

Abstract
In the brain, the human flavoprotein d-amino acid oxidase (hDAAO) is involved in the degradation of the gliotransmitter d-serine, an important modulator of NMDA-receptor-mediated neurotransmission; an increase in hDAAO activity (that yields a decrease in d-serine concentration) was recently proposed to be among the molecular mechanisms leading to the onset of schizophrenia susceptibility. This human flavoenzyme is a stable homodimer (even in the apoprotein form) that distinguishes from known d-amino acid oxidases because it shows the weakest interaction with the flavin cofactor in the free form. Instead, cofactor binding is significantly tighter in the presence of an active site ligand. In order to understand how hDAAO activity is modulated, we investigated the FAD binding process to the apoprotein moiety and compared the folding and stability properties of the holoenzyme and the apoprotein forms. The apoprotein of hDAAO can be distinguished from the holoenzyme form by the more “open” tertiary structure, higher protein fluorescence, larger exposure of hydrophobic surfaces, and higher sensitivity to proteolysis. Interestingly, the FAD binding only slightly increases the stability of hDAAO to denaturation by urea or temperature. Taken together, these results indicate that the weak cofactor binding is not related to protein (de)stabilization or oligomerization (as instead observed for the homologous enzyme from yeast) but rather should represent a means of modulating the activity of hDAAO. We propose that the absence in vivo of an active site ligand/substrate weakens the cofactor binding, yielding the inactive apoprotein form and thus avoiding excessive d-serine degradation.
Keywords: flavoproteins, neurotransmission, FAD binding, regulation mechanism, folding-stability, conformational change, protein structure/folding, stability and mutagenesis, enzymes, correlation of structure with spectra and other properties, circular dichroism, fluorescence, kinetics, mechanism-enzymes, d-serine
Introduction
d-Amino acid oxidase (DAAO, EC 1.4.3.3) is a flavoenzyme that catalyzes the oxidative deamination of d-amino acids to the corresponding α-keto acids, hydrogen peroxide, and ammonia; for a review see Ref. 1. The physiological role of DAAO differs in different organisms and tissues. In yeasts, DAAO is involved in the metabolic utilization of d-amino acids; in mammals its function is tissue specific. Several experimental findings suggest that in brain this flavoenzyme degrades the gliotransmitter d-serine, a neuromodulator that binds the glycine modulatory site on the NMDA-receptor and that is needed (together with glutamate) to activate the receptor.2 Interestingly, genes coding for human DAAO (hDAAO), as well as its putative binding partner pLG72 (the product of the G72 primate-specific gene), have been linked to schizophrenia susceptibility.3
With the goal of contributing to a better understanding of the correlation between modulation of the enzymatic activity in this flavoprotein and its role in neurotransmission and schizophrenia susceptibility, we recently investigated the major properties of hDAAO.4 hDAAO shows the main properties of the dehydrogenase-oxidase class of flavoproteins (it reacts quickly with oxygen in the reduced state, stabilizes the anionic red semiquinone, and binds sulfite covalently) and shares these properties with both pig kidney (pkDAAO) and yeast DAAOs. Analogously to pkDAAO, hDAAO also possesses a low turnover number and follows a ternary complex (sequential) kinetic mechanism. In contrast to other known DAAOs, the human enzyme is a stable homodimer even in the apoprotein form and binding of the FAD cofactor in the absence of an active site ligand is very weak (Kd is 8 μM).4 It is noteworthy that a 20-fold tighter interaction between FAD and apoprotein is observed in the presence of a substrate analogue, such as benzoate.4 When comparing the 3D structure of human and porcine DAAOs, a structural explanation for the aforementioned differences is not obvious.5 hDAAO and pkDAAO show the same mode of monomer–monomer interaction (“head-to-head”) but a higher degree of substitution is evident at the dimer interface (30% vs. 15% for the overall substitution frequency), yielding a significantly different electrostatic surface potential.5 Concerning the interaction between the FAD cofactor and the apoprotein moiety, the only remarkable difference is represented by the conformation of the V47-L51 hydrophobic stretch on the si-side of the flavin coenzyme, showing a deviation from RMS of ∼0.9 Å when compared with pkDAAO.
We recently demonstrated that hDAAO specifically interacts with pLG72 yielding a time-dependent loss of hDAAO activity due to alteration of the tertiary structure of hDAAO, and that the cellular concentration of d-serine decreases in U87 glioblastoma cells transfected with hDAAO.6 Therefore, we proposed that a decrease in the synaptic concentration of d-serine as the result of an anomalous increase in hDAAO activity (e.g., related to pLG72 hypoexpression) might represent a molecular mechanism by which hDAAO and pLG72 are involved in schizophrenia susceptibility. The in vivo regulation of hDAAO is still unknown, as is the actual fraction of active enzyme; in fact, and because of weak cofactor binding and low affinity for d-serine (Km,app is 7.5 mM), the amount of active holoenzyme in the cell, and thus on the DAAO-induced oxidation of this gliotransmitter, might be at a low level.
In the absence of the structure of the apoprotein form of hDAAO (no crystals of this enzyme form have been obtained so far) and to clarify the function–structure relationships in this flavoprotein related to d-serine catabolism in human brain, we compared the holo- and the apoprotein of hDAAO and examined the consequences of cofactor binding on stability and on the folding process of this flavooxidase. On the basis of these results, we propose that there is a correlation between the unique characteristics of human DAAO (i.e., the weak cofactor binding and stable dimeric state) and its physiological function in brain.
