

Abstract
Dystrophia myotonica protein kinase (DMPK) is a serine/threonine kinase composed of a kinase domain and a coiled-coil domain involved in the multimerization. The crystal structure of the kinase domain of DMPK bound to the inhibitor bisindolylmaleimide VIII (BIM-8) revealed a dimeric enzyme associated by a conserved dimerization domain. The affinity of dimerisation suggested that the kinase domain alone is insufficient for dimerisation in vivo and that the coiled-coil domains are required for stable dimer formation. The kinase domain is in an active conformation, with a fully-ordered and correctly positioned αC helix, and catalytic residues in a conformation competent for catalysis. The conserved hydrophobic motif at the C-terminal extension of the kinase domain is bound to the N-terminal lobe of the kinase domain, despite being unphosphorylated. Differences in the arrangement of the C-terminal extension compared to the closely related Rho-associated kinases include an altered PXXP motif, a different conformation and binding arrangement for the turn motif, and a different location for the conserved NFD motif. The BIM-8 inhibitor occupies the ATP site and has similar binding mode as observed in PDK1.
Keywords: kinase, myotonic dystrophy, crystallization, DMPK, bisindolylmaleimide, BIM, enzymes, active sites, structure, crystallography, protein crystallization, enzyme inhibitors, protein structures
Introduction
Dystrophia myotonica protein kinase (DMPK) is a protein strongly linked to myotonic dystrophy type 1 (DM1), the most prevalent muscular dystrophy in adults. Genetic defects in DM1 cause amplification of a trinucleotide repeat in the 3' untranslated region of DMPK.1,2 The severity of the disease depends on the number of repeats which can range from normal individuals that have 5–30 repeats, mildly affected persons (50–80 repeats), and severely affected individuals that have 2000 or more copies. It is thought that the disease results from both gain-of-function of the dmpk RNA and from decreased DMPK expression in the cytoplasm (reviewed in Ref. 3). Of these, the RNA repeats are most likely the primary causative agent: DM2 is caused by a similar gain-of-function of a CCTG repeat within intron 1 of znf9,4 and transgenic mice expressing an artificial CUG repeat have a DM phenotype.5 In addition, mice with the muscleblind-homologue protein MBNL knocked out display DM phenotypes6; in humans, there are three muscleblind-homologue proteins, which specifically bind CUG repeats, and these co-localize with nuclear foci in DM cells.7,8 An inducible CTG repeat mouse model confirmed these findings and showed that the CTG repeats themselves were causative of severe skeletal muscle wasting.9 The CTG repeats also result in mRNA mis-splicing of the insulin receptor.10 Therefore it appears that DM is primarily caused by sequestering of RNA/DNA binding proteins by the overlong nucleotide repeats.
However, reduced DMPK activity is thought to be responsible for certain disease phenotypes of DM1. DMPK−/− mice develop late onset mild myopathy,11,12 while DMPK−/− and DMPK+/− mice displayed cardiac conduction defects.13 In addition, homozygous knockout mice exhibited impaired insulin signaling, suggesting that DMPK has a role in susceptibility to type-2 diabetes.14 DMPK substrates identified in vitro include phospholemman, a membrane-bound protein involved in ion transport,15 the myosin-binding subunit of myosin phosphatase16 and phospholamban, a regulator of the calcium pump in cardiac muscle cells.17
DMPK is an AGC family kinase and is most closely related by sequence to the myotonic dystrophy kinase-related Cdc42-binding kinases alpha, beta and gamma (MRCKα/MRCKβ/MRCKγ) which all share about 60% sequence identity with the DMPK kinase domain. MRCKγ is also known as DMPK2. DMPK is also closely related to the Rho-associated kinases I and II (ROCK1/ROCK2), with 45 and 43% identity, respectively, and to citron kinase (CRIK). There are six different DMPK isoforms, of which four are distributed independently of tissue type while two are predominantly present in smooth muscle.18 Four isoforms have molecular weights of ∼74 kDa, while the two smooth muscle-specific isoforms are only approximately 68 kDa in size, being C-terminally truncated relative to the larger isoforms.
In addition to the kinase domain, all isoforms of DMPK possess an N-terminal region which is leucine-rich in some isoforms, and a C-terminal α-helical domain that forms coiled-coil structures; the structure of the isolated coiled-coil domain of DMPK has been reported recently.19 Both DMPK and the ROCKs are known to form dimers, and studies on DMPK, the ROCKs, and MRCK have shown that dimerisation is mediated not by the coiled-coil domain, but by the regions immediately N- and C-terminal to the kinase domain.19,20–22 The crystal structures of ROCK1 and ROCK2 confirmed these observations, showing that the N- and C-terminal regions of the kinase domain combine to form the dimerisation interface.22,23
AGC kinases are regulated by activation loop phosphorylation as well as phosphorylation of the hydrophobic motif. MRCK requires dimerisation and trans-autophosphorylation for activation, with likely phosphorylation sites at Ser234, Thr240, and Thr403.20 These residues are all conserved in DMPK, and while DMPK undergoes autophosphorylation,24 as yet no definite sites have been identified. Autophosphorylation activity differs between different isoforms and this activity is modulated by the VSGGG motif present only in some splice variants.24
To examine the structural basis of DMPK activity we determined the structure of the kinase and flanking association domain of DMPK isoform 2 (which includes the VSGGG motif) in complex with the ATP-competitive inhibitor bisindolylmaleimide VIII (BIM-8). The structure revealed an active kinase conformation in the absence of activation loop and hydrophobic motif phosphorylation. In solution, association studies determined dissociation constants in the low μM region suggesting that under physiological condition DMPK requires additional association domains for effective dimerization.
