

Abstract
We present here the 2.6Å resolution crystal structure of the pT26-6p protein, which is encoded by an ORF of the plasmid pT26-2, recently isolated from the hyperthermophilic archaeon, Thermococcus sp. 26,2. This large protein is present in all members of a new family of mobile elements that, beside pT26-2 include several virus-like elements integrated in the genomes of several Thermococcales and Methanococcales (phylum Euryarchaeota). Phylogenetic analysis suggested that this protein, together with its nearest neighbor (organized as an operon) have coevolved for a long time with the cellular hosts of the encoding mobile element. As the sequences of the N and C-terminal regions suggested a possible membrane association, a deletion construct (739 amino acids) was used for structural analysis. The structure consists of two very similar β-sheet domains with a new topology and a five helical bundle C-terminal domain. Each of these domains corresponds to a unique fold that has presently not been found in cellular proteins. This result supports the idea that proteins encoded by plasmid and viruses that have no cellular homologues could be a reservoir of new folds for structural genomic studies.
Keywords: extra chromosomal elements, structural genomics, function, domain repeat, plasmid, archaea, protein fold
Introduction
The origin of viruses, plasmids, and related elements is presently a highly controversial topic in evolutionary biology.1,2 A typical feature of viral genomes and plasmid sequences is to encode a much higher proportion of orphan proteins than cellular genomes.2 Furthermore, viral genomes and plasmids also encode many proteins that are not true ORFans (without any homologues in current databases) but have only homologues in other viruses or plasmids.1,3 Two hypotheses can be proposed to explain these observations. In the first one, viral- and plasmid-specific proteins have in fact cellular homologues, but these homologies cannot be recognized through sequence comparison, because they evolved more rapidly than their cellular counterparts. Alternatively, viral and plasmid specific proteins directly originated in ancient viral world or in ancient cellular lineages that have not left descendents in the present biosphere. If the first hypothesis is correct, one should expect that many viral- or plasmid-specific proteins should have cellular homologues detectable by structural similarity only (with already known folds), that will be finally identified once their three dimensional structure will be solved. On the contrary, if viral and plasmid specific proteins have really no homologue in modern cells, one should expect that many of them will exhibit totally new folds. In the latter hypothesis, viruses and plasmids might represent an immense reservoir of new protein folds, outnumbering by far folds already identified through structural genomics projects. This is an important question, since the number of new protein folds discovered through structural genomic projects targeting cellular proteins is continuously decreasing.4 We present here the first results of a small scale structural genomics project to start testing these ideas. We focused on several plasmids recently isolated from hyperthermophilic archaea of the genus Thermococcus.5 We focus on a large protein (pT26-6p) encoded by one of these plasmids, pT26-2, from the Thermococcus strain 26-2.We have solved the structure of the protein pT26-6p and found that it consists of three domains each with a unique fold. This protein has closely related “cellular” homologues in several genomes of Thermococcales and Methanococcales (phylum Euryarchaea). However, pT26-6p can be considered as a virus/plasmid specific protein since all these homologues are systematically present in virus-like elements integrated into these genomes.
Results and Discussion
The plasmid pT26-2 (21,566 bp long) which has been isolated from the strain Thermococcus sp 26-2 encodes 32 putative proteins, including an integrase, and will be described in detail elsewhere (Soler et al., in preparation). Homologues of several contiguous genes encoded by pT26-2 are present in the genomes of several archaea of the orders Thermococcales and Methanococcales, also forming continuous stretches of genes organized in the same order and transcribed in the same direction. Two of these structures have been already noticed in the genome of the hyperthermophilic archaeon Thermococcus kodakaraensis and dubbed TKV2 and TKV3 for Thermococcus Kodakaraensis Virus-like 2 and 3.6 Figure 1(a) illustrates the structure of the conserved portion of these elements in Thermococcales. In all cases, they are located nearby a tRNA gene located at the right end of the element, indicating that they probably correspond to integrated mobile elements. The C and N terminal domains of the pT26-2 integrase homologues are located at each side of the element, indicating that integration occurs by recombination between a region of a tRNA gene and a homologous region within the integrase coding gene. All mobile elements of this new family encode two large proteins that are encoded by contiguous genes. We have identified 14 couples of these two proteins in 10 archaeal genomes of the order Thermococcales and Methanococcales. A phylogenetic analysis of the concatenation of these two proteins shows a clear distinction between these two archaeal orders [Fig. 1(b)]. Interestingly, Thermococcales and Methanococcales are closely related in consensus archaeal phylogenetic trees based on ribosomal proteins or RNA polymerase subunits.9 These data indicate that proteins, pT26-5p and pT26-6p are essential components of the pT26-2 plasmid/virus family and that they have probably coevolved with their hosts since a very long time. These proteins can thus be considered as viral-/plasmid-specific (viral hallmark proteins sensu Koonin).3
Figure 1.
