibit arteriolar transglutaminase activity, thereby suppressing the propensity for vasoconstriction and “inward remodeling”.
10. ATHEROSCLEROSIS AND TRANSGLUTAMINASES
Since atherosclerosis is now recognized to be a disease in which inflammation plays a significant role (99,100), the modulation of inflammation by transglutaminases is surely an important factor in the contributions of factor XIII and TG2 to this disease process. However, there are several other potential roles for transglutaminase in atherosclerosis. Both factor XIIIa (101) and TG2 (102,103) are present in human atherosclerotic plaque. Furthermore, the transglutaminase-generated isodipeptide epsilon(gamma-glutamyl) lysine is elevated in fibro-lipid plaque obtained from diseased aortas of humans and hyperlipidemic rabbits (104). Transglutaminases may have a variety of roles in regulating both the chronic progression of atherosclerosis as well as its acute manifestations. In the atheroma, factor XIII is expressed principally by macrophages (105), while TG2 is also expressed by endothelial cells and smooth muscle cells. The most direct evidence for a role for Factor XIIIa in atherosclerosis comes from the experiments in which expression of a factor XIIIa inhibitor reduced monocyte entry into the artery wall and inhibited the development of atherosclerosis in hypercholesterolemic Apo E−/− mice (90).
The expression and activity of TG2 increases in the aortas of cholesterol-fed rabbits (106). This observation likely reflects increased macrophage content of the atherosclerotic aorta, although other cellular sources of TG are also possible. In contrast to Factor XIIIa, there is evidence that leukocyte TG2 expression limits the progression of atherosclerosis. When TG−/− or TG+/+ bone marrow was transplanted into lethally irradiated LDL receptor knockout mice, animals with TG−/− bone marrow had larger aortic valve lesions (107). TG2 expression in macrophages has been hypothesized to regulate atherosclerotic lesions size by: (1) controlling the phagocytosis of apoptotic cells (2) modulating TGF-beta activity and (3) altering ABCA1 expression, among other roles (107).
Extracellular TG2 expressing transglutaminase activity stimulates TGF-beta which induces matrix deposition and inhibits MMP production (108). This effect could increase plaque mass but also potentially reduce the likelihood of plaque rupture. On the other hand, upregulation of the intracellular G-protein function of TG2 has been reported to inhibit migration of human aortic smooth muscle cells (109), an effect that might reduce the thickness of the fibrous cap, predisposing to plaque rupture but also potentially retarding the growth of the atheroma. The stimulated expression of ABCA1 in response to alltrans retinoic acid in TG2−/− macrophages is reduced compared to controls (107). ABCA1 mediates reverse cholesterol transport, thereby providing and antiatherogenic effect (1). ABCA1 also has roles in controlling inflammation (111) and in regulating apoptosis (112).
Factor XIII, bound to fibrinogen, can diffuse across the endothelial cell barrier into the subintimal space (113), where it functions as an extracellular enzyme. Even the cellular sources of FXIII (platelets and monocytes) can contribute to extracellular FXIII. Factor XIII A subunits are expressed on the cell surface of macrophages where they can be activated to cross link ECM proteins (114). For example, Factor XIIIa cross-links Lp(a) to fibrinogen (101). Lp(a) that is cross-linked in the atheroma could exert a local anti-fibrinolytic and pro-atherogenic effect (101).
TG2 can be secreted and is detected in the extracellular space of a variety of tissues including the coronary artery (103). TG2 is expressed in the fibrous caps and near “shoulder regions” (102) of plaques where it possibly contributes to plaque stability. Interestingly, the HMG-CoA-reductase inhibitor atorvastatin induced the expression of TG2 in endothelial cells (115). This effect could contribute to the clinical benefit observed with “statins”, by promoting stabilization of the atherosclerotic plaque.
An example of an extracellular TG2 substrate in atherosclerotic plaque is elafin, an inhibitor of elastase that has a prosegment rich in glutamyl and lysyl residues (116). TG2 and elafin expression largely overlapped and increased with more severe CAD, but was diminished at the lipid core (103). Elafin and its precursor trappin-2 are inhibitors of the neutrophil serine proteases leukocyte elastase and proteinase 3. Both trappin-2 and elafin are crosslinked by TG to ECM proteins where they retain their SERPIN activity (117).
11. ASSOCIATION BETWEEN FACTOR XIII LEVELS, CARDIOVASCULAR RISK FACTORS AND ATHEROSCLEROSIS
A gene profile study of peripheral blood cells from patients with coronary artery disease demonstrated a 2.1-fold increase in factor XIII A subunit expression compared with healthy controls (118). Consistent with this observation, a case control study of patients with established CAD showed higher factor XIII A subunit levels compared with control, but there was no association between FXIII A subunit and MI (119). Similarly, elevated levels of factor Xlll have been reported in patients with obliterative atherosclerosis (120). Factor XIIIa activity has been positively correlated with carotid IMT (16).
In contrast to some studies correlating higher factor XIII levels with vascular events, a nested case-control study from the Second Northwick Park Heart Study (NPHSII) found the inverse relationship. In this study, patients who were free of CAD but later suffered an MI had lower FXIII A-subunit antigen levels compared with controls (121). Furthermore, lower factor XIII A subunit levels predicted larger stroke and increased stroke mortality in patients with acute stroke (121). Low factor XIII A subunit antigen concentrations were felt to be reflective of higher in vivo thrombin generation. Prothrombin fragment F1+2 levels showed a significant inverse correlation with A subunit levels (121). Incubation of plasma with thrombin reduced Factor XIII A but not B subunit levels (121).
Elevated factor XIII levels have been reported in patients with risk factors for atherosclerosis, including diabetes, hyperlipidemia and the metabolic syndrome. Elevated levels of factor XIII have been reported in patients with type 2 diabetes mellitus (122) and the crosslinking of plasmin inhibitor to fibrin from subjects with Type 2 diabetes is enhanced compared with controls (123). Furthermore, the extent of cross-linking of plasmin inhibitor of fibrin correlated with HbA1c and fasting glucose levels (123). Interestingly, dimethylbiguanide (metformin) interferes with factor XIII activation and with fibrin polymerization (122). In patients treated with metformin over a 12 week period, factor XIII antigen and activity levels were reduced (124).
Hyperlipidemic subjects have elevated factor XIII activity (125,126). Patients with elevated triglyceride levels, in particular, were likely to have elevated FXIIIa activity, possibly due to increased hepatic synthesis (126). Several studies have found correlations between insulin resistance and factor XIII A (122) or Factor XIII B (127) subunits. In addition to insulin resistance, Factor XIII B subunit levels have been positively correlated with other components of the metabolic syndrome, including waist:hip ratio, fasting triglycerides, and PAI-1 antigen (128).
Factor XIII levels are partly determined by heritable factors (h2 = 0.48 p<0.0001) (129). The structure of the fibrin clot (pore and fiber size) is influenced by fibrinogen levels and factor XIII subunit levels, and these levels are, in turn, under genetic as well as environmental control (130). The male relatives of patients with premature CAD had earlier onset of clot formation with smaller pores, lower fiber mass-length ratios, and contained thicker fibers compared to unrelated healthy controls (131).
12. TRANSGLUTAMINASES AND SIGNAL TRANSDUCTION
Tissue transglutaminase is identical to a high molecular weight G protein called Ghalpha (132). In this capacity, TG2 is involved as a signal transducing protein, mediating the activation of the delta1 isoform phospholipase C by the alpha1B- and alpha1D-adrenergic receptors (132,133) thereby increasing inositol phosphate turnover in TG2-transfected cells. However, although mice overexpressing TG2 in the myocardium develop a phenotype of LVH, there was no evidence in this model for a direct link between TG2 and inositol phosphate hydrolysis (134). TG2 not only enhances signaling of subtypes of alpha1-AR, but also thromboxane A2 receptor α subtype135 and the oxytocin receptor (136,137). TG2 also associates with the cytoplasmic tails of several integrins including alpha5, alphav, and alphaIIb (138).
Activating the RhoA/ROCK pathway is another cell signaling activity of tissue transglutaminase. RhoA, a small GTPase, is a member of the Ras superfamily and has roles in cell adhesion, migration, cytoskeletal organization as well as a variety of other cellular functions (139). TG2 can activate RhoA by two mechanisms. In the first, TG2 irreversibly activates RhoA by transamidating amino acid residue Gln63 in RhoA (140). An alternate mechanism, which is independent of the enzymatic activity of TG2 has been described in which cell surface TG2 enhances integrin-dependent signaling to RhoA and its downstream effector ROCK by inducing integrin clustering (91).
The intracellular function of TG2 is tightly regulated by intracellular calcium and GTP concentrations which have opposing influences on TG2 activities. When TG2 binds to GTP, its transglutaminase function is inhibited, but this inhibition is abrogated by calcium, which activates the transglutaminase enzymatic activity of TG2. The activation of TG2 during the increase in intracellular calcium and the reduced GTP generation that occurs with apoptosis leads to cross-linking of cellular proteins stabilizing the apoptotic body, thereby potentially limiting the release of damaging intracellular contents to neighboring cells and tissues (141). The sensitivity of TG2 activity to inhibition by GTP is increased by nitrosylation of cysteine residues (97) whereas the calcium-dependent transglutaminase activity of TG2 is increased by sphingosylphosphocholine, which is increased in cells undergoing apoptosis (142). The calcium binding protein calreticulin inhibits both the GTP binding and transglutaminase activity of TG2 (143,144). Thus, there appears to be tight regulation of TG2 activity, including a finely-tuned switch between its G protein and transglutaminase functions.
13. ROLE OF TRANSGLUTAMINASES IN VASCULAR PERMEABILITY
The loss of endothelial barrier function may contribute to the development of early atherosclerotic lesions (100). Factor XIII has a role in endothelial barrier function (145), reducing endothelial permeability, apparently by modifying paracellular transport in endothelial cell monolayers. In an animal model, factor XIII blocked enhanced permeability induced by antiendothelial cell antiserum (146).
Factor XIII administration may prevent capillary leak syndrome in certain clinical scenarios. Acquired factor XIII deficiency is common following cardiopulmonary bypass (147-150) and children may be particularly susceptible to the deficiency. In children undergoing open heart surgery for congenital cardiac defects, those with low factor XIII levels have a higher risk for developing myocardial edema requiring delayed sternal closure (151). In a small randomized study, administering factor XIII reduced myocardial edema formation (151). Similarly, increased pleural effusions correlate with lower factor XIII levels in children undergoing open heart surgery. Administration of recombinant factor XIII reduced pleural fluid accumulation (152).