Results
Spectral properties
Because of the weak interaction with the FAD cofactor, hDAAO exists as an equilibrium of holo- and apoprotein forms in solution (e.g., at a 1 mg/mL protein concentration the solution contains similar amounts of holo- and apoprotein species). This is made evident by the increase in activity observed by adding exogenous FAD to the assay solution4 and by isoelectrophoretic analyses. In fact, while the apoprotein gives a single band whose pI (6.3 ± 0.2) is very close to the theoretical value (=6.36), the holoenzyme yields three bands at pI = 6.3 ± 0.2, 5.9 ± 0.2 and 5.4 ± 0.35: only the more acidic ones show enzymatic activity in the zymogram (data not shown). As both holo- and apoprotein forms are dimers, a possible explanation is that the two additional bands observed for the holoenzyme preparation could be dimeric forms resulting from the combination of two holo-monomers (the band at more acidic pH) and from the combination of one holo- and one apoprotein monomer (the band at the intermediate value). This hypothesis is supported by the absence of post-translational modifications as indicated by mass spectrometry of holoenzyme and apoprotein forms that both yield a peak of ∼39,476 Da (theoretical value is 39,474 Da). For the sake of clarity, the hDAAO solution at the end of the purification was named “holo-apo” solution (because it contains both protein forms) and that to which 40 μM FAD was added was named “holo” (because the holoenzyme represents >95% of the protein).
The visible absorbance spectra of the holo- and apoprotein forms of recombinant hDAAO show evident differences because the flavin cofactor has been eliminated in the latter;4 however, some other spectral properties related to protein conformation also distinguish the two hDAAO forms. Tryptophan fluorescence (following excitation at 280 nm or at 298 nm) shows an emission maximum at 332 nm for holo- and at 338 nm for apo-hDAAO, respectively. Emission intensity is five-fold higher in the hDAAO apoprotein than in the holoprotein [Fig. 1(A)], indicating a different relevance of quenching interactions between tryptophans and surrounding side chains in the two protein forms. Interestingly, the structure of hDAAO holoenzyme shows the close proximity of Trp52 and Trp107, as well as of Trp185 and Trp247 at the interface between the flavin- and substrate-binding domains.5 For both hDAAO species, unfolding by urea is accompanied by a shift in the emission maximum (at 347 and 355 nm for holo- and apoprotein, respectively) and by an increase in the intensity of tryptophan fluorescence (see later).
Figure 1.
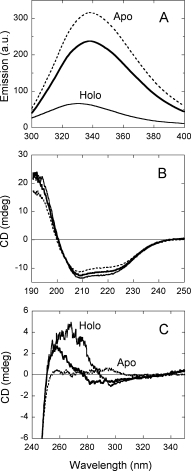
Comparison of spectral properties of holoenzyme in the absence (holo-apo, –––) and in the presence (holo, –––) of 40 μM FAD, and apoprotein (- - - -) of hDAAO. (A) Protein fluorescence (excitation at 280 nm, 0.1 mg protein/mL). (B) Far-UV CD spectrum (0.1 mg protein/mL). (C) Near-UV CD spectrum (0.4 mg protein/mL). Proteins were in 50 mM potassium pyrophosphate, pH 8.0, 5% glycerol; measurements were performed at 15°C.
The far-UV CD spectrum of hDAAO apoprotein is similar to that of the holoprotein [Fig. 1(B)]: estimation of the protein secondary structure by means of K2D2 software7 indicates that the α-helix content is 28 and 37% in apo- and holoprotein form of hDAAO. The far-UV CD signals of both hDAAO forms are abolished at ≥7M urea (see later). Differences are most evident in the near-UV CD spectra of the two protein forms [Fig. 1(C)], pointing to an alteration of the tertiary structure in the apoprotein. Interestingly, the spectrum of holo-apo hDAAO is not the simple combination of the ones determined for the holo- and the apoprotein forms, pointing to conformational changes which are apparent only when the enzyme is fully saturated by the FAD cofactor. In fact, the increase in secondary structure following FAD binding (as shown by the far-UV CD spectrum) could affect the exposition of tryptophan residues and thus also explain the change in protein fluorescence and near-UV CD spectra [Fig. 1(A,C)].
The hydrophobic fluorescent probe ANS was used to investigate the exposure of hydrophobic regions. Binding of this probe to hDAAO holoprotein results in a marked increase in ANS fluorescence and in a blue shift in its emission fluorescence maximum (not shown). From the plot of the emission change at 495 nm versus ANS concentration, a Kd value of 110 ± 10 μM and 90 ± 10 μM is determined for the holo-apo and the holoenzyme, respectively (the ΔEmax is ∼50 in the absence of exogenous FAD). At a saturating and comparable probe concentration, ANS fluorescence intensity is two-fold higher in the apo- than in the holoprotein, showing a much greater red shift in the holoprotein (maximum at 510 and 492 nm for holo- and apoprotein of hDAAO); a higher affinity for ANS binding to the apoprotein is evident (Kd = 65 ± 4 μM). The ΔEmax/Kd ratio is three-fold higher in apo- than in holoprotein of hDAAO, indicating that larger hydrophobic surfaces are exposed in the apoprotein form under native conditions. These regions could be to some extent attributed to the unoccupied flavin binding site which is most likely partially solvent accessible in the apoprotein form, as also suggested by the observed competition between ANS and FAD for binding to pig DAAO.8
Equilibrium unfolding studies
At first, the intrinsic fluorescence of tryptophan residues (a sensitive marker of protein conformation and of changes in hydrophobic regions) was used to monitor protein unfolding. At increasing urea concentration, the apoprotein of hDAAO shows an increase in emission intensity indicative of a two-step, three-state process [Fig. 2(A)]. In contrast, the same analysis performed on the holoprotein form suggests a simple two-state transition that approximately corresponds to the first change observed for the apoprotein [Fig. 2(A)]. In both cases, changes were complete at 7M urea: Cm values are reported in Table I. Addition of urea also caused the emission maximum of tryptophan fluorescence of hDAAO to shift from ∼332 nm to 347 nm (holoprotein) and from 338 nm to 355 nm (apoprotein) at >6M urea [Fig. 2(B)]: the fluorescence red shift stems from the transfer of tryptophan side chains to a more polar environment upon protein unfolding.9 It is noteworthy that the first change in fluorescence intensity is observed at lower denaturant concentration than the wavelength shift for both hDAAO forms [Fig. 2(A,B) and Table I]. Because of the large amount of free FAD in solution, flavin fluorescence could not be used as a probe to detect changes in holoprotein conformation; in fact, treatment with increasing urea concentrations causes the fluorescence intensity to rise linearly, similarly to that observed for free flavin alone (not shown).