Results and Discussion
The DMPK structure shows the typical fold of a protein kinase, with two lobes connected by a hinge region (see Fig. 1). The extended C-terminus typical of an AGC-family kinase wraps around the N-terminal lobe before packing between the N-terminal dimerisation helices and the kinase domain itself. The structure of DMPK superimposes with a 1.43 Å root mean squared deviation over 295 Cα atoms (out of 406 Cα atoms in total) with the structure of the related family member ROCK1 with which it shares 45% sequence identity over the kinase domain (see Supplementary Fig. 1 for a sequence alignment). A region of substantially different backbone conformation is in the extended C-terminus typical of AGC-family kinases, spanning from the conserved PXXP motif (residues 353–356) of the C-lobe tether (CLT)25 until the conserved C-terminal FXXΦ(S/T)Φ hydrophobic motif (Φ = hydrophobic, residues 400–405: FVGYSY in DMPK) that binds in a groove in the N-terminal lobe (the N-lobe tether, NLT).
Figure 1.
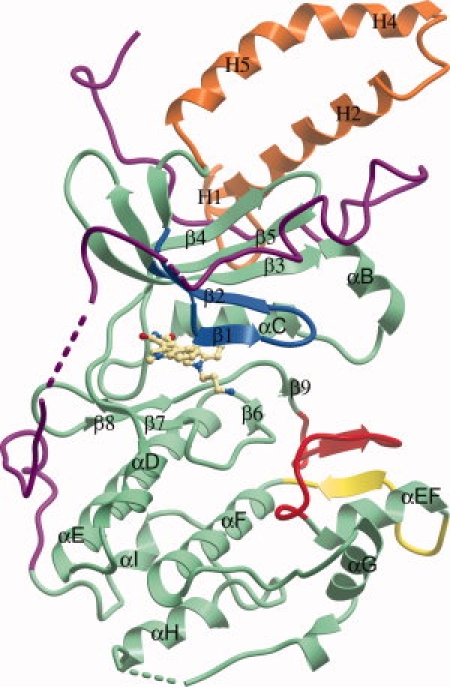
Domain arrangement of DMPK. The BIM-8 inhibitor in the active site is shown as a ball-and-stick representation. The N-terminal kinase lobe is mostly above the BIM-8 inhibitor and the C-terminal kinase lobe mostly below. The C-terminal section of the protein is colored purple, the activation loop is colored red, the αEF/αF loop is colored yellow, and the glycine rich loop colored blue. The N-terminal helices involved in the dimerisation interface are colored orange.
C-tail PXXP motif (C-lobe tether)
The PXXP motif that anchors the C-terminal extension to the loop between helices αD and αE [Fig. 2(A)] conserves the hydrophobic nature of this interaction as seen in the ROCK structures. The difference in the surface of the PXXP motif binding pocket, which comprises Trp174 and Tyr168 in ROCK1, and Met171 and Phe163 in DMPK, is accounted for by an alteration of the PXXP motif, from PVVP in ROCK1 to PFTP in DMPK resulting in a good shape complementary of the PXXP binding motifs and the binding pocket in both enzymes.
Figure 2.
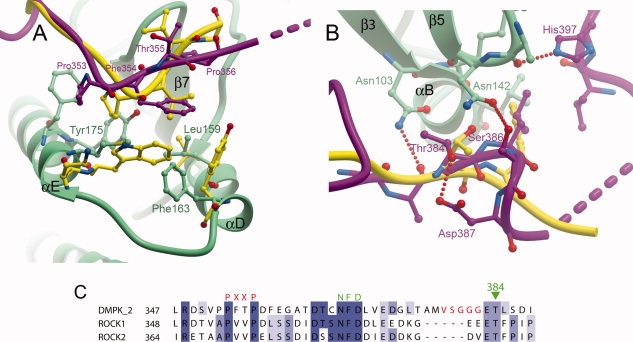
Comparison of the binding of the C-termini of DMPK and ROCK1 to the N-terminal lobe. (A) The PXXP motif, the ROCK1 C-terminus is in yellow, and DMPK is colored as in Figure 1. (B) The turn motif. (C) Sequence alignment of DMPK and the Rho kinases over the region covering the PXXP, active-site tether and turn motifs. The VSGGG insertion present in isoforms 1 and 2 of DMPK is indicated with red lettering.
C-tail conserved NFD motif (active-site tether)
In ROCK1, the region linking the two conserved AGC motifs contains a phenylalanine (ROCK1 Phe368) which points into the active site and is adjacent to bound inhibitors. This arrangement is reminiscent of the one described for protein kinase A26 and protein kinase C theta (PKCθ) with NVP-XAA228 (PDB ID 2JED). This region has been termed the active-site tether (AST),25 which may have a role in nucleotide binding and release, and substrate recognition. The NFD motif is completely conserved in the DMPK family (see Supplementary Fig. 1). However, although reasonably well-conserved in other AGC kinases this AST region is often disordered in AGC kinase structures. In DMPK, residues from Phe358 to The362 were disordered; however, in contrast to ROCK1, PKA, or PKCθ in complex with NVP-XAA228, the NFD motif (residues 366–368) was clearly defined by electron density in a position not adjacent to the active site and forming an intermolecular disulphide bond between Cys365 and Cys206. Previous work on PKA showed this region adjacent to the ATP-binding site in binary complexes with nucleotide, but disordered in the absence of nucleotide. The AST region has sufficient flexibility to allow this movement without effecting changes to the kinase conformation or the binding of the hydrophobic motif. However, while the intermolecular disulphide bond which presumably stabilizes the NFD motif away from the active site is a feature of crystallization, it is also possible that the equivalent phenylalanine in DMPK (Phe367) was prevented from binding adjacent to the active site by the bulky BIM-8 inhibitor, which projects further out of the active site compared to the inhibitors used for crystallization with ROCK1. It is worth noting that in the structure of PKCθ with staurosporine, the inhibitor on which BIM-8 was based, the entire AST region is disordered.27
C-tail turn motif
Despite the differing backbone conformations of DMPK and ROCK1 between the CLT and NLT, the side-chain of Thr384 in this linker region occupies a similar position to the equivalent conserved threonine in ROCK1 [Fig. 2(B)]. In PKA Ser338 is a known phosphorylation site where the phosphate group serves to stabilize a tight turn through interactions with PKA residues Asn340 and Lys342. These phosphoserine-binding residues are not conserved in DMPK or other members of the DMPK family. The side-chain hydroxyl of the un-phosphorylated Thr384 does form a hydrogen bond with the side-chain of Asp387, however, which stabilizes a different tight turn that allows a hydrogen bond to be formed between Ser386 from this part of the C-terminal region and Asn142 from the β4/β5 loop. Ser386 and Asp387 are not conserved in other members of the DMPK family [Fig. 2(C)] but of possibly greater importance, the hydrophobic residue present immediately C-terminal to Thr384 (Leu385 in DMPK) is conserved in all members apart from citron kinase. In both DMPK and ROCK1, this residue binds into a hydrophobic pocket at the junction between the conserved hydrophobic residue at the start of helix αB (Trp105 in DMPK), the β4/β5 loop and the start of the NLT, and appears important for stabilizing this part of the structure.