In adult patients undergoing CABG, factor XIII levels fell to a nadir value of approximately 55% at 30 minutes after the onset of extracorporeal circulation (153). Patients were randomized in a double blinded study to placebo, 1250 units or 2500 units of human coagulation factor XIII. The group that received 2500 units achieved levels of factor XIII equivalent to pre-operative values. Patients treated with placebo had the most bleeding and those treated with 2500 units had the least with the 1250 unit group being intermediate, but these differences were not statistically significant. Patients who had post-administration levels of factor XIII <70% had increased chest tube drainage and blood product transfusion during the first 48 hours (153). The authors concluded that FXIII administration would be justified if levels after extracorporeal circulation are <70% (153).
TG2 has a less well-defined role in promoting endothelial barrier function. TGI, but not TG2, is translocated to the intercellular junctions as the monolayer matures (154). Specific silencing of TGI with RNAi inhibited the maturation-dependent changes in the barrier properties of endothelial monolayers. Of interest is the finding that some types of endothelial cells (e.g., mouse myocardial) express ample TG I whereas others (mouse lung microvascular) do not (154).
14. TRANSGLUTAMINASE ACTIVITY AND ANGIOGENESIS
Human transglutaminase (TG2) enhances angiogenesis and wound healing in a rat dorsal skin flap chamber155. However, there has also been a report of an inhibitory effect of TG2 on angiogenesis (156). In a mouse CT26 tumor model, the injection of TG2 reduced tumor growth and induced tumor regression in some cases (156). This effect appeared to be due to a fibrotic response with increased collagen accumulation that represented a physical barrier to tumor invasion as well as to a destabilization of tumor angiogenesis (156).
Factor XIIIa can support endothelial cell adhesion and modulate EC tube formation (38). Factor XIIIa binds to alphavbeta3 on EC, enhancing the noncovalent association between alphavbeta3 and VEGFR-2 (157). Factor XIIIa also cross-links the two receptors (157). Platelets that are activated by thrombin release thymosin beta4 which is cross-linked by factor XIIIa to collagen and fibrin (158). Thymosin beta4 has roles in promoting angiogenesis by supporting endothelial cell attachment, spreading and migration (158). Furthermore, FXIIIa downregulates thrombospondin-1, an inhibitor of angiogenesis (159). As predicted from these biological effects, factor XIIIa has been shown to have a direct proangiogenic effect in vitro (reviewed in reference 160) and in vivo (160-162).
In an angiogenesis model in which matrigel containing bFGF was injected subcutaneously, FXIII null mice had reduced angiogenic response compared to wild type mice. This deficit was restored to near normal levels with the addition of FXIIIa to the matrigel (163). In a neonatal cardiac heterotropic allograft model, wild-type mice injected with FXIIIa had increased vessel formation around the implanted heart allografts and improved contractile performance compared with controls (163).
15. LVH AND HEART FAILURE
In addition to regulating the microvascular blood supply to the heart, the transglutaminases could also influence myocardial hypertrophy, extracellular matrix abundance and composition, responses to ischemia/reperfusion and infarct healing. TG2 could also regulate contractile performance by its function as an adrenergic receptor-coupled G protein. There is evidence for alterations in TG2 expression and function in animals and patients with heart failure. Tissue transglutaminase expression is upregulated with cardiac hypertrophy and heart failure in animal models (164). In contrast, activity (GTP binding and GTPase) of TG2 was found to be downregulated in patients with ischemic and dilated cardiomyopathies (165).
Mice that overexpress TG2 in a cardiac-specific manner have cardiac hypertrophy, interstitial fibrosis, increased apoptosis, depressed LV systolic and diastolic function, elevated heart rate, but preserved responsiveness to beta adrenergic receptor stimulation (134). Cardiomyocytes from TG2 transgenic mice had evidence of increased lipid peroxidation and cyclo-oxygenase 2 activation (166). The heart failure of TG2 overexpressing mice was partly dependent on COX-2 derived thromboxane A2 and the phenotype could be rescued by treatment with COX-2 inhibitors (166).
Interestingly, patients who develop autoantibodies to tissue transglutaminase may develop cardiac disease. The presence of IgA autoantibodies to TG2 is nearly pathognomonic for celiac disease (83). Celiac Disease has been described in 4.4% of patients with myocarditis, a prevalence that is 14-fold higher than in control subjects (167). Similarly, CD occurs in up to 5.7% of patients with idiopathic dilated cardiomyopathy (168,169). A common auto-antigen in celiac disease and its associated autoimmune cardiomyopathy is TG2 (167,170). These patients may respond to a gluten-free diet alone or with immunosuppression (171).
Autoantibodies to tissue TG may occur in patients with heart failure even in the absence of symptoms of celiac disease. In a study of 288 patients with end stage heart failure awaiting cardiac transplantation, nearly half were found to have positive IgA and IgG antibodies directed at recombinant human TG2 (172). These patients had no clinical evidence of celiac disease. The authors speculated that high TG2 levels in heart failure, playing a role in fibrogenesis and apoptosis in this disorder, might secondarily induce antibodies to TG2. In a general population survey in southern Germany, 63/4633 (1.4%) subjects had elevated IgA antibody titers to TG2. All cause mortality was significantly elevated in these subjects, with heart failure and cancer being the most common causes of death (173).
16. EFFECT OF TG2 ON CARDIAC ISCHEMIA/REPERFUSION INJURY
Reperfusion of an ischemic tissue may result in paradoxical cellular damage with myocardial stunning, microvascular injury and apoptosis or necrosis. The proteolysis of thin filament proteins, especially troponin I has been shown to be a key event in cardiac stunning. McDonough et al (174). have shown that moderate ischemia/reperfusion injury in rats is accompanied by proteolysis of 17 residues from the C-terminus of cardiac troponin I. The resultant fragment (cTnI193) forms covalent complexes with N-terminal residues in cTnC and with C-terminal residues of TnT (174). Gorza et al. also reported covalent complexes containing cTnT in ischemia/reperfusion models (174). These covalent complexes appear to be mediated by TG2-mediated isopeptide bond formation (174,176). Although the physiological significance of these interactions is not understood, it is possible that the cross-linking of cTnc and cTnT alters Ca++ dynamics within the sarcomere, which could, in turn, alter myocardial contractile function.
Prolonged ischemia can result in cardiomyocyte apoptosis or infarction. The cross-linking of TnT by TG2 occurs with apoptosis (177). Infarct size and the occurrence of ventricular fibrillation is significantly increased in TG2−/− ischemia/reperfused isolated hearts as compared to ischemia/reperfused wild-type hearts (178). The increased sensitivity of TG2 null mice to I/R injury is not due to impaired adrenergic receptor signaling. Instead, the increased susceptibility to injury appeared to be due to lower baseline stores of high energy phosphates in the TG2 null mice. TG2−/− mice have a mitochondrial functional defect with impaired ATPase function (178). These studies provide evidence for a novel role of TG2 in maintenance of mitochondrial respiratory function. It has been previously shown that TG2 colocalizes with mitochondria and its overexpression leads to mitochondrial hyper-polarization (179).
17. EFFECTS OF FACTOR XIII ON MYOCARDIAL HEALING AFTER MI
Factor XIII deficient mice have a normal cardiac phenotype, but exhibit impaired healing and fatal rupture of the LV after myocardial infarction (180). Mice with even relatively mild deficiency of factor XIII (70% activity in FXIII+/− mice) displayed this phenotype. Cardiac rupture of factor XIII deficient mice (FXIII−/− and FXIII −/+) occurred between 3 and 5 days after MI, consistent with clinical observations of this complication (180). Cardiac ruptures were likely due to an imbalance in ECM turnover, since MMP-9 expression was 7-fold higher and collagen-1 production was 53% lower in FXIII−/− mice (180). Replenishing Factor XIII levels in FXIII−/− mice reduced cardiac rupture and death rates to those observed in wild type mice. FXIII administration also reduced MMP-9 expression but failed to improve collagen-1 production. The authors speculated that the lack of increased collagen syntheses with intravenous supplemental FXIII could be due to failure of this therapy to replenish intracellular factor XIII in platelets and monocytes/macrophages (180).
The clinical importance of these findings require further investigation. However, the finding of a 25% decrease in FXIII levels during the first week after an MI or stroke raises the possibility that some patients may be susceptible to myocardial rupture due to low factor XIII levels (181).
18. DIAGNOSTIC AND THERAPEUTIC IMPLICATIONS OF TRANSGLUTAMINASES
The D-dimer antigen assay has been widely used as a screen for venous thromboembolic events (182). The finding of a negative D-dimer with low clinical effectively excludes the diagnosis of deep vein thrombosis (182). The D-dimer is generated by factor XIII-mediated ligation of aligned fibrin gamma chains, with cross-linking of glutamine residues 398 or 399 of one gamma-chain to lysine residue 406 of another gamma-chain (183). Factor XIIIa generates soluble cross-linked complexes that contain the D-dimer antigen before a fibrin gel is formed (184). The D-dimer antigen is released from soluble and insoluble fibrin by plasmin proteolysis (185).
Low molecular weight substrates of factor XIIIa (186) and TG2 (187) have been developed to localize transglutaminase activity within blood clots, atherosclerotic plaques or other tissues. The substrates used for these studies consist of a peptide derived from alpha2 antiplasmin coupled to either gadolinium for magnetic resonance imaging or to a near-infrared fluorescence probe.
There are several potential clinical applications of the transglutaminases. Infusions of factor XIII to reduce bleeding during cardiopulmonary bypass surgery has been discussed. Transglutaminase-modified matrices could be used to improved physical and biological properties or to incorporate specific agents into engineered tissues (188-190).