Figure 2.
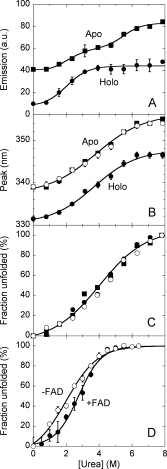
Equilibrium denaturation curves of holoenzyme in the absence (open circles) and in the presence (filled circles) of 40 μM FAD and apoprotein (squares) of hDAAO as detected by using different techniques. (A,B) Tryptophan fluorescence (excitation at 280 nm, 0.02 mg protein/mL): the fraction of unfolded hDAAO was determined from the fluorescence intensity at ∼340 nm using the value for the untreated and 8M urea-treated proteins as reference, (A); position of the emission peak maximum, (B). (C) Far-UV CD measurements at 220 nm (proteins were 0.1 mg/mL). (D) Activity of hDAAO holoenzyme as a function of urea concentration following incubation with the denaturant for 60 min at 15°C. Lines represent the best fit obtained using a two-state denaturation model with the only exception of changes in fluorescence intensity for the apoprotein (panel A), for which a three-state model was used. The reported values have been corrected for readings prior to protein addition.
Table I.
Comparison of Melting Concentration Values (Cm) for Urea-Induced Unfolding and of Melting Temperature Values (Tm) as Determined by Different Approaches on Holo and Apoprotein Forms of hDAAO
| Holo | Holo-apo | Apoprotein | |
|---|---|---|---|
| Urea-induced unfolding |
Cm (M) |
||
| Protein fluorescence: intensity | 2.0 ± 0.2 | — | First: 2.4 ± 0.1 |
| (2.8 ± 0.1)a | Second: 5.6 ± 0.3 | ||
| Wavelength maximum | 3.7 ± 0.2 | — | 3.8 ± 0.2 |
| Far-UV CD (220 nm) | 4.2 ± 0.2 | — | 3.9 ± 0.3 |
| Temperature-induced unfolding |
Tm (°C) |
||
| Trp fluorescence^ | 57.7 ± 0.3 | 52.4 ± 1.2 | 48.6 ± 1.0 |
| FAD fluorescence | — | 53.5 ± 1.1 | — |
| Far-UV CD (220 nm) | 57.0 ± 1.0 | 57.0 ± 1.0 | 51.0 ± 1.0 |
Protein samples were in 20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 5% glycerol and 5 mM 2-mercaptoethanol or, only for CD measurements, in 50 mM sodium pyrophosphate, pH 8.0, 5% glycerol, at the following concentrations: fluorescence measurements, 0.02 mg protein/mL; far-UV CD and temperature ramp experiments, 0.1 mg protein/mL. Temperature ramp experiments were performed at an identical heating rate (0.5°C/min). Tm values were obtained by calculating the first derivative of the spectroscopic signals and are uncorrected for delay effects. The values determined in the presence of 40 μM FAD are indicated as holo form.
Value determined from the loss of catalytic activity.
The urea-induced loss of secondary structure elements was monitored by following the changes in ellipticity at 220–225 nm: the CD signals of holo- and apoprotein of hDAAO show a similar sensitivity to the denaturant [Cm of 4.2 and 3.9M, respectively, see Table I and Fig. 2(C)].
The effect of urea on the activity of hDAAO was investigated by measuring the enzyme activity on holoprotein samples previously incubated for 60 min at 15°C in the presence of different urea concentrations by using the standard assay mixture. As reported in Figure 2(D), the enzyme activity of the holoenzyme starts to decline at >1M urea and is lost at 4.5M urea. The Cm value is significantly affected by the presence of free FAD in the incubation mixture: it is 1.9 and 2.8M in the absence and in the presence of 40 μM FAD, respectively (Table I).
All the unfolding data reported in Figure 2 fit excellently using a two-state mechanism of unfolding, in which only native and denatured molecules are taken into account.10 The only exception is given by the changes in fluorescence intensity of the apoprotein that shows two transitions [Fig. 2(A)]. Anyway, and because of the partial reversibility of the unfolding process (see later), data analysis according to a thermodynamic model based on equilibrium process is not feasible. In the case of the hDAAO holoenzyme a series of events occur as the urea concentration is increased: the intensity of fluorescence signals was most sensitive to the denaturant concentration (Cm = 2.0M), followed by the activity (Cm = 2.8M), and finally by the change in wavelength at maximal protein fluorescence and in far-UV CD signal (Cm = 4.0M). The same transitions (at similar urea concentrations) were observed for the apoprotein form together with a second change in protein fluorescence at a high urea concentration (Cm = 5.6M, Table I). The addition of exogenous FAD to holo-apo hDAAO only affects the urea sensitivity of the enzymatic activity to a significant extent.