Isoforms 1 and 2 of DMPK have a five residue insertion (VSGGG) directly N-terminal to Thr384, relative to isoforms 3 and 4, but a similar position for Thr384 would still be possible in isoforms without this insertion as demonstrated by ROCK1 which also does not have this insertion. The VSGGG motif is unique among DMPK-related kinases and has been shown to modulate autophosphorylation activity.24 Whether presence of the motif increases the kinase activity of DMPK, perhaps by stabilizing the active conformation, or if the presence of the motif makes a turn-motif serine/threonine more available for autophosphorylation is unknown.
From these observations it appears that the region of the DMPK family kinases between the CLT and the NLT can assume a variety of different conformations, despite the very high level of sequence conservation. The exception to this allowed flexibility is the necessity for the loop to bind at the junction between helix αB, the β4/β5 loop, and the start of the NLT motif.
Conserved dimerisation domain
The crystallographic asymmetric unit contains two molecules of the DMPK kinase domain. These two molecules are not related by a simple rotation and therefore are unlikely to constitute the genuine biological dimer. An examination of the crystallographic symmetry revealed a conserved inter-molecule interaction that represents the biological dimer interface [Fig. 3(A)] conserved in the structures of ROCK1 and ROCK222,23 [Fig. 3(B–D)]. The regions of DMPK involved in the dimer interface are the four N-terminal helices, and the C-terminus of the kinase domain, which fits with the observed necessity of these regions for dimerization in DMPK19, the ROCKs21,22 and MRCK.20
Figure 3.
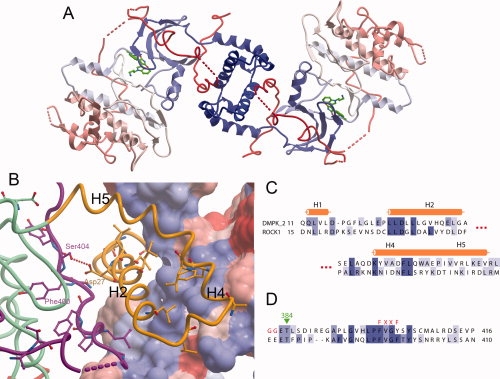
DMPK dimer. (A) The DMPK biological dimer, with each molecule colored blue to red from N- to C-terminus. (B) The dimerisation interface of DMPK. The surface is shown for one DMPK monomer, colored according to hydrophobicity, with blue more hydrophobic and red less hydrophobic. The other DMPK monomer is shown as a ribbon, with residues that are conserved between ROCK1 and DMPK illustrated. The DMPK ribbon is colored as in Figure 1. (C) Partial sequence alignment of the N-terminal regions of DMPK and ROCK1 involved in the dimer interface. (D) Partial sequence alignment of the C-terminal regions of DMPK and ROCK1 involved in the dimer interface.
The dimeric state of DMPK was confirmed by size-exclusion chromatography (data not shown) and by analytical ultracentrifugation (AUC). The data obtained from AUC sedimentation velocity experiments identified a mixture of monomer and dimer present in solution (see Fig. 4). From a monomer-dimer model used to fit sedimentation equilibrium data, a dissociation constant, Kd, in the range 1–5 μM was calculated (see Fig. 4). It is therefore likely that under physiological concentrations of DMPK the coiled-coil region of DMPK contributes and is required for effective dimerisation. The surface area of interaction of the DMPK dimer is 1266 Å2, while that of ROCK1 is 2115 Å2 (calculated by the MSD-PISA server: http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html). Some of this difference may be accounted for by the N-terminal truncation of the DMPK construct; in the ROCK structures there is an additional N-terminal α-helix that contributes to the dimerization interface. Therefore, our measurement of the dimerization affinity of the kinase domain may be an underestimate.
Figure 4.
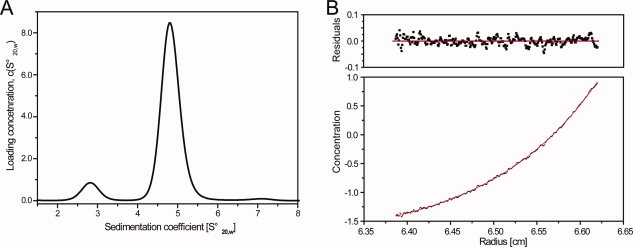
Self association of DMPK studied by analytical ultracentrifugation. (A) Sedimentation velocity experiment, the concentration of DMPK was 25 μM. At that concentration the protein was mainly populated as a dimer as indicated by the peak at 4.8 Swedberg units. The molecular weights determined from the velocity data were ∼48 and ∼100 kDa, respectively, consistent with the expected molecular weight of a DMPK monomer and dimer. (B) Sedimentation equilibrium experiment of DMPK. The upper panel shows residuals to a nonlinear least-squares fit to a monomer–dimer model, shown as a solid line. The determined association constant was in the range 1–5 μM.