19. PERSPECTIVE
Despite the panoply of interactions already described for FXIII and TG2 in this arena, there are likely many new discoveries yet to be made. It is probable that other members of the transglutaminase family also have relevant functions for cardiovascular disease. Indeed, we have already discussed the role of keratinocyte TG (TG1), expressed in endothelial cells, as an important contributor to endothelial barrier function. We have considered the cytoplasmic, cell-surface and matrix function of transglutaminases, but little is currently known about the roles for FXIII and TG2 in the nucleus. It is known that TG2 modifies participants in transcriptional regulation including histones (191), retinoblastoma gene product (192), and Sp1 (193). However the implication of these activities for heart and vascular health are currently unknown. Although we have considered the actions of TG2 in cross-linking, acting as a G-protein and as an integrin co-receptor, there are other functions of this enzyme that are only beginning to be understood. The roles of TG2-mediated polyamination or deamidation of glutamine residues and acylation of lysine residues are not fully known. Furthermore, TG2 also acts as a protein disulfide isomerase (194) and as a protein kinase (195-197) for certain target proteins. It is clear that future studies of transglutaminases represent an exciting frontier that may advance the diagnosis and treatment of cardiovascular diseases.
20. REFERENCES
- 1.Greenberg CS, Birckbichler PJ, Rice RH. Transglutaminases: multifunctional cross-linking enzymes that stabilize tissues. FASEB J. 1991;5:3071–3077. doi: 10.1096/fasebj.5.15.1683845. [DOI] [PubMed] [Google Scholar]
- 2.Board PG, Losowsky MS, Miloszewski KJ. Factor XIII: inherited and acquired deficiency. Blood Rev. 1993;7:229–242. doi: 10.1016/0268-960x(93)90010-2. [DOI] [PubMed] [Google Scholar]
- 3.Bhattacharya M, Biswas A, Ahmed RP, Kannan M, Gupta M, Mahapatra M, Choudhry VP, Saxena R. Clinicohematologic profile of factor XIII-deficient patients. Clin Appl Thromb Hemost. 2005;11:475–480. doi: 10.1177/107602960501100416. [DOI] [PubMed] [Google Scholar]
- 4.Lauer P, Metzner HJ, Zettlmeissl G, Li M, Smith AG, Lathe R, Dickneite G. Targeted inactivation of the mouse locus encoding coagulation factor XIII-A: hemostatic abnormalities in mutant mice and characterization of the coagulation deficit. Thromb Haemost. 2002;88:967–974. [PubMed] [Google Scholar]
- 5.Koseki-Kuno S, Yamakawa M, Dickneite G, Ichinose A. Factor XIII A subunit-deficient mice developed severe uterine bleeding events and subsequent spontaneous miscarriages. Blood. 2003;102:4410–4412. doi: 10.1182/blood-2003-05-1467. [DOI] [PubMed] [Google Scholar]
- 6.Reed GL, Houng AK. The contribution of activated factor XIII to fibrinolytic resistance in experimental pulmonary embolism. Circulation. 1999;99:299–304. doi: 10.1161/01.cir.99.2.299. [DOI] [PubMed] [Google Scholar]
- 7.Shebuski RJ, Sitko GR, Claremon DA, Baldwin JJ, Remy DC, Stern AM. Inhibition of factor XIIIa in a canine model of coronary thrombosis: effect on reperfusion and acute reocclusion after recombinant tissue-type plasminogen activator. Blood. 1990;75:1455–1459. [PubMed] [Google Scholar]
- 8.Leidy EM, Stern AM, Friedman PA, Bush LR. Enhanced thrombolysis by a factor XIIIa inhibitor in a rabbit model of femoral artery thrombosis. Thromb Res. 1990;59:15–26. doi: 10.1016/0049-3848(90)90267-g. [DOI] [PubMed] [Google Scholar]
- 9.Robinson BR, Houng AK, Reed GL. Catalytic life of activated factor XIII in thrombi. Implications for fibrinolytic resistance and thrombus aging. Circulation. 2000;102:1151–1157. doi: 10.1161/01.cir.102.10.1151. [DOI] [PubMed] [Google Scholar]
- 10.Siebenlist KR, Mosesson MW. Progressive cross-linking of fibrin γ chains increases resistance to fibrinolysis. J Biol Chem. 1994;28:28414–28419. [PubMed] [Google Scholar]
- 11.Gaffney PJ, Brasher M, Lord K, Strachan CJ, Wilkinson AR, Kakkar VV, Scully MF. Fibrin subunits in venous and arterial thromboemobolism. Cardiovasc Res. 1976;10:421–426. doi: 10.1093/cvr/10.4.421. [DOI] [PubMed] [Google Scholar]
- 12.Sakata Y, Aoiki N. Crosslinking α2-plasmin inhibitor of fibrin by fibrin-stabilizing factor. J Clin Invest. 1980;65:290–297. doi: 10.1172/JCI109671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valnickova Z, Enghild JJ. Human procarboxypeptidase U, or thrombin-activable fibrinolysis inhibitor, is a substrate for transglutaminases. J Biol Chem. 1998;273:27220–27224. doi: 10.1074/jbc.273.42.27220. [DOI] [PubMed] [Google Scholar]
- 14.Matsuka YV, Migliorini MM, Ingham KC. Cross-linking of fibronectin to C-terminal fragments of the fibrinogen alpha-chain by factor XIIIa. J Protein Chem. 1997;16:739–745. doi: 10.1023/a:1026307731751. [DOI] [PubMed] [Google Scholar]
- 15.Cho J, Mosher DF. Enhancement of thrombogenesis by plasma fibronectin crosslinked to fibrin and assembled in platelet thrombi. Blood. 2006;107:3555–3563. doi: 10.1182/blood-2005-10-4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ames PR, Iannaccone L, Alves JD, Margarita A, Lopez LR, Brancaccio V. Factor XIII in primary antiphospholipid syndrome. J Rheumatol. 2005;32:1058–1062. [PubMed] [Google Scholar]
- 17.Wilhelmsen L, Svardsudd K, Korsan-Bengtsen K, Larsson B, Welin L, Tibblin G. Fibrinogen as a risk factor for stroke and myocardial infarction. N Engl J Med. 1984;311:501–505. doi: 10.1056/NEJM198408233110804. [DOI] [PubMed] [Google Scholar]
- 18.Siebenlist KR, Meh DA, Mosesson MW. Plasma factor XIII binds specifically to fibrinogen molecules containing γ' chains. Biochemistry. 1996;35:10448–10453. doi: 10.1021/bi9606206. [DOI] [PubMed] [Google Scholar]
- 19.Lovely RS, Falls LA, Al-Mondhiry HA, Chambers CE, Sexton GJ, Ni H, Farrell DH. Association of the γA/γ' fibrinogen levels and coronary artery disease. Thomb Haemost. 2002;88:26–31. [PubMed] [Google Scholar]
- 20.Mosesson MW. Fibrinogen and fibrin structure and functions. J Thromb Haemost. 2005;3:1894–1904. doi: 10.1111/j.1538-7836.2005.01365.x. [DOI] [PubMed] [Google Scholar]
- 21.Naito M, Nomura H, Iguchi A, Thompson WD, Smith EB. Effect of crosslinking by factor XIIIa on the migration of vascular smooth muscle cells into fibrin gels. Thromb Res. 1998;90:111–116. doi: 10.1016/s0049-3848(98)00027-9. [DOI] [PubMed] [Google Scholar]
- 22.Inbal A, Lubetsky A, Krapp T, Castel D, Shaish A, Dickneite G, Modis L, Muszbek L, Inbal A. Impaired wound healing in factor XIII deficient mice. Thromb Haemost. 2005;94:432–437. doi: 10.1160/TH05-04-0291. [DOI] [PubMed] [Google Scholar]
- 23.Zaman AG, Helft G, Worthley SG, Badimon JJ. The role of plaque rupture and thrombosis in coronary artery disease. Atherosclerosis. 2000;149:251–266. doi: 10.1016/s0021-9150(99)00479-7. [DOI] [PubMed] [Google Scholar]
- 24.Muszbek L, Adany R, Mikkola H. Novel aspects of blood coagulation factor XIII.I. structure, distribution, activation, and function. Crit Rev Clin Lab Sci. 1996;33:357–421. doi: 10.3109/10408369609084691. [DOI] [PubMed] [Google Scholar]
- 25.Puszkin EG, Raghuraman V. Catalytic properties of a calmodulin-regulated transglutaminase from human platelet and chicken gizzard. J Biol Chem. 1985;260:16012–16020. [PubMed] [Google Scholar]
- 26.Balduini A, Apolito MD', Arcelli D, Conti V, Pecci A, Pietra D, Danova M, Benvenuto F, Perotti C, Zelante L, Volinia S, Balduini CL, Sovoias A. Cord blood in vitro expanded CD41+ cells: identification of novel components of megakaryocytopoiesis. J Thromb Haemost. 2006;4:848–860. doi: 10.1111/j.1538-7836.2006.01802.x. [DOI] [PubMed] [Google Scholar]
- 27.Adany R, Bardos H. Factor XIII subunit A as an intracellular transglutaminase. Cell Mol Life Sci. 2003;60:1049–1060. doi: 10.1007/s00018-003-2178-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muszbek L, Polgar J, Boda Z. Platelet factor XIII becomes active without the release of activation peptide during platelet activation. Thromb Haemost. 1993;69:282–285. [PubMed] [Google Scholar]
- 29.Kreager JA, Devine DV, Greenberg CS. Cytofluorometric identification of plasmin-sensitive factor XIIIa binding to platelets. Thromb Haemost. 1988;60:88–93. [PubMed] [Google Scholar]
- 30.Dale GL, Friese P, Batar P, Hamilton SF, Reed GL, Jackson KW, Clemetson KL, Alberio L. Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell surface. Nature. 2002;415:175–179. doi: 10.1038/415175a. [DOI] [PubMed] [Google Scholar]