The refolding of urea-denatured hDAAO apo- and holoprotein was studied by monitoring the changes following a 10-fold dilution of the urea-treated proteins in plain buffer containing a 10-fold excess of free FAD and 5% glycerol. The holoenzyme recovered (∼50% of the initial specific activity after unfolding with 4M urea, a concentration at which >90% of the initial activity is lost [Fig. 2(D)] but the activity recovery is negligible at higher denaturant concentrations. Analogously, the unfolding of hDAAO holoenzyme is for the most part reversible up to 4M urea in terms of its overall secondary and tertiary structure as inferred from protein fluorescence and far-UV CD spectra. In contrast, a partial recovery of secondary and tertiary structure is observed for the refolding of 2, 4, and 6M urea-denatured hDAAO apoprotein (not shown). The recovery of enzymatic activity for the urea-treated apoprotein represents only 5–15% of the activity retrieved for the holoprotein refolded under the same experimental conditions. Yields of holo- and apoprotein refolding are not modified by the presence of 20% glycerol as flavoprotein chemical chaperone.8 In terms of recovery of the enzymatic activity the refolding process is not modified by the presence of GroEL either; however, the protein fluorescence and the far-UV CD spectra of the reconstituted holoprotein obtained in the presence of GroEL closely resemble those of native hDAAO (e.g., the fluorescence intensity at 338 nm at the end of the reconstitution process of 6M-treated apoprotein is 238 and 165 arbitrary units in the absence and in the presence of GroEL, respectively, vs. 156 arbitrary units for the holoenzyme).
Spectroscopic studies on the thermal stability
The temperature sensitivity of specific structural features was compared in the holo- and apoprotein forms of hDAAO using temperature ramp experiments. Studies on the temperature sensitivity of tryptophan and FAD fluorescence (taken as a reporter of tertiary structure modifications) and of the CD signal at 220 nm (taken as a reporter of the change in secondary structure) show midpoint transition temperatures that are ∼6–9°C higher for the holo- than for the apoprotein of hDAAO (see Table I and Fig. 3 for protein fluorescence). Comparison of the Tm values indicates that the temperature sensitivity of secondary and tertiary structure elements is similar in the holoenzyme form.
Figure 3.
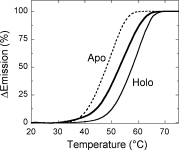
Comparison of the temperature dependence of tryptophan fluorescence spectroscopic signals for the holo- (holo-apo, in the absence –––, and holo in the presence ––– of 40 μM FAD) and apoprotein (- - - -) of hDAAO. All experiments were performed in 20 mM Tris-HCl, pH 8.0, 100 mM sodium chloride, 5% glycerol and 5 mM 2-mercaptoethanol, and at 0.1 mg/mL protein concentration. Spectral signals were monitored continuously during progressive heating from 20 to 80°C at a heating rate of 0.5°C/min and are given as percent of the total observed change regardless of the direction of the change.
The FAD-binding process
The kinetics of reconstituting the hDAAO apoprotein-FAD complex was studied under pseudo-first-order conditions and the results are reported in the Supporting Information. Briefly, the dimeric apoprotein of hDAAO reversibly binds FAD, yielding a Holo* complex in which the flavin fluorescence is largely quenched and the enzymatic activity is recovered. Subsequently, this intermediate slowly converts into the final holoenzyme, as made evident by the changes in protein fluorescence: this step requires >60 min at 15°C to complete.
The changes in conformation between apoprotein and holoenzyme of hDAAO were probed by limited proteolysis experiments using 10% (w/w) trypsin and at 25°C. SDS-PAGE analysis of the time course of holo-apo hDAAO trypsinolysis clearly shows that the 40-kDa band corresponding to the intact protein is quickly degraded [∼20% of the intact protein is present after 30 min, Fig. 4(A,B)]. The addition of a 10-fold excess of free FAD protects the enzyme from trypsin cleavage: more than 75% of the intact hDAAO is present after 30 min of incubation. Under the same experimental conditions, but in the absence of FAD, the apoprotein form of hDAAO is fully degraded in 5–10 min [Fig. 4(A,B)]. These data indicate that proteolytic sites of the apoprotein are more accessible to trypsin than in the corresponding holoenzyme form and suggest that the observed proteolysis of the holoenzyme is mainly due to the FAD dissociation. Indeed, under the same experimental conditions, the apoprotein is largely protected from proteolysis by prior incubation for 5 min with the coenzyme [see sample Apo + FAD in Fig. 4(A,B)]. This result indicates that a significant change in the apoprotein conformation is obtained within a few minutes after adding the coenzyme, a period corresponding to the time required to complete the first step in the cofactor binding process, as indicated by the flavin fluorescence studies and the full recovery of enzymatic activity [see Supporting Information Fig. 1(A)].
Figure 4.
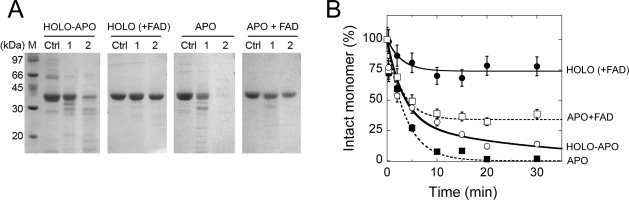
Time course of trypsin digestion of different hDAAO forms (10 μM, 0.4 mg protein/mL) with 10% (w/w) trypsin at 25°C. (A) SDS-PAGE analysis of hDAAO samples at different times: (lane Ctrl) control before trypsin addition; (lane 1) immediately after trypsin addition (∼30 s); (lane 2) 30 min after trypsin addition. (B) Time courses of the amount of the 40-kDa band (corresponding to the intact hDAAO monomer) as determined by densitometric analysis of gels such as those reported in panel A. HOLO (+ FAD) is the sample obtained following the addition of 100 μM free FAD to the holo-apo hDAAO. APO + FAD is the apoprotein sample to which 100 μM free FAD was added 5 min before adding trypsin. The intensity of the 40-kDa band of the sample before adding trypsin (sample in lane Ctrl, panel A) is indicated as 100%. Bars indicate S.E. as determined for three independent experiments.