A number of residues involved in the dimer interface are strictly conserved between DMPK and the ROCKs (see Fig. 3). Most of the conserved residues are hydrophobic, but of particular interest is the conserved Asp27, which forms a hydrogen bond to the hydrophobic motif Ser404. This may be a method of stabilization of the hydrophobic motif in this family, instead of the phosphorylation observed in other families of AGC kinase. Asp27 is conserved in the ROCKs (and the same interaction is seen in the ROCK structures), and in MRKCα and MRCKβ. Interestingly however, it is not conserved in MRCKγ or in citron kinase (CRIK). MRCKγ does have an adjacent glutamate (Glu15), C-terminal to the conserved position, which may perform a similar function.
Conserved binding of the C-terminal tail to the N-terminal lobe - the hydrophobic motif
The dimerization interface is made possible by the binding of the C-terminal tail of the kinase domain to a groove in the N-terminal lobe (see Fig. 3). This binding arrangement is a general feature of AGC kinases,25,28 where a groove is created in the N-terminal lobe by insertion of a small α-helix, which causes a separation of the αC helix and the β4 strand. The C-terminus of the protein wraps around the N-terminal lobe to allow the binding of the hydrophobic motif on the C-terminal tail between αC and β4. This binding has occurred despite the lack of phosphorylation at the hydrophobic motif, with Asp27 from the N-terminal part of the protein involved in the dimer interface providing a hydrogen bond partner for the hydrophobic motif serine. In the recent crystal structure of inactive serum and glucocorticoid-regulated kinase 1 (SGK1), also with an un-phosphorylated hydrophobic motif, the C-terminal tail does not bind the N-lobe and is disordered.29 In the AGC kinase family, there are a number of variations to the hydrophobic motif that do not have a phosphorylation site, for example FXXFN as in STK32A,B,C (YANK1-3), FXXFD as in PKN1-3, FXXFE as in PKCι and PKCζ, or FXXF-COOH as in PRKG1,2 and PRKX and PRKY. In some of these the variation may act as a phosphomimetic (as seen in the structure of PKCι30) while in others such as the YANKs an alternative interaction may occur.
Active conformation despite lack of phosphorylation
Mass spectrometry of the recombinant DMPK confirmed that the crystallized protein was unphosphorylated. However, the activation loop occupies a well-ordered conformation that does not impede access to the nucleotide or substrate binding sites (see Fig. 5). This loop is in the same conformation in both independent molecules in the structure and resembles an active conformation. As in the ROCK structures, an extension in the length of the activation loop relative to that in PKA allows a small antiparallel β-sheet to be formed from a part of the activation loop, and a part of the αEF/αF loop, which is also extended in DMPK and ROCK1. In DMPK, the αEF/αF loop is extended by a further three residues compared with that of ROCK1. The conservation of these structural features between DMPK and ROCK1 confirms that the interactions between the activation loop and the αEF/αF loop are important in stabilizing the activation loop in this active conformation. The active conformation of the activation segment is additionally stabilized by a hydrogen bond formed by the catalytic loop (HRD) arginine (Arg194) with the backbone carbonyl of Ser216 located C-terminally to the DFG motif. The glycine-rich loop was ordered due to the presence of the BIM-8 inhibitor but high temperature factors in particular in the tip of the loop suggest that this structural element is still quite flexible.
Figure 5.
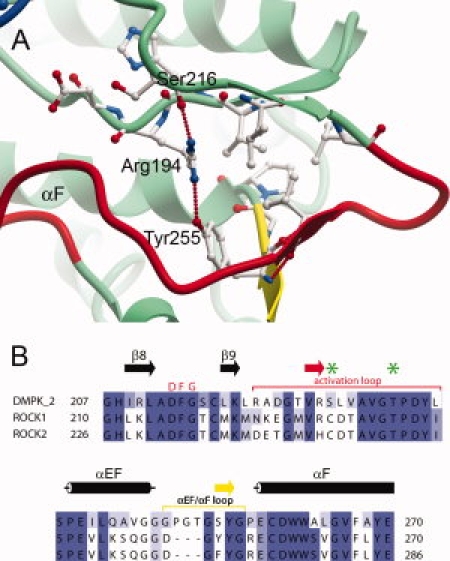
(A) Activation segment of DMPK, coloured as in Figure 1. (B) Partial sequence alignment of the activation loop and αEF/αF loop regions of DMPK against the Rho kinases 1 and 2, which are colored as in (A). The residues equivalent to those phosphorylated in MRCK are indicated with green stars. A full sequence alignment of the DMPK subfamily can be seen as Supplementary Figure 1.
The catalytic aspartate of the HRD motif (Asp195) is in a suitable position for catalysis, and forms a hydrogen bond to Asn200. Also important for catalysis is the position of helix αC in close proximity to the active site. This helix is completely ordered in the structure, unlike the structures of some inactive kinases, and the salt bridge between αC and β3 that is an indicator of an active kinase conformation is present (residues Glu119 and Lys100 in DMPK).
The combined observations of the dimerisation interface that stabilizes the bound, unphosphorylated, hydrophobic motif, and the active conformation of the unphosphorylated activation loop suggest that DMPK does not require phosphorylation for activation. This has also been proposed for the ROCKs.23
Although DMPK is active without phosphorylation, there is still the question of what the structural impact of phosphorylation at the conserved phosphorylation sites would be, if indeed they do get phosphorylated. With regard to potential kinases that phosphorylate DMPK, a known example is Raf-1 kinase which can bind to DMPK and also phosphorylate it, stimulating both its auto-phosphorylation and trans-phosphorylation activity.31 It is unknown which sites on DMPK are phosphorylated in this activation. DMPK does not have the typical basic residues at the start of αC and on β9 to bind a phosphorylation at the activation loop position of Ser228. The arginine of the HRD motif that would bind such a phosphorylation is present, but taken together with the activated position of the activation loop in the absence of phosphorylation, this suggests that phosphorylation of the activation loop may not increase the activity of DMPK.