- 31.Dale GL. Coated-platelets: an emerging component of the procoagulant response. J Thromb Haemost. 2005;3:2185–2192. doi: 10.1111/j.1538-7836.2005.01274.x. [DOI] [PubMed] [Google Scholar]
- 32.Szasz R, Dale GL. Thrombospondin and fibrinogen bind serotonin-derivatized proteins on COAT-platelets. Blood. 2002;100:2827–2831. doi: 10.1182/blood-2002-02-0354. [DOI] [PubMed] [Google Scholar]
- 33.Jobe SM, Leo L, Eastvold JS, Dickneite G, Ratliff TL, Lentz SR, Di Paola J. Role of FcRgamma and factor XIIIA in coated platelet formation. Blood. 2005;106:4146–4151. doi: 10.1182/blood-2005-03-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walther DJ, Peter JU, Winter S, Holtje M, Paulmann N, Grohmann M, Vowinckel J, Alamo-Bethencourt V, Wilhelm CS, Ahnert-Hilger G, Bader M. Serotonylation of small GTPases is signal transduction pathway that triggers platelet alpha-granule release. Cell. 2003;115:851–862. doi: 10.1016/s0092-8674(03)01014-6. [DOI] [PubMed] [Google Scholar]
- 35.Serrano K, Devine DV. Intracellular factor XIII crosslinks platelet cytoskeletal elements upon platelet activation. Thromb Haemost. 2002;88:315–320. [PubMed] [Google Scholar]
- 36.Kulkarni S, Jackson SP. Platelet factor XIII and calpain negatively regulate integrin αIIbβ3 adhesive function and thrombus growth. J Biol Chem. 2004;279:30697–30706. doi: 10.1074/jbc.M403559200. [DOI] [PubMed] [Google Scholar]
- 37.Dardik R, Shenkman B, Tamarin I, Eskaraev R, Harsfalvi J, Varon D, Inbal A. Factor XIII mediates adhesion of platelets to endothelial cells through alpha(v)beta(3) and glycoprotein IIb/IIIa integrins. Thromb Res. 2002;105:317–323. doi: 10.1016/s0049-3848(02)00014-2. [DOI] [PubMed] [Google Scholar]
- 38.Dallabrida SM, Falls LA, Farrell DH. Factor XIIIa supports microvascular endothelial cell adhesion and inhibits capillary tube formation in fibrin. Blood. 2000;95:2586–2892. [PubMed] [Google Scholar]
- 39.Cox AD, Devine DV. Factor XIIIa binding to activated platelets is mediated through activation of glycoprotein IIb-IIIa. Blood. 1994;83:1006–1016. [PubMed] [Google Scholar]
- 40.Devine DV, Bishop PD. Platelet-associated factor XIII in platelet activation, adhesion, and clot stabilization. Semin Thromb Hemost. 1996;22:409–413. doi: 10.1055/s-2007-999039. [DOI] [PubMed] [Google Scholar]
- 41.Stephens G, Yan Y, Jandrot-Perrus M, Villeval JL, Clemetson KJ, Phillips DR. Platelet activation induces metalloproteinase-dependent GP VI cleavage to down-regulate platelet reactivity to collagen. Blood. 2005;105:186–191. doi: 10.1182/blood-2004-07-2842. [DOI] [PubMed] [Google Scholar]
- 42.Massberg S, Konrad I, Bultmann A, Schulz C, Munch G, Peluso M, Lorenz M, Schneider S, Besta F, Muller I, Hu B, Langer H, Kremmer E, Rudelius M, Heinzmann U, Ungerer M, Gawaz M. Soluble glycoprotein VI dimer inhibits platelet adhesion and aggregation to the injured vessel wall in vivo. FASEB J. 2004;18:397–399. doi: 10.1096/fj.03-0464fje. [DOI] [PubMed] [Google Scholar]
- 43.Bergmeier W, Burger PC, Piffath CL, Hoffmeister KM, Hartwig JH, Nieswandt B, Wagner DD. Metalloproteinase inhibitors improve the recovery and hemostatic function of in vitro-aged or -injured mouse platelets. Blood. 2003;102:4229–4235. doi: 10.1182/blood-2003-04-1305. [DOI] [PubMed] [Google Scholar]
- 44.Andre P, Hartwell D, Hrachovinova I, Saffaripour S, Wagner DD. Pro-coagulant state resulting from high levels of soluble P-selectin in blood. Proc Natl Acad Sci U S A. 2000;97:13835–13840. doi: 10.1073/pnas.250475997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berger G, Hartwell DW, Wagner DD. P-Selectin and platelet clearance. Blood. 1998;92:4446–4452. [PubMed] [Google Scholar]
- 46.Belkin AM, Akimov SS, Zaritskaya LS, Ratnikov BI, Deryugina EI, Strongin AY. Matrix-dependent proteolysis of surface transglutaminase by membrane-type metalloproteinase regulates cancer cell adhesion and locomotion. J Biol Chem. 2001;276:18415–18422. doi: 10.1074/jbc.M010135200. [DOI] [PubMed] [Google Scholar]
- 47.Belkin AM, Zemskov EA, Hang J, Akimov SS, Sikora S, Strongin AY. Cell-surface-associated tissue transglutaminase is a target of MMP-2 proteolysis. Biochemistry. 2004;43:11760–11769. doi: 10.1021/bi049266z. [DOI] [PubMed] [Google Scholar]
- 48.Akimov SS, Krylov D, Fleischman LF, Belkin AM. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J Cell Biol. 2000;148:825–838. doi: 10.1083/jcb.148.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Akimov SS, Belkin AM. Cell-surface transglutaminase promotes fibronectin assembly via interaction with the gelatin-binding domain of fibronectin: a role in TGFbeta-dependent matrix deposition. J Cell Sci. 2001;114:2989–3000. doi: 10.1242/jcs.114.16.2989. [DOI] [PubMed] [Google Scholar]
- 50.Akimov SS, AM Belkin AM. Cell surface tissue transglutaminase is involved in adhesion and migration of monocytic cells on fibronectin. Blood. 2001;98:1567–1576. doi: 10.1182/blood.v98.5.1567. [DOI] [PubMed] [Google Scholar]
- 51.Balklava Z, Verderio E, Collighan R, Gross S, Adams J, Griffin M. Analysis of tissue transglutaminase function in the migration of Swiss 3T3 fibroblasts: the active-state conformation of the enzyme does not affect cell motility but is important for its secretion. J Biol Chem. 2002;277:16567–16575. doi: 10.1074/jbc.M109836200. [DOI] [PubMed] [Google Scholar]
- 52.Xu L, Begum S, Hearn JD. GPR56, an atypical G protein-coupled receptor, binds tissue transglutaminase, TG2, and inhibits melanoma tumor growth and metastasis. Proc Natl Acad Sci USA. 2006;103:9023–9028. doi: 10.1073/pnas.0602681103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wolpl A, Lattke H, Board PG, Arnold R, Schmeiser T, Kubanek B, Robin-Winn M, Pichelmayr R, Goldmann SF. Coagulation factor XIII A and B subunits in bone marrow and liver transplantation. Transplantation. 1987;43:151–153. doi: 10.1097/00007890-198701000-00032. [DOI] [PubMed] [Google Scholar]
- 54.Thomazy V, Fesus L. Differential expression of tissue transglutaminase in human cells. An immunohistochemical study. Cell Tissue Res. 1989;255:215–224. doi: 10.1007/BF00229084. [DOI] [PubMed] [Google Scholar]
- 55.Lee SK, Chi JG, Park SC, Chung SI. Transient expression of transglutaminase C during prenatal development of human muscles. J Histochem Cytochem. 2000;48:1565–1574. doi: 10.1177/002215540004801113. [DOI] [PubMed] [Google Scholar]
- 56.Kashima K, Yokoyama S, Daa T, Nakayama I, Iwaki T. Immunohistochemical study on tissue transglutaminase and copper-zinc superoxide dismutase in human myocardium: its relevance to apoptosis detected by the nick end labelling method. Virchows Arch. 1997;430:333–338. doi: 10.1007/BF01092757. [DOI] [PubMed] [Google Scholar]
- 57.El-Maadawy S, Kaartinen MT, Schinke T, Murshed M, Karsenty G, McKee MD. Cartilage formation and calcification in arteries of mice lacking matrix Gla protein. Connect Tissue Res. 2003;44(suppl 1):272–278. [PubMed] [Google Scholar]
- 58.Tyson KL, Reynolds JL, McNair R, Zhang Q, Weissberg PL, Shanahan CM. Oste/chondrocytic transcription factors and their target genes exhibit distinct patterns of expression in human arterial calcification. Arterioscler Thromb Vasc Biol. 2003;23:489–494. doi: 10.1161/01.ATV.0000059406.92165.31. [DOI] [PubMed] [Google Scholar]
- 59.Rosenthal AK, Mosesson MW, Gohr CM, Masuda I, Heinkel D, Seibenlist KR. Regulation of transglutaminase activity in articular chondrocytes through thrombin receptor-mediated factor XIII synthesis. Thromb Haemost. 2004;91:558–568. doi: 10.1160/TH03-07-0462. [DOI] [PubMed] [Google Scholar]
- 60.Johnson KA, Terkeltaub RA. External GTP-bound transglutaminase 2 is a molecular switch for chondrocyte hypertrophic differentiation and calcification. J Biol Chem. 2005;280:15004–15012. doi: 10.1074/jbc.M500962200. [DOI] [PubMed] [Google Scholar]
- 61.Nurminskaya M, Magee C, Faverman L, Linsenmayer TF. Chondrocyte-derived transglutaminase promotes maturation of preosteoblasts in periosteal bone. Develop Biol. 2003;263:139–152. doi: 10.1016/s0012-1606(03)00445-7. [DOI] [PubMed] [Google Scholar]
- 62.Mohan K, Pinto D, Issekutz TB. Identification of tissue transglutaminase as a novel molecule involved in human CD8+ T cell transendothelial migration. J Immunology. 2003;171:3179–3186. doi: 10.4049/jimmunol.171.6.3179. [DOI] [PubMed] [Google Scholar]
- 63.Seiving B, Ohlsson K, Linder C, Stenberg P. Transglutaminase differentiation during maturation of human blood monocytes to macrophages. Eur J Haematol. 1991;46:263–271. doi: 10.1111/j.1600-0609.1991.tb01537.x. [DOI] [PubMed] [Google Scholar]