Associative behavior
We previously reported that both holo- and apoprotein forms of hDAAO elute in size-exclusion chromatography as a single peak corresponding to a 80-kDa homodimer protein in the 0.1–10 mg protein/mL concentration range.4 Under the same conditions, the addition of 40 μM FAD in the elution buffer does not modify the elution volume of hDAAO (not shown).
The effects of urea and thiocyanate on oligomerization state and/or the molecular size of hDAAO were also analyzed by the same method. At increasing urea concentration, the holo- and apoprotein forms elute as multiple peaks at a lower retention volume, indicating an increase in molecular dimensions (higher aggregation state or increased hydrodynamic radius, see Fig. 5). The eluted peaks had retention volumes corresponding to hDAAO aggregates comprising four to eight subunits, as determined using a calibration curve obtained with standard proteins. The main difference between hDAAO holo- and apoprotein forms is the significant decrease in the total peak area in the chromatograms at a 4M urea concentration: this reduction is more evident in the presence of 40 μM free FAD in the elution buffer (to ∼30% of the area measured for the untreated enzyme, see Fig. 5). This result clearly indicates that the solubility of the holoenzyme is minimal at this urea concentration, corresponding to the condition at which an unfolding intermediate is evident for the apoprotein [see Fig. 2(A)]. When the urea concentration is increased further, a large fraction of the insoluble aggregates are converted into soluble polymeric forms, as seen by the increase in the total area of the eluted peaks. Consistent with low affinity of hDAAO for FAD, the peak at a very high elution volume (≥20 mL) contains the released flavin and its intensity is only identical using a elution buffer with or without 40 μM FAD at urea concentrations >6M (see Fig. 5). It is noteworthy that the urea-induced dissociation of dimeric hDAAO into monomers produces “adhesive” protein conformers which are prone to aggregate; this process is not due to disulfide bond formation since it was also observed in the presence of reducing agents during gel-permeation experiments.
Figure 5.
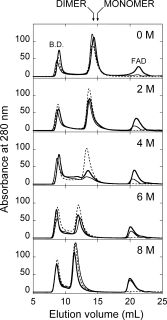
Effect of increasing urea concentrations on the molecular dimension of holo- (holo-apo, in the absence –––, and holo, in the presence ––– of 40 μM FAD) and apoprotein (- - - -) of hDAAO. Size exclusion chromatographic profile of hDAAO samples (1 mg protein/mL incubated for 60 min at 15°C in the presence of different urea concentration) on a Superdex 200 column equilibrated in 20 mM Tris-HCl buffer, pH 8.5, 150 mM sodium chloride, 5% glycerol, 5 mM 2-mercaptoethanol without (holo-apo and apoprotein samples) or with 40 μM FAD (holo sample), and containing the same urea concentration used for preincubation.
Thiocyanate is a lipophilic ion that interferes with ionic pairs located either on the protein surface or in the hydrophobic core of folded proteins: we previously reported that the dimeric holoenzyme of yeast DAAO is fully converted into its monomeric counterpart at 0.5M thiocyanate.11 The effect of NH4SCN on the quaternary structure of holoenzyme of hDAAO was investigated by means of size-exclusion chromatography on a Superdex 200 column (see Fig. 6). The elution profiles of the different hDAAO forms (at a concentration of 1 mg protein/mL) at different NH4SCN concentrations show the presence of a peak corresponding to a monomeric form only at a concentration of the lipophilic ion ≥1.5M. In any case, the area of this peak is less than 15% of the original starting material, which indicates aggregation of a large part of the protein under these conditions (see Fig. 6).
Figure 6.
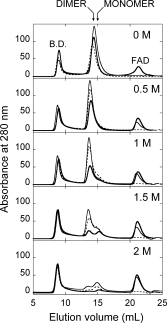
Effect of thiocyanate on the molecular dimension of holo- (holo-apo, in the absence –––, and holo, in the presence ––– of 40 μM FAD) and apoprotein (- - - -) of hDAAO. Size exclusion chromatographic profile of hDAAO samples (1 mg protein/mL incubated for 60 min at 15°C in the presence of different thiocyanate concentrations) on a Superdex 200 column equilibrated in 20 mM Tris-HCl buffer, pH 8.5, 150 mM sodium chloride, 5% glycerol, 5 mM 2-mercaptoethanol without (holo-apo and apoprotein samples) or with 40 μM FAD (holo sample), and containing the same thiocyanate concentration used for pre-incubation.
Discussion
DAAO is a catabolic enzyme (strictly specific for d-isomers of amino acids) destined for the peroxisome.1 hDAAO is a homodimer containing one molecule of weakly bound coenzyme FAD per 40 kDa protein monomer; cofactor binding is mandatory for the enzymatic activity. The human enzyme is the only member of the DAAO family which is a stable homodimer even in the apoprotein form.1 In solution, and because of the weak interaction with the FAD cofactor, hDAAO exists in equilibrium between the holoenzyme (active) form and the corresponding apoprotein (inactive) counterpart. Our results highlight differences in conformation between hDAAO forms: the CD spectra differ, the protein emission intensity is five-fold higher and more hydrophobic surfaces are exposed in the apoprotein form than in the holoenzyme. The different conformation is further demonstrated by the higher sensitivity to proteolytic cleavage of hDAAO apoprotein with respect to the holoenzyme.