Phosphorylation of the hydrophobic motif Ser404 might increase DMPK activity; DMPK has a glutamine residue on β4 (Gln139) equivalent to the glutamine in PKCθ that binds a phosphoserine at the hydrophobic motif. However, the conformation of Gln139 is same in the non-phosphorylated DMPK as in the phosphorylated PKCθ.
Although Raf-1 is known to bind DMPK, it is unknown exactly where on DMPK this binding takes place although it is on the kinase domain, in contrast to the binding and increase in activation of DMPK with constitutively active Rac1,31 which is likely due to GTPase binding the coiled-coil domain, as is the case with ROCK1 and RhoA.32 This interaction also explains the activity increase in response to the G-protein activator GTPγS.33 It is unknown whether there are any negative modulators of DMPK corresponding to the interaction of RhoE/Rnd3 with the kinase domain of ROCK1.34
The BIM-8 inhibitor occupies the ATP binding site
The ATP-competitive inhibitor BIM-8 (bisindolylmaleimide VIII) occupies the DMPK ATP binding site (see Fig. 6). The inhibitor forms hydrogen bonds to the backbone of residues Glu149 and Tyr151 in a typical ATP mimetic binding mode, while the hydrophilic primary amine group is directed towards the solvent space, forming a hydrogen bond with the side-chain of Asp213, which would be expected to coordinate Mg2+ in a structure with bound ATP. Hydrophobic interactions on both faces of the inhibitor provide the majority of the binding interactions. Key interactions are with Ile77 and Val85 on the ‘top’ face of the inhibitor, and Leu202 on the “bottom” face.
Figure 6.
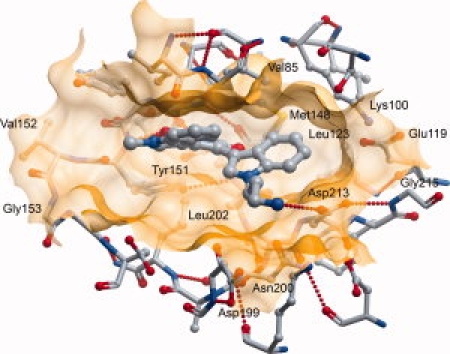
The active site of DMPK with inhibitor BIM-8 bound.
The inhibitor BIM-8 is bulkier than any of the inhibitors that were shown bound to ROCK1,22 and it also binds deeper in the ATP binding site. The extra depth in the ATP binding site is made possible by the smaller size of DMPK1 Leu123 compared with the equivalent Met128 in ROCK1. This allows a different conformation of DMPK1A Met148 compared with the equivalent Met153 in ROCK1, in which the side-chain of Met148 points away from the ATP binding site. Another difference which increases the volume of the ATP binding site comes from the conserved Lys100 and Glu119, which form a salt bridge as in activated structures, and are both displaced away from the active site by more than 1 Å compared to their positions in the ROCK1: inhibitor complexes.
The BIM-8 compound has also been cocrystallized with PDK1.35 In both structures, BIM-8 has essentially the same orientation. The only difference is in the position of the primary amine. In PDK1 this moiety forms a hydrogen bond with Glu166; the equivalent residue in DMPK is Asp155, which is not long enough to reach the inhibitor decoration. Instead, in DMPK, the primary amine group forms a hydrogen bond with the side-chain of Asp213. Eleven other kinases, out of sixty that were analyzed for ligand binding by a thermal shift assay, showed a ΔTm of greater than 5°C and, therefore, a significant interaction with BIM-8.36
Materials and Methods
Cloning
DNA for DMPK1 isoform two residues Gln11 to Pro420 was amplified by PCR from template DNA in the Mammalian Gene Collection (IMAGE Consortium Clone ID 6480902). The PCR product was incorporated into a homemade vector containing an N-terminal hexahistidine tag and TEV protease tag cleavage site (sequence MHHHHHHSSGVDLGTENLYFQSM), by ligation-independent cloning. The resulting plasmid was transformed into E. coli BL21 (DE3) cells containing the pRARE2 plasmid from commercial Rosetta II (DE3) cells.
Protein purification
Cultures were grown in shaker flasks with 1 L of LB medium containing 50 μg/mL kanamycin. 2L of culture were combined for a typical purification. After inoculation, the cells were grown at 37°C to an OD600 of 0.4 when the temperature was reduced to 18°C. When the OD600 reached 0.8 protein expression was induced by addition of 1 mM isopropyl-β-D-thiogalactopyranoside. Expression was continued overnight before the cells were harvested by centrifugation. The cells were resuspended in binding buffer (50 mM Hepes pH 7.5, 500 mM NaCl, 5% Glycerol, 5 mM Imidazole, 0.5 mM TCEP and frozen at −20°C).
The resuspended cells were thawed, lysed by sonication, and the cell debris was removed by centrifugation. The DMPK1 protein was purified from the clarified cell lysate by immobilized metal ion chromatography followed by size-exclusion chromatography: The clarified lysate was passed through a drip column containing DE52 resin (pre-equilibrated in binding buffer) to bind the DNA, followed by a drip column containing Ni2+ resin. When the lysate had passed through the Ni2+ resin, the resin was washed with binding buffer, and then with binding buffer containing 30 mM, 50 mM and finally 250 mM imidazole. The DMPK1 eluted in both the 50 mM and 250 mM imidazole fractions. The eluted DMPK1 was concentrated to a volume of 8 mL using a spin concentrator and, twice, 4 mL was injected onto an S200 16/60 size-exclusion column pre-equilibrated in gel filtration buffer. The eluted fractions containing DMPK1 were pooled and TEV protease was added. The digestion was left overnight at 4°C and when the digestion had completed the TEV protease and hexahistidine tag were removed by binding to Ni2+ resin.