- 64.Torocsik D, Bardos H, Nagy L, Adany R. Identification of factor XIII-A as a marker of alternative macrophage activation. Cell Mol Life Sci. 2005;62:2132–2139. doi: 10.1007/s00018-005-5242-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nagy JA, Henriksson P, McDonagh J. Biosynthesis of factor XIII B subunit by human hepatoma cell lines. Blood. 1986;68:1272–1279. [PubMed] [Google Scholar]
- 66.Adany R, Antal M. Three different cell types can synthesize factor XIII subunit A in the human liver. Thromb Haemost. 1996;76:74–79. [PubMed] [Google Scholar]
- 67.Anwar R, Miloszewski KJ. Factor XIII deficiency. Br J Haematol. 1999;107:468–484. doi: 10.1046/j.1365-2141.1999.01648.x. [DOI] [PubMed] [Google Scholar]
- 68.Lee J, Kim Y-S, Choi D-H, Bang M-S, Han T-R, Joh T-H, Kim S-Y. Transglutaminase 2 induces nuclear factor-κB activation via a novel pathway in BV-2 microglia. J Biol Chem. 2004;279:53725–53735. doi: 10.1074/jbc.M407627200. [DOI] [PubMed] [Google Scholar]
- 69.Mirza A, Liu SL, Frizell E, Zhu J, Maddukuri S, Martinez J, Davies JPJA, Schwarting R, Norton P, Zern MA. A role for tissue transglutaminase in hepatic injury and fibrogenesis and regulation by NF-κB. Am J Physiol. 1997;272:G281–288. doi: 10.1152/ajpgi.1997.272.2.G281. [DOI] [PubMed] [Google Scholar]
- 70.Kim S-Y, Jeong E-J, Steinert PM. INF-γ induces transglutaminase 2 expression in rat small intestinal cells. J Interferon Cytokine Res. 2002;22:677–682. doi: 10.1089/10799900260100169. [DOI] [PubMed] [Google Scholar]
- 71.Quan G, Choi J-Y, Lee D-S, Lee S-C. TGF-β1 up-regulates transglutaminase two and fibronectin in dermal fibroblasts: a possible mechanism for the stabilization of tissue inflammation. Arch Dermatol Res. 2005;297:84–90. doi: 10.1007/s00403-005-0582-8. [DOI] [PubMed] [Google Scholar]
- 72.Nunes I, Gleizes P-E, Metz CN, Rifkin DB. Latent transforming growht factor-β binding protein domains involved in activation and transglutaminase-dependent cross-linking of latent transforming growth factor-β. J Cell Biol. 1997;136:1151–1163. doi: 10.1083/jcb.136.5.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, Annunziata N, Doetschman T. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Szondy Z, Sarang Z, Molnar P, Nemeth T, Piacentini M, Mastroberardino PG, Falasca L, Aeschlimann D, Kovacs J, Kiss I, Szegezdi E, Lakos G, Rajnavolgyi E, Birckbichler PJ, Melino G, Fesus L. Transglutaminase 2−/− mice reveal a phagocytosis-associated crosstalk between macrophages and apoptotic cells. Proc Natl Acad Sci U S A. 2003;100:7812–7817. doi: 10.1073/pnas.0832466100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Miele L. New weapons against inflammation: dual inhibitors of phospholipase A2 and transglutaminase. J Clin Invest. 2003;111:19–21. doi: 10.1172/JCI17506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cordella-Miele E, Miele L, Mukkerjee AB. A novel transglutaminase-mediated post-translational modification of phospholipase A2 dramatically increases its catalytic activity. J Biol Chem. 1990;265:17180–17188. [PubMed] [Google Scholar]
- 77.Cordella-Miele E, Miele L, Beninati S, Mukherjee AB. Transglutaminase-catalyzed incorporation of polyamines into phospholipase A2. J Biochem (Tokyo) 1993;113:164–173. doi: 10.1093/oxfordjournals.jbchem.a124021. [DOI] [PubMed] [Google Scholar]
- 78.Sohn J, Kim T-I, Yoon Y-H, Kim J-Y, Kim S-Y. Novel transglutaminase inhibitors reverse the inflammation of allergic conjunctivitis. J Clin Invest. 2003;111:121–128. doi: 10.1172/JCI15937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shu GY, Ham HS, Lee S, Choi JC, Koh W-J, Kim S-Y, Lee J, Han J, Kim HP, Choi AMK, Kwon OJ. A peptide with anti-transglutaminase activity decreases lipopolysaccharide-induced lung inflammation in mice. Exp Lung Res. 2006;32:43–53. doi: 10.1080/01902140600691514. [DOI] [PubMed] [Google Scholar]
- 80.Nunes I, Shapiro RL, Rifkin DB. Characterization of latent TGF-beta activation by murine peritoneal macrophages. J Immunol. 1995;155:1450–1459. [PubMed] [Google Scholar]
- 81.Balajthy Z, Csomós K, Vámosi G, Szánto A, Lanotte M, Fésüs L. Tissue-transglutaminase contributes to neutrophil granulocyte differentiation and functions. Blood. doi: 10.1182/blood-2004-02-007948. prepublished online June 8, 2006 DOI: 10.1182/blood-2004−02−007948. [DOI] [PubMed] [Google Scholar]
- 82.Kim S-Y, Jeitner TM, Steinert PM. Transglutaminases in disease. Neurochemistry International. 2002;40:85–103. doi: 10.1016/s0197-0186(01)00064-x. [DOI] [PubMed] [Google Scholar]
- 83.Dieterich W, Ehnis T, Bauer M, Donner P, Volta U, Riecken EO, Schuppan D. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med. 1997;3:797–801. doi: 10.1038/nm0797-797. [DOI] [PubMed] [Google Scholar]
- 84.Reunala T, Collin P. Diseases associated with dermatitis herpetiformis. Br J Dermatol. 1997;136:315–318. [PubMed] [Google Scholar]
- 85.Bao F, Yu L, Babu S, Wang T, Hoffenberg EJ, Rewers M, Eisenbarth GS. One third of HLA DQ2 homozygous patients with type 1 diabetes express celiac disease associated transglutaminase autoantibodies. J Autoimmun. 1999;13:143–148. doi: 10.1006/jaut.1999.0303. [DOI] [PubMed] [Google Scholar]
- 86.Sanchez D, Tuckova L, Sebo P, Michalak M, Whelan A, Sterzl I, Jelinkova L, Havrdova E, Imramovska M, Benes Z, Krupickova S, Tlaskalova-Hogenova H. Occurrence of IgA and IgG autoantibodies to calreticulin in coeliac disease and various autoimmune diseases. J Autoimmun. 2000;15:441–449. doi: 10.1006/jaut.2000.0452. [DOI] [PubMed] [Google Scholar]
- 87.De Laurenzi V, Melino G. Gene disruption of tissue transglutaminase. Mol Cell Biol. 2001;21:148–155. doi: 10.1128/MCB.21.1.148-155.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nanda N, Iismaa SE, Owens WA, Husain A, Mackay F, Graham RM. Targeted inactivation of Gh/tissue transglutaminase II. J Biol Chem. 2001;276:20673–20678. doi: 10.1074/jbc.M010846200. [DOI] [PubMed] [Google Scholar]
- 89.Bernassola F, Federici M, Corazzari M, Terrinoni A, Hribal ML, De Laurenzi V, Ranalli M, Massa O, Sesti G, McLean WH, Citro G, Barbetti F, Melino G. Role of transglutaminase 2 in glucose tolerance: knockout mice studies and a putative mutation in a MODY patient. FASEB J. 2002;16:1371–1378. doi: 10.1096/fj.01-0689com. [DOI] [PubMed] [Google Scholar]
- 90.AbdAlla S, Lother H, Langer A, el Faramawy Y, Quitterer U. Factor XIIIA transglutaminase crosslinks AT1 receptor dimers of monocytes at the onset of atherosclerosis. Cell. 2004;119:343–354. doi: 10.1016/j.cell.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 91.Janiak A, Zemskov EA, Belkin AM. Cell surface transglutaminase promotes RhoA activation via integrin clustering and suppression of the Src-p190RhoGAP signaling pathway. Mol Biol Cell. 2006;17:1606–1619. doi: 10.1091/mbc.E05-06-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Langille BL, Dajnowiec D. Cross-linking vasomotor tone and vascular remodeling: a novel function for tissue transglutaminase? Circ Res. 2005;96:9–11. doi: 10.1161/01.RES.0000153883.55971.81. [DOI] [PubMed] [Google Scholar]
- 93.Mulvany MJ, Baumbach GL, Aalkjaer C, Heagerty AM, Korsgaard N, Schiffrin EL, Heistad DD. Vascular remodeling. Hypertension. 1996;28:505–506. [PubMed] [Google Scholar]
- 94.Bakker EN, Buus CL, Spaan JA, Perree J, Ganga A, Rolf TM, Sorop O, Bramsen LH, Mulvany MJ, Vanbavel E. Small artery remodeling depends on tissue-type transglutaminase. Circ Res. 2005;96:119–126. doi: 10.1161/01.RES.0000151333.56089.66. [DOI] [PubMed] [Google Scholar]
- 95.Chade AR, Zhu X, Mushin OP, Napoli C, Lerman A, Lerman LO. Simvastatin promotes angiogenesis and prevents microvascular remodeling in chronic renal ischemia. FASEB J. 2006;20:E1014–E1023. doi: 10.1096/fj.05-5680fje. [DOI] [PubMed] [Google Scholar]
- 96.Bakker EN, Piestea A, Spaan JAE, Rolf T, de Vries CJ, van Rooijen N, Candi E, VanBavel E. Flow-dependent remodeling of small arteries in mice deficient for tissue-type transglutaminase. Circ Res. 2006;99:86–92. doi: 10.1161/01.RES.0000229657.83816.a7. [DOI] [PubMed] [Google Scholar]
- 97.Lai TS, Hausladen A, Slaughter TF, Eu JP, Stamler JS, Greenberg CS. Calcium regulates S-nitrosylation, denitrosylation, and activity of tissue transglutaminase. Biochemistry. 2001;40:4904–4910. doi: 10.1021/bi002321t. [DOI] [PubMed] [Google Scholar]
- 98.Bernassola F, Rossi A, Melino G. Regulation of transglutaminases by nitric oxide. Ann NY Acad Sci. 1999;887:83–91. doi: 10.1111/j.1749-6632.1999.tb07924.x. [DOI] [PubMed] [Google Scholar]