A further conclusion from the present study is that binding of the flavin cofactor only slightly increases the structural stability of hDAAO towards chemical denaturation (see Table I). In contrast, a most significant effect is evident for the thermal unfolding: the Tm value for the unfolding of the apoprotein is ∼6–9°C lower than that of the holoenzyme (Table I). However, the stabilization due to FAD binding in hDAAO is lower than that observed for the homologous enzyme from yeast: Tm values are 50 and 36°C for the holo- and the apoprotein of R. gracilis DAAO, respectively (unpublished data). The urea-induced unfolding process of both holo- and apoprotein of hDAAO can be considered as a two-step (three-state) process, analogously to that reported for the yeast enzyme.12 The presence of intermediate(s) in the unfolding process is demonstrated by the changes in the protein fluorescence intensity for the apoprotein [Fig. 2(A)] and is also revealed by the different urea sensitivity of the spectral probes used (Table I). The chaotropic effect of urea disrupts the solvating water structure of hDAAO and stabilizes an expanded, fully unfolded, and soluble multimeric aggregate (see Fig. 5). Gel-permeation experiments show that at 4M urea the apoprotein produces a structural intermediate that retains part of the secondary and tertiary structure: this intermediate can be distinguished from the one obtained for the holoenzyme form by its higher solubility. Refolding activity recovery data indicate that chemical denaturation of hDAAO holoenzyme is partially reversible (50% of the initial activity is recovered starting with the refolding from 4M urea-denatured holoprotein) while the refolding of apoprotein is largely irreversible even at 2M urea (15–20% of recovery of enzymatic activity vs. 90–100% for the holoenzyme) and even in the presence of the cofactor. Protein fluorescence and far-UV CD data are indicative of structural differences between the native and the refolded and reconstituted holoprotein: even minor modifications in the conformation, such as those observed following refolding in the presence of GroEL, can prevent the full recovery of the native conformation of hDAAO.
The recombination process of hDAAO holoenzyme is represented by the sequence of steps reported in Supporting Information Eq. (1), in which a comparatively rapid binding of FAD related to attainment of the enzyme activity is followed by a slower secondary conformational change of the reconstituted holoprotein. The reconstitution process of hDAAO contrasts with that of the homologous enzymes from different sources: in the porcine enzyme a lag in the appearance of enzymatic activity was also observed but the secondary conformational change was required to acquire enzymatic activity.13 Limited proteolysis data demonstrate that FAD binding to the hDAAO apoprotein quickly produces a compact conformation similar to that of the holoenzyme. Since the apoprotein of hDAAO is also present in solution as a dimer, as the holoenzyme is, we can rule out a dimerization step in the recombination process. Instead, previous investigations on yeast DAAO, whose apoprotein is monomeric while the holoenzyme is dimeric and whose mode of monomer-monomer interaction is different than that of mammalian DAAOs,1 showed that the dimerization step is involved in the kinetics of forming the intermediate observed in the reconstitution process14 and that monomerization, obtained by site-directed mutagenenesis or thiocyanate treatment, destabilizes the holoenzyme (of ∼6°C).11,12
Elucidation of the interaction between hDAAO apoprotein and FAD and its effect on the catalytic and stability properties of this flavoprotein represents an important issue even from a physiological and pathological point of view. In fact, hDAAO is involved in the catabolism of the gliotransmitter d-serine, an allosteric activator of the NMDA-type glutamate receptors in human brain, and thus, was associated with the onset of schizophrenia.1,3,6 We recently established that the binding of the protein pLG72 does not affect the kinetic properties of hDAAO or the affinity for FAD but rather induces inactivation faster.6 We now demonstrate that FAD binding to hDAAO apoprotein does not increase its stability to a significant extent and does not modify its oligomeric state, as instead observed for the homologous enzyme from yeast.11,12 The weak interaction between hDAAO apoprotein moiety and its cofactor may represent a means of evolving an enzyme that is largely present in the holoenzyme (active) form only in the presence of an active site ligand, for example, of the substrate d-serine (in the millimolar range) or of an unknown physiological ligand. In fact, in extracellular fluid d-serine concentration is ∼6 μM in forebrain areas and the overall concentration in brain is ∼300 μM15 values significantly lower than the Km of hDAAO for d-serine (7.5 mM)4 Furthermore, overall FAD concentration in brain is estimated ∼5 μM,16 a value close to the Kd for coenzyme binding to hDAAO apoprotein. Therefore, at physiological substrate and FAD concentrations hDAAO should be present in the uncomplexed form that is in equilibrium with the corresponding inactive apoprotein. The low affinity of hDAAO for its coenzyme thus appears to be evolved in the human flavoenzyme to control d-serine concentration and avoid an excessive decrease of this gliotransmitter (which is the underlying cause of a number of neurological disorders). Noteworthy, this hypothesis also correlates with the long half-life of d-serine in brain (∼16 h).2
Materials and Methods
Enzymes
The recombinant hDAAO is expressed in E. coli cells and purified as reported in Ref. 4; 40 μM free FAD is present during all purification steps. For some experiments, this enzyme sample was further purified by gel-permeation chromatography on a HiLoad Superdex 200 column; the final enzyme preparation was stored in 20 mM Tris-HCl buffer, pH 8.0, 100 mM NaCl, 5% glycerol, 5 mM 2-mercaptoethanol, and 40 μM FAD and diluted in plain buffer without FAD before use. hDAAO apoprotein was prepared by overnight dialysis of the holoenzyme against 1M KBr.4 Enzyme concentration was determined by using a known extinction coefficient at 445 nm (12.2 mM−1cm−1) for the holoenzyme and at 280 nm (75.2 mM−1cm−1) for the apoprotein.
DAAO activity assay
DAAO activity was assayed with an oxygen electrode at pH 8.5, air saturation, and 25°C, using 28 mM d-alanine as substrate in the presence of 0.2 mM FAD.4 The effect of urea concentration on the enzymatic activity was determined using the same assay on protein samples previously incubated at 15°C for 60 min in the presence of different concentrations of denaturant.