For crystallization the gel filtration buffer was 50 mM BisTrisPropane pH 6.5, 500 mM KCl, 0.5 mM TCEP, and subsequently the protein was exchanged into 50 mM BisTrisPropane pH 6.5, 100 mM KCl, 0.5 mM TCEP. The protein was then concentrated to 11 mg/mL in a spin concentrator.
Crystallization and structure solution
Crystals were grown at 4°C using the sitting drop vapor diffusion method. The BIM-8 inhibitor was added to the protein solution (11 mg/mL in 50 mM BisTrisPropane pH 6.5, 100 mM KCl, 0.5 mM TCEP) to a concentration of 0.5 mM. This protein:inhibitor solution was mixed in a 3:1 ratio with the reservoir solution (1 M (NH4)2SO4, 1.5% PEG3350, 0.1M Bis-Tris pH 7.0) in a total drop size of 300 nL and equilibrated against reservoir solution. Crystals appeared after a few weeks.
Crystals were cryoprotected by transfer into reservoir solution diluted with ethylene glycol to a final ethylene glycol concentration of 25%, and then flash-frozen in liquid nitrogen. X-ray data was collected at the Swiss Light Source beamline X10 using a cryostream to maintain the crystal at 100 K.
The data was processed with MOSFLM37 and the CCP4 suite.38 The structure was solved by molecular replacement using PHASER39 and the structure of the ROCK1 kinase domain minus dimerisation regions (PDB: 2ESM) as a search model. There were two molecules in the asymmetric unit of a P3221 unit cell. The model was rebuilt using COOT40 and refined using REFMAC5.41 Data collection and refinement statistics are in Table I.
Table 1.
Data Collection and Refinement Statistics
| Data collection | |
|---|---|
| Space group | P3221 |
| Unit cell [a, b, c (Å)] | 122.6, 122.6, 134.3 |
| Resolution rangea (Å) | 45.27–2.80 (2.95.2.80) |
| No. of unique observationsa | 29,215 (4185) |
| No. of total observationsa | 163,620 (23,719) |
| Completenessa (%) | 100.0 (100.0) |
| Multiplicitya | 5.6 (5.7) |
| Rmergea (%) | 10.0 (85.1) |
| 〈I/σ(I)〉a | 16.7 (3.0) |
| Refinement | |
| R factor (%) | 19.6 |
| Rfree (%) | 24.3 |
| Rmsd bond length (Å) (angle (°)) | 0.010 (1.3) |
| PDB ID | 2VD5 |
Values in parentheses are for the highest resolution shell.
Analytical ultracentrifugation
Samples were studied in a solution of 25 mM HEPES pH 7.5, 150 mM NaCl, 0.5 mM TCEP, at a temperature of 8°C. Sedimentation of particles was monitored using the interference optics in a Beckman XL-I Analytical Ultracentrifuge equipped with a Ti-50 rotor. Sedimentation velocity experiments were carried out on samples at concentrations of 8, 16, and 25 μM, in two-sector cells, employing a rotor speed of 50,000 rpm Radial scans were collected in 30-s intervals. Data were analyzed using SEDFIT42 to calculate c(s) distributions. The software package SEDNTERP was used in order to normalize the obtained sedimentation coefficient values to the corresponding values in water at 20°C,  . Sedimentation equilibrium experiments were performed at 8°C and a protein concentration of 8, 14, and 25 μM. The dissociation constant was calculated from global fitting of all data to a monomer–dimer equilibrium model, using HETEROANALYSIS (University of Connecticut Bioservices Center). The data was also analyzed using ULTRASPIN (MRC Centre for Protein Engineering) for comparison, resulting in a similar dissociation constant.
. Sedimentation equilibrium experiments were performed at 8°C and a protein concentration of 8, 14, and 25 μM. The dissociation constant was calculated from global fitting of all data to a monomer–dimer equilibrium model, using HETEROANALYSIS (University of Connecticut Bioservices Center). The data was also analyzed using ULTRASPIN (MRC Centre for Protein Engineering) for comparison, resulting in a similar dissociation constant.
Conclusions
The BIM compounds were originally designed to be more effective, and also more selective, versions of the general kinase inhibitor staurosporine, as well as providing access to a range of synthetically accessible derivatives of the basic scaffold and allowing flexibility between the indole rings. For many protein kinases it is desirable to be able to have specific inhibitors, however for DMPK it seems more likely that avoidance of inhibition is the aim. Nevertheless, inhibitor design for drug targets in the AGC family of kinases is aided by additional structural knowledge of this family. There is still much to learn about the role of the AGC C-terminus, especially with regard to the NFD motif (which Kannan et al. called the active-site tether, AST25) and its potential role in nucleotide binding or release.
The structure of DMPK illustrates that the most structurally varied part of AGC kinase proteins is the long C-terminus; compared with the structure of the closely related ROCKs, this is the only region of substantial structural divergence. Furthermore, the difference in the location of the conserved C-terminal NFD motif, adjacent to the active site in the ROCKs and away from it in DMPK, can be compared with the similarity in position of the only partially conserved C-terminal turn motif. In designing specific inhibitors for AGC family kinases, it may be necessary to incorporate the C-terminus into the design. Inhibitors that can occupy the ATP-binding site while also offering moieties capable of binding divergent motifs on the C-terminal extension may be a worthwhile future direction.
Coordinates
Coordinates and structure factors have been deposited in the Protein Data Bank with the accession code 2VD5.
Acknowledgments
The authors thank Frank von Delft and members of the SGC crystallography group for assistance with X-ray data collection. This work is based on diffraction data collected at beamline PXII of the Swiss light source SLS, Paul Scherrer Institute, Villigen, Switzerland.
Glossary
Abbreviations:
- DMPK
Dystrophia myotonica protein kinase
- ROCK
Rho-associated kinase.