- 99.Libby P. Inflammation and cardiovascular disease mechanisms. Am J Clin Nutr. 2006;83(suppl):456S–460S. doi: 10.1093/ajcn/83.2.456S. [DOI] [PubMed] [Google Scholar]
- 100.Falk E. Pathogenesis of atherosclerosis. J Am Coll Cardiol. 2006;47(8 suppl):C7–C12. doi: 10.1016/j.jacc.2005.09.068. [DOI] [PubMed] [Google Scholar]
- 101.Romanic AM, Arleth AJ, Willette RN, Ohlstein EH. Factor XIIIa cross-links lipoprotein(a) with fibrinogen and is present in human atherosclerotic lesions. Circ Res. 1998;83:264–269. doi: 10.1161/01.res.83.3.264. [DOI] [PubMed] [Google Scholar]
- 102.Haroon ZA, Wannenburg T, Gupta M, Greenberg CS, Wallin R, Sane DC. Localization of tissue transglutaminase in human carotid and coronary artery atherosclerosis: implications for plaque stability and progression. Lab Invest. 2001;81:83–93. doi: 10.1038/labinvest.3780214. [DOI] [PubMed] [Google Scholar]
- 103.Sumi Y, Inoue N, Azumi H, Seno T, Okuda M, Hirata K, Kawashima S, Hayashi Y, Itoh H, Yokoyama M. Expression of tissue transglutaminase and elafin in human coronary artery: implication for plaque instability. Atherosclerosis. 2002;160:31–39. doi: 10.1016/s0021-9150(01)00542-1. [DOI] [PubMed] [Google Scholar]
- 104.Bowness JM, Marcello V, Tarr AH, Taylor JR. Incease in ε(γ-glutamyl) lysine cross-links in atherosclerotic aortas. Atherosclerosis. 1994;111:247–253. doi: 10.1016/0021-9150(94)90099-x. [DOI] [PubMed] [Google Scholar]
- 105.Wilcox JN, Nelken NA, Coughlin SR, Gordon D, Schall TJ. Local expression of inflammatory cytokines in human atherosclerotic plaques. J Atheroscler Thromb. 1994;(Suppl 1):S10–S13. doi: 10.5551/jat1994.1.supplemment1_s10. [DOI] [PubMed] [Google Scholar]
- 106.Wiebe RI, Tarr AH, Bowness JM. Increased transglutaminase in the aortas of cholesterol-fed rabbits: occurrence of buffer soluble and insoluble forms and an inhibitor. Biochem Cell Biol. 1991;69:821–827. doi: 10.1139/o91-122. [DOI] [PubMed] [Google Scholar]
- 107.Boisvert WA, Rose DM, Boullier A, Quehenberger O, Sydlaske A, Johnson KA, Curtiss LK, Terkeltaub R. Leukocyte transglutaminase 2 expression limits atherosclerotic lesion size. Arterioscler Thromb Vasc Biol. 2006;26:563–569. doi: 10.1161/01.ATV.0000203503.82693.c1. [DOI] [PubMed] [Google Scholar]
- 108.Mauviel A. Transforming growth factor-beta: a key mediator of fibrosis. Methods Mol Med. 2005;117:69–80. doi: 10.1385/1-59259-940-0:069. [DOI] [PubMed] [Google Scholar]
- 109.Kang SK, Yi KS, Kwon NS, Park K-H, Kim U-H, Baek KJ, Im M-J. α1B-adrenoceptor signaling and cell motility. J Biol Chem. 2004;279:36593–3660. doi: 10.1074/jbc.M402084200. [DOI] [PubMed] [Google Scholar]
- 110.Attie AD, Kastelein JP, Hayden MR. Pivotal role of ABCA1 in reverse cholesterol transport influencing HDL levels and susceptibility to atherosclerosis. J Lipid Res. 2001;42:1717–1726. [PubMed] [Google Scholar]
- 111.Schmitz G, Kaminski WE, Porsch-Ozcurumez M, Klucken J, Orso E, Bodzioch M M, Buchler C, Drobnik W. ATP-binding cassette transporter A1 (ABCA 1) in macrophages: a dual function in inflammation and lipid metabolism. Pathobiology. 1999;67:236–240. doi: 10.1159/000028100. [DOI] [PubMed] [Google Scholar]
- 112.Hamon Y, Chambenoit O, Chimini G. ABCA1 and the engulfment of apoptotic cells. Biochem Biophys Acta. 2002;1585:64–71. doi: 10.1016/s1388-1981(02)00325-6. [DOI] [PubMed] [Google Scholar]
- 113.Shainoff JR, Page IH. Deposition of modified fibrinogen within the aortic intima. Atherosclerosis. 1972;16:287–305. doi: 10.1016/0021-9150(72)90079-2. [DOI] [PubMed] [Google Scholar]
- 114.Conkling PR, Achyuthan KE, Greenberg CS, Newcomb TF, Weinberg JB. Human mononuclear phagocyte transglutaminase activity cross-links fibrin. Thromb Res. 1989;55:57–68. doi: 10.1016/0049-3848(89)90456-8. [DOI] [PubMed] [Google Scholar]
- 115.Soecnlein O, Eskafi S, Schmeisser A, Kloos H, Daniel WG, Garlichs CD. Atorvastatin induces tissue transglutaminase in human endothelial cells. Biochem Biophys Res Comm. 2004;322:105–109. doi: 10.1016/j.bbrc.2004.07.087. [DOI] [PubMed] [Google Scholar]
- 116.Schalkwijk J, Wiedow O, Hirose S. The trappin gene family: proteins defined by an N-terminal transglutaminase substrate domain and a C-terminal four-disulphide core. Biochem J. 1999;340:569–577. [PMC free article] [PubMed] [Google Scholar]
- 117.Guyot N, Zani ML, Maurel MC, Dallet-Choisy S, Moreau T. Elafin and its precursor trappin-2 still inhibit neutrophil serine proteinases when they are covalently bound to extracellular matrix proteins by tissue transglutaminase. Biochemistry. 2005;44:15610–15618. doi: 10.1021/bi051418i. [DOI] [PubMed] [Google Scholar]
- 118.Ma J, Liew CC. Gene profiling identifies secreted protein transcripts from peripheral blood cells in coronary artery disease. J Mol Cell Cardiol. 2003;35:993–998. doi: 10.1016/s0022-2828(03)00179-2. [DOI] [PubMed] [Google Scholar]
- 119.Kohler HP, Ariens RA, Mansfield MW, Whitaker P, Grant PJ. Factor XIII activity and antigen levels in patients with coronary artery disease. Thromb Haemost. 2001;85:569–570. [PubMed] [Google Scholar]
- 120.Kloczko J, Wojtukiewicz M, Bielawiec M, Zuch A. Alterations of haemostasis parameters with special reference to fibrin stabilization, factor XIII and fibronectin in patients with obliterative atherosclerosis. Thromb Res. 1988;51:575–581. doi: 10.1016/0049-3848(88)90141-7. [DOI] [PubMed] [Google Scholar]
- 121.Kohler HP, Ariens RA, Catto AJ, Carter AM, Miller GJ, Cooper JA, Mansfield MW, Standeven KF, Grant PJ. Factor XIII A-subunit concentration predicts outcome in stroke subjects and vascular outcome in healthy, middle-aged men. Br J Haematol. 2002;118:825–832. doi: 10.1046/j.1365-2141.2002.03707.x. [DOI] [PubMed] [Google Scholar]
- 122.Mansfield MW, Kohler HP, Ariens RA, McCormack LJ, Grant PJ. Circulating levels of coagulation factor XIII in subjects with type 2 diabetes and in their first-degree relatives. Diabetes Care. 2000;23:703–705. doi: 10.2337/diacare.23.5.703. [DOI] [PubMed] [Google Scholar]
- 123.Dunn EJ, Philippou H, Ariens RA, Grant PJ. Molecular mechanisms involved in the resistance of fibrin to clot lysis by plasmin in subjects with type 2 diabetes mellitus. Diabetologia. 2006;49:1071–1080. doi: 10.1007/s00125-006-0197-4. [DOI] [PubMed] [Google Scholar]
- 124.Standeven KF, Ariens RA, Whitaker P, Ashcroft AE, Weisel JW, Grant PJ. The effect of dimethylbiguanide on thrombin activity, FXIII activation, fibrin polymerization, and fibrin clot formation. Diabetes. 2002;51:189–197. doi: 10.2337/diabetes.51.1.189. [DOI] [PubMed] [Google Scholar]
- 125.Cucuianu MP, Vasile VW, Popescu TA, Opincaru A, Crisnic I, Tapalaga D. Clinical studies concerning factor XIII; with special reference to hyperlipemia. Thromb Diath Haemorrh. 1973;30:480–493. [PubMed] [Google Scholar]
- 126.Cucuianu MP, Miloszewski K, Porutiu D D, Losowsky MS. Plasma factor XIII and platelet factor XIII in hyperlipaemia. Thromb Haemost. 1976;36:542–550. [PubMed] [Google Scholar]
- 127.Kain K, Catto AJ, Grant PJ. Associations between insulin resistance and thrombotic risk factors in high-risk South Asian subjects. Diabet Med. 2003;20:651–655. doi: 10.1046/j.1464-5491.2003.00958.x. [DOI] [PubMed] [Google Scholar]
- 128.Warner D, Mansfield MW, Grant PJ. Coagulation factor XIII and cardiovascular disease in UK Asian patients undergoing coronary angiography. Thromb Haemost. 2001;85:408–411. [PubMed] [Google Scholar]
- 129.Moskau S, Golla A, Grothe C, Boes M, Pohl C, Klockgether T. Heritability of carotid artery atherosclerotic lesions: an ultrasound study in 154 families. Stroke. 2005;36:5–8. doi: 10.1161/01.STR.0000149936.33498.83. [DOI] [PubMed] [Google Scholar]
- 130.Dunn EJ, Ariens RA, de Lange M, Snieder H, Turney JH, Spector TD, Grant PJ. Genetics of fibrin clot structure: a twin study. Blood. 2004;103:1735–1740. doi: 10.1182/blood-2003-07-2247. [DOI] [PubMed] [Google Scholar]
- 131.Mills JD, Ariens RA, Mansfield MW, Grant PJ. Altered fibrin clot structure in the healthy relatives of patients with premature coronary artery disease. Circulation. 2002;106:1938–1942. doi: 10.1161/01.cir.0000033221.73082.06. [DOI] [PubMed] [Google Scholar]
- 132.Nakaoka H, Perez DM, Baek KJ, Das T, Husain A, Misono K, Im MJ, Graham RM. Gh: a GTP-binding protein with transglutaminase activity and receptor signaling function. Science. 1994;264:1593–1596. doi: 10.1126/science.7911253. [DOI] [PubMed] [Google Scholar]