Spectral measurements
Flavin and protein fluorescence measurements were routinely performed in a Jasco FP-750 instrument. For rapid reactions (e.g., the investigation of the kinetics of FAD binding to hDAAO apoprotein), a Biologic SFM-300 stopped-flow apparatus was employed and connected to the same Jasco fluorimeter.4 Protein emission spectra were taken from 300 to 400 nm (excitation at 280 or 298 nm); flavin emission spectra were recorded from 475 to 600 nm (excitation at 450 nm). Steady-state fluorescence measurements were performed at a 0.1 mg/mL protein concentration and corrected for buffer contributions. Fixed wavelength emission measurements were taken at 340 and 526 nm for tryptophan and flavin fluorescence, respectively. Circular dichroism (CD) spectra were recorded on a J-815 Jasco spectropolarimeter. Cell path length was 1 cm for measurements above 250 nm and 0.1 cm for measurements in the 190–250 nm region. All spectral measurements were carried out at 15°C. For temperature ramp experiments, both the fluorimeter and the spectropolarimeter were equipped with a software-driven Peltier-based temperature controller producing the same temperature gradient (0.5°C/min); data were analyzed by means of Jasco software.
Unfolding-refolding equilibrium and ANS binding experiments
The unfolding equilibrium of holoenzyme and apoprotein of hDAAO was determined by following the changes in protein (and flavin, for the holoenzyme only) fluorescence, catalytic activity, and far-UV CD signal as detailed elsewhere17 and by primarily using the “consensus” set of experimental conditions suggested by.18 Each point in the urea denaturation curves was determined at 15°C on an individual sample, prepared by mixing appropriate volumes of a concentrated protein, of 8M urea in buffer, and of 50 mM sodium pyrophosphate, pH 8.0, 5% glycerol (for CD measurements) or 20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 5% glycerol, and 5 mM 2-mercaptoethanol (for fluorescence measurements). The unfolding curves were analyzed by fitting to a two-state or three-state mechanism as described previously.10,12 A least-squares curve-fitting analysis was used to calculate the Cm values by using a software routine. ANS binding experiments were carried out at 15°C and at a 0.1 mg/mL protein concentration. ANS fluorescence emission spectra were recorded in the 450- to 600 nm range with excitation at 370 nm. To investigate the refolding process, holo- and apoprotein samples of hDAAO were incubated for 60 min at 15°C in buffer containing different concentrations of urea (0–6M) and then refolded by 10-fold dilution in 50 mM sodium pyrophosphate, pH 8.0, 5% glycerol and 15°C, in the presence of a 10-fold molar excess of FAD. The refolding yield was determined by monitoring protein fluorescence, far-UV CD spectra, and the recovery of enzymatic activity. Refolding experiments were also performed in the presence of 20% glycerol, or of GroEL tetradecamer (0.25 μM with respect to 2.5 μM apoprotein) in 1 mM ATP, 25 μM FAD, and 5% glycerol.
Limited proteolysis
Various hDAAO forms (0.4 mg protein/mL, corresponding to ∼10 μM) were incubated at 25°C in 20 mM Tris-HCl, pH 8.5, 150 mM NaCl, 5% glycerol, and 5 mM 2-mercaptoethanol, with 10% (w/w) trypsin. Protein samples (7 μg) taken at various times after adding trypsin were diluted in sample buffer for SDS-PAGE, heated at 100°C for 3 min, and analyzed electrophoretically. The intensity of the protein bands was determined by densitometric analysis using the QuantityOne software (BioRad Lab.). Changes in apoprotein conformation during the cofactor binding process were analyzed by proteolysis of 10 μM apoprotein samples pre-incubated for different times (5–60 min) with 100 μM FAD.
Size-exclusion chromatography
Size-exclusion chromatography was performed at room temperature on a Superdex 200 column using an Äkta chromatographic system (GE Healthcare), using 20 mM Tris-HCl buffer, pH 8.5, 150 mM sodium chloride, 5% glycerol, 5 mM 2-mercaptoethanol, and the appropriate concentrations of FAD/urea/ammonium thiocyanate as eluant. The column was calibrated with suitable standard proteins.
Acknowledgments
The authors thank the support from Consorzio Interuniversitario per le Biotecnologie (CIB) and Sara Pesenti for technical support.
Glossary
Abbreviations:
- ANS
8-anilino-1-napthalene-sulphonic acid
- CD
circular dichroism
- Cm
concentration of urea to give half-unfolded protein
- hDAAO
human d-amino acid oxidase
- pkDAAO
pig kidney d-amino acid oxidase
- Tm
melting temperature.