References
- 1.Ranum LPW, Day JW. Myotonic dystrophy: RNA Pathogenesis Comes into Focus. Am J Hum Genet. 2004;74:793–804. doi: 10.1086/383590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Day JW, Ranum LPW. RNA pathogenesis of the myotonic dystrophies. Neuromuscular Disord. 2005;15:5–16. doi: 10.1016/j.nmd.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 3.Wansink DG, Wieringa B. Transgenic mouse models for myotonic dystrophy type 1 (DM1) Cytogenet Genome Res. 2003;100:230–242. doi: 10.1159/000072859. [DOI] [PubMed] [Google Scholar]
- 4.Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LPW. Myotonic Dystrophy Type 2 Caused by a CCTG Expansion in Intron 1 of ZNF9. Science. 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 5.Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D, Krym M, Thornton CA. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 2000;289:1769–1773. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- 6.Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, Swanson MS. A Muscleblind Knockout Model for Myotonic Dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- 7.Miller JW, Urbinati CR, Teng-umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS. Recruitment of human muscleblind proteins to (CUG)n expansions associated with myotonic dystrophy. EMBO J. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fardaei M, Rogers MT, Thorpe HM, Larkin K, Hamshere MG, Harper PS, Brook JD. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum Mol Genet. 2002;11:805–814. doi: 10.1093/hmg/11.7.805. [DOI] [PubMed] [Google Scholar]
- 9.Orengo JP, Chambon P, Metzger D, Mosier DR, Snipes GJ, Cooper TA. Expanded CTG repeats within the DMPK 3' UTR causes severe skeletal muscle wasting in an inducible mouse model for myotonic dystrophy. PNAS. 2008;105:2646–2651. doi: 10.1073/pnas.0708519105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guiraud-Dogan C, Huguet A, Gomes-Pereira M, Brisson E, Bassez G, Junien C, Gourdon G. DM1 CTG expansions affect insulin receptor isoforms expression in various tissues of transgenic mice. Biochim Biophys Acta. 2007;1772:1183–1191. doi: 10.1016/j.bbadis.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 11.Jansen G, Groenen PJTA, Bächner D, Jap PHK, Coerwinkel M, Oerlemans F, van den Broek W, Gohlsch B, Pette D, Plomp JJ, Molenaar PC, Nederhoff MGJ, van Echteld CJA, Dekker M, Berns A, Hameister H, Wieringa B. Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat Genet. 1996;13:316–324. doi: 10.1038/ng0796-316. [DOI] [PubMed] [Google Scholar]
- 12.Reddy S, Smith DBJ, Rich MM, Leferovich JM, Reilly P, Davis BM, Tran K, Rayburn H, Bronson R, Cros D, Balice-Gordon RJ, Housman D. Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat Genet. 1996;13:325–335. doi: 10.1038/ng0796-325. [DOI] [PubMed] [Google Scholar]
- 13.Berul CI, Maguire CT, Aronovitz MJ, Greenwood J, Miller C, Gehrmann J, Housman D, Mendelsohn ME, Reddy S. DMPK dosage alterations result in atrioventricular conduction abnormalities in a mouse myotonic dystrophy model. J Clin Invest. 1999;103:R1–R7. doi: 10.1172/JCI5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Llagostera E, Catalucci D, Marti L, Liesa M, Camps M, Ciaraldi TP, Kondo R, Reddy S, Dillmann WH, Palacín M, Zorzano A, Ruiz-Lozano P, Gomis R, Kaliman P. Role of myotonic dystrophy protein kinase (DMPK) in glucose homeostasis and muscle insulin action. PLo S ONE. 2007;2:e1134. doi: 10.1371/journal.pone.0001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mounsey JP, John JE, III, Helmke SM, Bush EW, Gilbert J, Roses AD, Perryman MB, Jones LR, Moorman JR. Phospholemman is a substrate for myotonic dystrophy protein kinase. J Biol Chem. 2000;275:23362–23367. doi: 10.1074/jbc.M000899200. [DOI] [PubMed] [Google Scholar]
- 16.Murányi A, Zhang R, Liu F, Hirano K, Ito M, Epstein HF, Hartshorne DJ. Myotonic dystrophy protein kinase phosphorylates the myosin phosphatase targetting subunit and inhibits myosin phosphatase activity. FEBS Lett. 2001;493:80–84. doi: 10.1016/s0014-5793(01)02283-9. [DOI] [PubMed] [Google Scholar]
- 17.Kaliman P, Catalucci D, Lam JT, Kondo R, Gutiérrez JCP, Reddy S, Palacín M, Zorzano A, Chien KR, Ruiz-Lozano P. Myotonic dystrophy protein kinase phosphorylates phospholamban and regulates calcium uptake in cardiomyocyte sarcoplasmic reticulum. J Biol Chem. 2005;280:8016–8021. doi: 10.1074/jbc.M412845200. [DOI] [PubMed] [Google Scholar]
- 18.Groenen PJTA, Wansink DG, Coerwinkel MM, van den Broek W, Jansen G, Wieringa B. Constitutive and regulated modes of splicing produce six major myotonic dystrophy protein kinase (DMPK) isoforms with distinct properties. Hum Mol Genet. 2000;9:605–616. doi: 10.1093/hmg/9.4.605. [DOI] [PubMed] [Google Scholar]
- 19.Garcia P, Ucurum Z, Bucher R, Svergun DI, Huber T, Lustig A, Konarev PV, Marino M, Mayans O. Molecular insights into the self-assembly mechanism of dystrophia myotonica kinase. FASEB J. 2006;20:1142–1151. doi: 10.1096/fj.05-5262com. [DOI] [PubMed] [Google Scholar]