- 133.Chen S, Lin F, Iismaa S, Lee KN, Birckbichler PJ, Graham RM. α1-adrenergic receptor signaling via Gh is subtype specific and independent of its transglutaminase activity. J Biol Chem. 1996;271:32385–32391. doi: 10.1074/jbc.271.50.32385. [DOI] [PubMed] [Google Scholar]
- 134.Small K, Feng JF, Lorenz J, Donnelly ET, Yu A, Im MJ, Dorn GW, 2nd, Liggett SB. Cardiac specific overexpression of transglutaminase II (G(h)) results in a unique hypertrophy phenotype independent of phospholipase C activation. J Biol Chem. 1999;274:21291–21296. doi: 10.1074/jbc.274.30.21291. [DOI] [PubMed] [Google Scholar]
- 135.Vezza R, Habib A, FitzGerald GA. Differential signaling by the thromboxane receptor isoforms via the novel GTP-binding protein, Gh. J Biol Chem. 1999;274:12774–12779. doi: 10.1074/jbc.274.18.12774. [DOI] [PubMed] [Google Scholar]
- 136.Baek KJ, Kwon NS, Lee HS, Kim MS, Muralidhar P, Im MJ. Oxytocin receptor couples to the 80 kDa Gh alpha family protein in human myometrium. Biochem J. 1996;315:739–744. [PMC free article] [PubMed] [Google Scholar]
- 137.Park E-S, Won JH, Han KJ, Suh P-G, Ryu SH, Lee HS, Yun H-Y, Kwon NS, Beak KJ. Phospholipase C-γ1 and oxytocin receptor signaling: evidence of its role as an effector. Biochem J. 1998;331:283–189. doi: 10.1042/bj3310283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kang SK, Yi KS, Kwon NS, Park K-H, Kim U-H, Baek KJ, Im M-J. α1B-adrenoceptor signaling and cell motility. J Biol Chem. 2004;279:36593–3660. doi: 10.1074/jbc.M402084200. [DOI] [PubMed] [Google Scholar]
- 139.Loirand G, Guerin P, Pacaud P. Rho kinases in cardiovascular physiology and pathophysiology. Circ Res. 2006;98:322–334. doi: 10.1161/01.RES.0000201960.04223.3c. [DOI] [PubMed] [Google Scholar]
- 140.Singh US, Kunar MT, Kao YL, Baker KM. Role of transglutaminase II in retinoic acid-induced activation of RhoA-associated kinase-2. EMBO J. 2001;20:2413–2423. doi: 10.1093/emboj/20.10.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Smethurst PA, Griffin M. Measurement of tissue transglutaminase activity in a permeabilized cell system: its regulation by Ca2+ and nucleotides. Biochem J. 1996;303:803–808. doi: 10.1042/bj3130803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lai T-S, Bielawska A, Peoples KA, Hannun YA, Greenberg CS. Sphingosylphosphocholine reduces the calcium ion requirement for activating tissue transglutaminase. J Biol Chem. 1997;272:16295–16300. doi: 10.1074/jbc.272.26.16295. [DOI] [PubMed] [Google Scholar]
- 143.Feng J-F, Readon M, Yadov SP, Im M-J. Calreticulin down-regulates both GTP binding and transglutaminase activities of transglutaminase II. Biochemistry. 1999;38:10743–10749. doi: 10.1021/bi9905009. [DOI] [PubMed] [Google Scholar]
- 144.Lee K-H, Lee N, Lim S, Jung H, Ko Y-G, Park H-Y, Jang Y, Lee H, Hwang K-C. Calreticulin inhibits the MEK1,2-ERK1,2 pathway in α1-adrenergic receptor/Gh-stimulated hypertrophy of neonatal rat cardiomyocytes. J Ster Biochem Mol Biol. 2003;84:101–107. doi: 10.1016/s0960-0760(03)00006-2. [DOI] [PubMed] [Google Scholar]
- 145.Noll T, Wozniak G, McCarson K, Hajimohammad A, Metzner HJ, Inserte J, Kummer W, Hehrlein FW, Piper HM. Effect of Factor XIII on Endothelial Barrier Function. J Exp Med. 1999;189:1373–1382. doi: 10.1084/jem.189.9.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hirahara K, Shinbo K, Takahashi M, Matsuishi T. Suppressive effect of human blood coagulation factor XIII on the vascular permeability induced by anti-guinea pig endothelial cell antiserum in guinea pigs. Thromb Res. 1993;71:139–148. doi: 10.1016/0049-3848(93)90180-v. [DOI] [PubMed] [Google Scholar]
- 147.Brody JI, Pickering NJ, Fink GB. Concentrations of factor VIII-related antigen and factor XIII during open heart surgery. Transfusion. 1986;26:478–480. doi: 10.1046/j.1537-2995.1986.26587020130.x. [DOI] [PubMed] [Google Scholar]
- 148.Chandler WL, Patel MA, Gravelle L, Soltow LO, Lewis K, Bishop PD, Spiess BD. Factor XIIIA and clot strength after cardiopulmonary bypass. Blood Coagul Fibrinolysis. 2001;12:101–108. doi: 10.1097/00001721-200103000-00003. [DOI] [PubMed] [Google Scholar]
- 149.Godje O, Haushofer M, Lamm P, Reichart B. The effect of factor XIII on bleeding in coronary surgery. Thorac Cardiovasc Surg. 1998;46:263–267. doi: 10.1055/s-2007-1010236. [DOI] [PubMed] [Google Scholar]
- 150.Shainoff JR, Estafanous FG, Yared JP, DiBello PM, Kottke-Marchant K, Loop FD. Low factor XIIIA levels are associated with increased blood loss after coronary artery bypass grafting J. Thorac Cardiovasc Surg. 1994;108:437–445. [PubMed] [Google Scholar]
- 151.Wozniak G, Noll T, Akinturk H, Thul J, Muller M. Factor XIII prevents development of myocardial edema in children undergoing surgery for congenital heart disease. Ann N Y Acad Sci. 2001;936:617–620. doi: 10.1111/j.1749-6632.2001.tb03549.x. [DOI] [PubMed] [Google Scholar]
- 152.Schroth M, Meissner U, Cesnjevar R, Weyand M, Singer H, Rascher W, Klinge J. Recombinant Factor XIII Reduces Severe Pleural Effusion in Children after Open-Heart Surgery. Pediatr Cardiol. 2006;27:56–60. doi: 10.1007/s00246-005-0993-5. [DOI] [PubMed] [Google Scholar]
- 153.Godje O, Gallmeier U, Schelian M, Grunewald M, Mair H. Coagulation factor XIII reduces postoperative bleeding after coronary surgery with extracorporeal circulation. Thorac Cardiovasc Surg. 2006;54:26–33. doi: 10.1055/s-2005-872853. [DOI] [PubMed] [Google Scholar]
- 154.Baumgartner W, Golenhofen N, Weth A, Hiiragi T, Saint R, Griffin M, Drenckhahn D. Role of transglutaminase 1 in stabilisation of intercellular junctions of the vascular endothelium. Histochem Cell Biol. 2004;122:17–25. doi: 10.1007/s00418-004-0668-y. [DOI] [PubMed] [Google Scholar]
- 155.Haroon ZA, Hettasch JM, Lai TS, Dewhirst MW, Greenberg CS. Tissue transglutaminase is expressed, active, and directly involved in rat dermal wound healing and angiogenesis. FASEB J. 1999;13:1787–1795. doi: 10.1096/fasebj.13.13.1787. [DOI] [PubMed] [Google Scholar]
- 156.Jones RA, Kotsakis P, Johnson TS, Chau DY, Ali S, Melino G, Griffin M. Matrix changes induced by transglutaminase 2 lead to inhibition of angiogenesis and tumor growth. Cell Death Differ. 2005 Nov 18; doi: 10.1038/sj.cdd.4401816. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 157.Dardik R, Loscalzo J, Eskaraev R, Inbal A. Molecular mechanisms underlying the proangiogenic effect of factor XIII. Arterioscler Thromb Vasc Biol. 2005;25:526–532. doi: 10.1161/01.ATV.0000154137.21230.80. [DOI] [PubMed] [Google Scholar]
- 158.Huff T, Otto AM, Müller CSG, Meier M, Hannappel E. Thymosin β4 is released from human blood platelets and attached by factor XIIIa (transglutaminase) to fibrin and collagen. FASEB J. 2002;16:691–696. doi: 10.1096/fj.01-0713com. [DOI] [PubMed] [Google Scholar]
- 159.Dardik R, Solomon A, Loscalzo J, Eskaraev R, Bialik A, Goldberg I, Schiby G, Inbal A. Novel proangiogenic effect of factor XIII associated with suppression of thrombospondin 1 expression. Arterioscler Thromb Vasc Biol. 2003;23:1472–1477. doi: 10.1161/01.ATV.0000081636.25235.C6. [DOI] [PubMed] [Google Scholar]
- 160.Dardik R, Loscalzo J, Inbal A. Factor XIII (FXIII) and angiogenesis. J Thromb Haemost. 2006;4:19–25. doi: 10.1111/j.1538-7836.2005.01473.x. [DOI] [PubMed] [Google Scholar]
- 161.Inbal A, Leor J, Skutelsky E, Castel D, Holbova R, Schibi G, Goldberg I, Eskaraev R, Dardik R. Evaluation of pro-angiogenic activity of factor XIII (FXIII) in ischemic tissue, heart transplantation and FXIII-deficient mice. Blood. 2004;104:Abstract 2987. ASH Annual Meeting Abstracts. [Google Scholar]
- 162.Kilian O, Fuhrmann R, Alt V, Noll T, Coskun S, Dingeldein E, Schnettler R, Franke RP. Plasma transglutaminase factor XIII induces microvessel ingrowth into biodegradable hydroxyapatite implants in rats. Biomaterials. 2005;26:1819–1827. doi: 10.1016/j.biomaterials.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 163.Dardik R, Leor J, Skutelsky E, Castel D, Holbova R, Schiby G, Shaish A, Dickneite G, Loscalzo J, Inbal A. Evaluation of the pro-angiogenic effect of factor XIII in heterotopic mouse heart allografts and FXIII-deficient mice. Thromb Haemost. 2006;95:546–550. doi: 10.1160/TH05-06-0409. [DOI] [PubMed] [Google Scholar]
- 164.Iwai N, Shimoike H, Kinoshita M. Genes up-regulated in hypertrophied ventricle. Biochem Biophys Res Commun. 1995;209:527–534. doi: 10.1006/bbrc.1995.1533. [DOI] [PubMed] [Google Scholar]