References
- 1.Pollegioni L, Piubelli L, Sacchi S, Pilone MS, Molla G. Physiological functions of D-amino acid oxidases: from yeast to humans. Cell Mol Life Sci. 2007;64:1373–1394. doi: 10.1007/s00018-007-6558-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martineau M, Baux G, Mothet JP. D-serine signalling in the brafriend and foe. Trends Neurosci. 2006;29:481–491. doi: 10.1016/j.tins.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 3.Chumakov I, Blumenfeld M, Guerassimenko O, Cavarec L, Palicio M, Abderrahim H, Bougueleret L, Barry C, Tanaka H, La Rosa P, Puech A, Tahri N, Cohen-Akenine A, Delabrosse S, Lissarrague S, Picard FP, Maurice K, Essioux L, Millasseau P, Grel P, Debailleul V, Simon AM, Caterina D, Dufaure I, Malekzadeh K, Belova M, Luan JJ, Bouillot M, Sambucy JL, Primas G, Saumier M, Boubkiri N, Martin-Saumier S, Nasroune M, Peixoto H, Delaye A, Pinchot V, Bastucci M, Guillou S, Chevillon M, Sainz-Fuertes R, Meguenni S, Aurich-Costa J, Cherif D, Gimalac A, Van Duijn C, Gauvreau D, Ouellette G, Fortier I, Raelson J, Sherbatich T, Riazanskaia N, Rogaev E, Raeymaekers P, Aerssens J, Konings F, Luyten W, Macciardi F, Sham PC, Straub RE, Weinberger DR, Cohen N, Cohen D. Genetic and physiological data implicating the new human gene G72 and the gene for D-amino acid oxidase in schizophrenia. Proc Natl Acad Sci USA. 2002;99:13675–13680. doi: 10.1073/pnas.182412499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Molla G, Sacchi S, Bernasconi M, Pilone MS, Fukui K, Pollegioni L. Characterization of human D-amino acid oxidase. FEBS Lett. 2006;580:2358–2364. doi: 10.1016/j.febslet.2006.03.045. [DOI] [PubMed] [Google Scholar]
- 5.Kawazoe T, Tsuge H, Pilone MS, Fukui K. Crystal structure of human D-amino acid oxidase: context-dependent variability of the backbone conformation of the VAAGL hydrophobic stretch located at the si-face of the flavin ring. Protein Sci. 2006;15:2708–2717. doi: 10.1110/ps.062421606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sacchi S, Bernasconi M, Martineau M, Mothet JP, Ruzzene M, Pilone MS, Pollegioni L, Molla G. pLG72 modulates intracellular D-serine levels through its interaction with D-amino acid oxidase: effect on schizophrenia susceptibility. J Biol Chem. 2008;283:22244–22256. doi: 10.1074/jbc.M709153200. [DOI] [PubMed] [Google Scholar]
- 7.Perez-Iratxeta C, Andrade-Navarro MA. K2D2: estimation of protein secondary structure from circular dichroism spectra. BMC Struct Biol. 2008;8:1–5. doi: 10.1186/1472-6807-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raibekas AA, Massey V. Glycerol-assisted restorative adjustment of flavoenzyme conformation perturbed by site-directed mutagenesis. J Biol Chem. 1997;272:22248–22252. doi: 10.1074/jbc.272.35.22248. [DOI] [PubMed] [Google Scholar]
- 9.Burstein EA, Vedenkina NS, Ivkova MN. Fluorescence and the location of tryptophan residues in protein molecules. Photochem Photobiol. 1973;18:263–279. doi: 10.1111/j.1751-1097.1973.tb06422.x. [DOI] [PubMed] [Google Scholar]
- 10.Pace CN. Measuring and increasing protein stability. Trends Biotechnol. 1990;8:93–98. doi: 10.1016/0167-7799(90)90146-o. [DOI] [PubMed] [Google Scholar]
- 11.Pollegioni L, Iametti S, Fessas D, Caldinelli L, Piubelli L, Barbiroli A, Pilone MS, Bonomi F. Contribution of the dimeric state to the thermal stability of the flavoprotein D-amino acid oxidase. Protein Sci. 2003;12:1018–1029. doi: 10.1110/ps.0234603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caldinelli L, Iametti S, Barbiroli A, Bonomi F, Piubelli L, Ferranti P, Picariello G, Pilone MS, Pollegioni L. Unfolding intermediate in the peroxisomal flavoprotein D-amino acid oxidase. J Biol Chem. 2004;279:28426–28434. doi: 10.1074/jbc.M403489200. [DOI] [PubMed] [Google Scholar]
- 13.Massey V, Curti B. A new method of preparation of D-amino acid oxidase apoprotein and a conformational change after its combination with flavin adenine dinucleotide. J Biol Chem. 1966;241:3417–3423. [PubMed] [Google Scholar]
- 14.Pollegioni L, Pilone MS. On the holoenzyme reconstitution process in native and truncated Rhodotorula gracilis D-amino acid oxidase. Arch Biochem Biophys. 1996;332:58–62. doi: 10.1006/abbi.1996.0316. [DOI] [PubMed] [Google Scholar]
- 15.Hashimoto A, Oka T, Nishikawa T. Extracellular concentration of endogenous free D-serine in the rat brain as revealed by in vivo microdialysis. Neurosci. 1995;66:635–643. doi: 10.1016/0306-4522(94)00597-x. [DOI] [PubMed] [Google Scholar]
- 16.Spector R. Riboflavin accumulation by rabbit brain slices. in vitro J Neurosci. 1980;34:1768–1771. doi: 10.1111/j.1471-4159.1980.tb11274.x. [DOI] [PubMed] [Google Scholar]
- 17.Caldinelli L, Iametti S, Barbiroli A, Bonomi F, Fessas D, Molla G, Pilone MS, Pollegioni L. Dissecting the structural determinants of the stability of cholesterol oxidase containing covalently bound flavin. J Biol Chem. 2005;280:22572–22581. doi: 10.1074/jbc.M500549200. [DOI] [PubMed] [Google Scholar]
- 18.Maxwell KL, Wildes D, Zarrine-Afsar A, De Los Rios MA, Brown AG, Friel CT, Hedberg L, Horng JC, Bona D, Miller EJ, Vallée-Bélisle A, Main ER, Bemporad F, Qiu L, Teilum K, Vu ND, Edwards AM, Ruczinski I, Poulsen FM, Kragelund BB, Michnick SW, Chiti F, Bai Y, Hagen SJ, Serrano L, Oliveberg M, Raleigh DP, Wittung-Stafshede P, Radford SE, Jackson SE, Sosnick TR, Marqusee S, Davidson AR, Plaxco KW. Protein folding: defining a “standard” set of experimental conditions and a preliminary kinetic data set of two-state proteins. Prot Sci. 2005;14:602–616. doi: 10.1110/ps.041205405. [DOI] [PMC free article] [PubMed] [Google Scholar]