- 20.Tan I, Seow KT, Lim L, Leung T. Intermolecular and intramolecular interactions regulate catalytic activity of myotonic dystrophy kinase-related cdc42-binding kinase α. Mol Cell Biol. 2001;21:2767–2778. doi: 10.1128/MCB.21.8.2767-2778.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doran JD, Liu X, Taslimi P, Saadat A, Fox T. New insights into the structure-function relationships of Rho-associated kinase: a thermodynamic and hydrodynamic study of the dimer-to-monomer transition and its kinetic implications. Biochem J. 2004;384:255–262. doi: 10.1042/BJ20040344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacobs M, Hayakawa K, Swenson L, Bellon S, Fleming M, Taslimi P, Doran J. The structure of dimeric ROCK1 reveals the mechanism for ligand selectivity. J Biol Chem. 2006;281:260–268. doi: 10.1074/jbc.M508847200. [DOI] [PubMed] [Google Scholar]
- 23.Yamaguchi H, Kasa M, Amano M, Kaibuchi K, Hakoshima T. Molecular mechanism for the regulation of rho-kinase by dimerization and its inhibition by fasudil. Structure. 2006;14:589–600. doi: 10.1016/j.str.2005.11.024. [DOI] [PubMed] [Google Scholar]
- 24.Wansink DG, van Herpen REMA, Coerwinkel-Driessen MM, Groenen PJTA, Hemmings BA, Wieringa B. Alternative splicing controls myotonic dystrophy protein kinase structure, enzymatic activity, and subcellular localization. Mol Cell Biol. 2003;23:5489–5501. doi: 10.1128/MCB.23.16.5489-5501.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kannan N, Haste N, Taylor SS, Neuwald AF. The hallmark of AGC kinase functional divergence is its C-terminal tail, a cis-acting regulatory module. PNAS. 2007;104:1272–1277. doi: 10.1073/pnas.0610251104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Narayana N, Cox S, Xuong N-H, Ten Eyck LF, Taylor SS. A binary complex of the catalytix subunit of cAMP-dependent protein kinase and adenosine further defines conformational flexibility. Structure. 1997;5:921–935. doi: 10.1016/s0969-2126(97)00246-3. [DOI] [PubMed] [Google Scholar]
- 27.Xu Z-B, Chaudhary D, Olland S, Wolfrom S, Czerwinski R, Malakian K, Lin L, Stahl ML, Joseph-McCarthy D, Benander C, Fitz L, Greco R, Somers WS, Mosyak L. Catalytic domain crystal structure of protein kinase C-θ (PKCθ) J Biol Chem. 2004;279:50401–50409. doi: 10.1074/jbc.M409216200. [DOI] [PubMed] [Google Scholar]
- 28.Gold MG, Barford D, Komander D. Lining the pockets of kinases and phosphatases. Curr Opin Struct Biol. 2006;16:693–701. doi: 10.1016/j.sbi.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 29.Zhao B, Lehr R, Smallwood AM, Ho TF, Maley K, Randall T, Head MS, Koretke KK, Schnackenberg CG. Crystal structure of the kinase domain of serum and glucocorticoid-regulated kinase 1 in complex with AMP-PNP. Prot Sci. 2007;16:1–9. doi: 10.1110/ps.073161707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Messerschmidt A, Macieira S, Velarde M, Bädeker M, Benda C, Jestel A, Brandstetter H, Neuefeind T, Blaesse M. Crystal structure of the catalytic domain of human atypical protein kinase C-iota reveals interaction mode of phosphorylation site in turn motif. J Mol Biol. 2005;352:918–931. doi: 10.1016/j.jmb.2005.07.060. [DOI] [PubMed] [Google Scholar]
- 31.Shimizu M, Wang W, Walch ET, Dunne PW, Epstein HF. Rac-1 and Raf-1 kinases, components of distinct signaling pathways, activate myotonic dystrophy protein kinase. FEBS Lett. 2000;475:273–277. doi: 10.1016/s0014-5793(00)01692-6. [DOI] [PubMed] [Google Scholar]
- 32.Dvorsky R, Blumenstein L, Vetter IR, Ahmadian MR. Structural insights into the interaction of ROCK1 with the switch regions of RhoA. J Biol Chem. 2004;279:7098–7104. doi: 10.1074/jbc.M311911200. [DOI] [PubMed] [Google Scholar]
- 33.Bush EW, Helmke SM, Birnbaum RA, Perryman MB. Myotonic dystrophy protein kinase domains mediate localization, oligomerization, novel catalytic activity, and autoinhibition. Biochemistry. 2000;39:8480–8490. doi: 10.1021/bi992142f. [DOI] [PubMed] [Google Scholar]
- 34.Komander D, Garg R, Wan PTC, Ridley AJ, Barford D. Mechanism of multi-site phosphorylation from a ROCK1:RhoE complex structure. EMBO J. 2008;27:3175–3185. doi: 10.1038/emboj.2008.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Komander D, Kular GS, Schüttelkopf AW, Deak M, Prakash KRC, Bain J, Elliott M, Garrido-Franco M, Kozikowski AP, Alessi DR, van Aalten DMF. Interactions of LY333531 and other bisindolyl maleimide inhibitors with PDK1. Structure. 2004;12:215–226. doi: 10.1016/j.str.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 36.Fedorov O, Marsden B, Pogacic V, Müller S, Bullock AN, Schwaller J, Sundström M, Knapp S. A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. PNAS. 2007;104:20523–20528. doi: 10.1073/pnas.0708800104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leslie AGW. Integration of macromolecular diffraction data. Acta Crystallogr. 1999;D55:1696–1702. doi: 10.1107/s090744499900846x. [DOI] [PubMed] [Google Scholar]
- 38.The CCP4 suite: programs for protein crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. Collaborative Computational Project Number 4. [DOI] [PubMed] [Google Scholar]
- 39.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr. 2004;D60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 40.Emsley P, Cowtan K. COOT: model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 41.Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr. 1999;D55:247–255. doi: 10.1107/S090744499801405X. [DOI] [PubMed] [Google Scholar]
- 42.Brown PH, Schuck P. Macromolecular size-and-shape distributions by sedimentation velocity analytical ultracentrifugation. Biophys J. 2006;90:4651–4661. doi: 10.1529/biophysj.106.081372. [DOI] [PMC free article] [PubMed] [Google Scholar]