- 165.Hwang KC, Gray CD, Sweet WE, Moravec CS, Im MJ. Alpha 1-adrenergic receptor coupling with Gh in the failing human heart. Circulation. 1996;94:718–726. doi: 10.1161/01.cir.94.4.718. [DOI] [PubMed] [Google Scholar]
- 166.Zhang Z, Vezza R, Plappert T, McNamara P, Lawson JA, Austin S, Pratico D, Sutton MS, FitzGerald GA. COX-2-dependent cardiac failure in Gh/TG2 transgenic mice. Circ Res. 2003;92:1153–1161. doi: 10.1161/01.RES.0000071749.22027.45. [DOI] [PubMed] [Google Scholar]
- 167.Frustaci A, Cuoco L, Chimenti C, Pieroni M, Fioravanti G, Gentiloni N, Maseri A, Gasbarrini G. Celiac disease associated with autoimmune myocarditis. Circulation. 2002;105:2611–2618. doi: 10.1161/01.cir.0000017880.86166.87. [DOI] [PubMed] [Google Scholar]
- 168.Curione M, Barbato M, De Biase L, Viola F, Lo Russo L, Cardi E. Prevalence of coeliac disease in idiopathic dilated cardiomyopathy. Lancet. 1999;354:222–223. doi: 10.1016/s0140-6736(99)01501-9. [DOI] [PubMed] [Google Scholar]
- 169.Chimenti C, Pieroni M, Frustaci A. Celiac disease in idiopathic dilated cardiomyopathy. Ital Heart J. 2001;2:658–659. [PubMed] [Google Scholar]
- 170.Sategna-Guidetti C, Franco E, Martini S, Bobbio M. Binding by serum IgA antibodies from patients with coeliac disease to monkey heart tissue. Scand J Gastroenterol. 2004;36:540–543. doi: 10.1080/00365520410008764. [DOI] [PubMed] [Google Scholar]
- 171.Bazzigaluppi E, Roggero P, Parma B, Brambillasca MF, Meroni F, Mora S, Bosi E, Barera G. Antibodies to recombinant human tissue-transglutaminase in coeliac disease: diagnostic effectiveness and decline pattern after gluten-free diet. Dig Liver Dis. 2006;38:98–102. doi: 10.1016/j.dld.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 172.Peracchi M, Trovato C, Longhi M, Gasparin M, Conte D, Tarantino C, Prati D, Bardella MT. Tissue transglutaminase antibodies in patients with end-stage heart failure. Am J Gastroenterol. 2002;97:2850–2854. doi: 10.1111/j.1572-0241.2002.07033.x. [DOI] [PubMed] [Google Scholar]
- 173.Metzger M-H, Heier M, Mäki M, Bravi E, Schneider A, Löwel H, Illig T, Schuppan D, Wichmann H-E. Mortality excess individuals with elevated IgA anti-transglutaminase antibodies: The KORA/MONICA Augsburg cohort study 1989−1998. Eur J Epidem. 2006;21:359–365. doi: 10.1007/s10654-006-9002-4. [DOI] [PubMed] [Google Scholar]
- 174.McDonough JL, Arrell DK, Van Eyk JE. Troponin I degradation and covalent complex formation accompanies myocardial ischemia/reperfusion injury. Circ Res. 1999;84:9–20. doi: 10.1161/01.res.84.1.9. [DOI] [PubMed] [Google Scholar]
- 175.Gorza L, Menabo R, Vitadello M, Bergamini CM, Di Lisa F. Cardiomyocyte troponin T immunoreactivity is modified by cross-linking resulting from intracellular calcium overload. Circulation. 1996;93:1896–1904. doi: 10.1161/01.cir.93.10.1896. [DOI] [PubMed] [Google Scholar]
- 176.RJ Solaro. Troponin I, stunning, hypertrophy, and failure of the heart. Circ Res. 1999;84:122–124. doi: 10.1161/01.res.84.1.122. [DOI] [PubMed] [Google Scholar]
- 177.Gorza L, Menabo R, Di Lisa F, Vitadello M. Troponin T cross-linking in human apoptotic cardiomyocytes. Am J Pathol. 1997;150:2087–2097. [PMC free article] [PubMed] [Google Scholar]
- 178.Szondy Z, Mastroberardino PG, Varadi J, Farrace MG, Nagy N, Bak I, Viti I, Wieckowski MR, Melino G, Rizzuto R, Tosaki A, Fesus L, Piacentini M. Tissue transglutaminase (TG2) protects cardiomyocytes against ischemia/reperfusion injury by regulating ATP synthesis. Cell Death Differ. 2006 March 10; doi: 10.1038/sj.cdd.4401889. Epub ahead of print DOI: 10.1038/sj.cdd.4401889. [DOI] [PubMed] [Google Scholar]
- 179.Piacentini M, Farrace MG, Piredda L, Matarrese P, Ciccosanti F, Falasca L, Rodolfo C, Giammarioli AM, Verderio E, Griffin M, Malorni W. Transglutaminase overexpression sensitizes neuronal cell lines to apoptosis by increasing mitochondrial membrane potential and cellular oxidative stress. J Neurochem. 2002;81:1061–1072. doi: 10.1046/j.1471-4159.2002.00898.x. [DOI] [PubMed] [Google Scholar]
- 180.Nahrendorf M, Hu K, Frantz S, Jaffer FA, Tung CH, Hiller KH, Voll S, Nordbeck P, Sosnovik D, Gattenlohner S, Novikov M, Dickneite G, Reed GL, Jakob P, Rosenzweig A, Bauer WR, Weissleder R, Ertl G. Factor XIII deficiency causes cardiac rupture, impairs wound healing, and aggravates cardiac remodeling in mice with myocardial infarction. Circulation. 2006;113:1196–1202. doi: 10.1161/CIRCULATIONAHA.105.602094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Alkjaersig N, Fletcher AP, Lewis M, Ittyerah R. Reduction of coagulation factor XIII concentration in patients with myocardial infarction, cerebral infarction, and other thromboembolic disorders. Thromb Haemost. 1977;38:863–873. [PubMed] [Google Scholar]
- 182.Wells PS, Owen C, Doucette S, Fergusson D, Tran H. Does this patient have deep vein thrombosis? JAMA. 2006;295:199–207. doi: 10.1001/jama.295.2.199. [DOI] [PubMed] [Google Scholar]
- 183.Mosesson MW, Siebenlist KR, Hernandez I, Wall JS, Hainfeld JF. Fibrinogen assembly and crosslinking on a fibrin fragment E template. Thromb Haemost. 2002;87:651–658. [PubMed] [Google Scholar]
- 184.Greenberg CS, Miraglia CC. The effect of fibrin polymers on catalyzed plasma factor XIIIa formation. Blood. 1985;66:466–469. [PubMed] [Google Scholar]
- 185.Dempfle CE. Validation, calibration, and specificity of quantitative D-dimer assays. Semin Vasc Med. 2005;5:315–320. doi: 10.1055/s-2005-922476. [DOI] [PubMed] [Google Scholar]
- 186.Tung C-H, Ho N-H, Zeng Q, Tang Y, Jaffer FA, Reed GL, Weissleder R. Novel factor XIII probes for blood coagulation imaging. Chem Bio Chem. 2003;14:897–899. doi: 10.1002/cbic.200300602. [DOI] [PubMed] [Google Scholar]
- 187.Mazooz G, Mehlman T, Lai T-S, Greenberg CS, Dewhirst MW. Development of magnetic resonance imaging contrast material for in vivo mapping of tissue transglutaminase activity. Cancer Res. 2005;65:1369–1375. doi: 10.1158/0008-5472.CAN-04-2269. [DOI] [PubMed] [Google Scholar]
- 188.Ito A, Mase A, Takizawa Y, Ai MS, Honda H, Hata K-I, Ueda M, Kobayashi T. Transglutaminase-mediated gelatin matrices incorporating cell adhesion factors as a biomaterial for tissue engineering. J Biosci and Bioeng. 2003;95:196–199. [PubMed] [Google Scholar]
- 189.Chau DYS, Collighan RJ, Verderio EAM, Addy VL, Griffin M. The cellular response to transglutaminase-cross-linked collagen. Biomaterials. 2003;26:6158–6529. doi: 10.1016/j.biomaterials.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 190.Jürgensen K, Aeschlimann D, Cavin V, Genge M, Hunziker EB. A new biological glue for cartilage-cartilage interfaces: tissue transglutaminase. J Bone Joint Surg Am. 1997;79:185–193. doi: 10.2106/00004623-199702000-00004. [DOI] [PubMed] [Google Scholar]
- 191.Shimizu T, Tako T, Hozumi K, Nunomura K, Ohta S, Shimonishi Y, Ikegami S. Structure of a covalently cross-linked form of core histones present in starfish sperm. Biochemistry. 1997;36:12071–12079. doi: 10.1021/bi970922n. [DOI] [PubMed] [Google Scholar]
- 192.Oliverio S, Amendola A, Di Sano F, Farrace MG, Fesus L, Nemes Z, Piredda L, Spinedi A, Piacentini M. Tissue transglutaminase-dependent posttranslational modification of the retinoblastoma gene product in promonocytic cells undergoing apoptosis. Mol Cell Biol. 1997;17:6040–6048. doi: 10.1128/mcb.17.10.6040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Han JA, Park SC. Transglutaminase-dependent modulation of transcription factor Sp1 activity. Mol Cells. 2000;10:612–618. doi: 10.1007/s10059-000-0612-5. [DOI] [PubMed] [Google Scholar]
- 194.Hasagawa G, Suwa M, Ichikawa Y, Ohtsuka T, Kumagai S, Kikuchi M, Sato Y, Saito Y. A novel function of tissue-type transglutaminase: protein disulphide isomerase. Biochem J. 2003;373:793–803. doi: 10.1042/BJ20021084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Mishra S, Murphy LJ. Tissue transglutaminase has intrinsic kinase activity: identification of transglutaminase 2 as an insulin-like growth factor-binding protein-3 kinase. J Biol Chem. 2004;279:23863. doi: 10.1074/jbc.M311919200. [DOI] [PubMed] [Google Scholar]
- 196.Mishra S, Murphy LJ. The p53 oncoprotein is a substrate for tissue transglutaminase kinase activity. Biocehm Biophys Res Commun. 2006;339:726–730. doi: 10.1016/j.bbrc.2005.11.071. [DOI] [PubMed] [Google Scholar]
- 197.Mishra S, Saleh A, Espino PS, Davie JR, Murphy LJ. Phosphorylation of histones by tissue transglutaminase. J Biol Chem. 2006;281:5532–5538. doi: 10.1074/jbc.M506864200. [DOI] [PubMed] [Google Scholar]