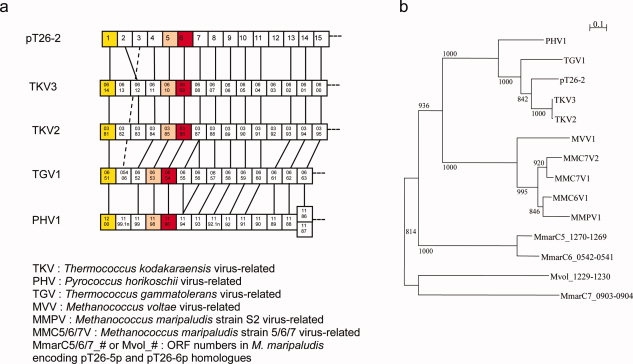
Genomic and phylogenetic repartition of pT26-5p and pT26-6p homologues. (a) Genomic organization of a part of virus-related integrated elements including pT26-6p homologues. Boxes represent putative ORFs, and numbers inside boxes correspond to the ORF numbers in the genomes. The yellow, pink and red boxes respectively represent the ORF encoding the C-terminal part of the integrases, the pT26-5 homologues and the pT26-6 homologues. Homologous ORFs are linked together (with doted line for a jumping link). (b) Unrooted maximum likelihood tree of concatenation of pT26-5p and pT26-6p homologues. 14 sequences from Thermococcales and Methanococcales were aligned by MUSCLE7 and 401 homologous positions were selected for tree calculation by PHYML8 using a JTT model of amino acid substitution. A gamma correction with four discrete classes of sites was used. The alpha parameter and the proportion of invariable sites were estimated. The robutness of the tree was estimated by non-parametric bootstrap analysis (1000 replicates) using PHYML. Scale bar represents the number of substitutions per site.
Overall structure of pT26-6p
Sequence analysis and membrane region prediction algorithms suggested the presence of membrane spanning segments at the N- and C-termini of the protein pT26-6p (not shown). To carry out structural studies, we decided to concentrate on a protein construct from which we removed these putative membrane spanning regions. We purified and crystallized a truncated protein construct missing 146 residues at the N- and 78 residues at the C-terminal (ΔN147–ΔC671). The protein crystallized in P65 space group and the structure was solved at 2.6Å resolution. The statistics on data collection and refinement are provided in Table I. The asymmetric unit contains two copies of pT26-6p related by a local two-fold symmetry axis.
Table I.
Statistics of Data Collection and Structure Refinement
| Space group | P65 |
|---|---|
| Unit-cell parameters | 133.15, 133.15, 164.21 |
| a, b, c (Å) | |
| Resolution (Å) | 47.14–2.60 (2.74–2.60) |
| Total number of refl. | 205,989 (27,568) |
| Total of unique refl. | 50,396 (6968) |
| Multiplicity | 4.1 (4.0) |
| Rmerge1 | 0.128 (0.031) |
| I/σ(I) | 4.6 (0.9) |
| Overall completeness (%) | 99.1 (99.1) |
| R/Rfree (%)b | 19.23/23.37 |
| R.m.s.d. bonds (Å) | 0.007 |
| R.m.s.d. angles (°) | 1.075 |
| Ramachandran plot (%) | |
| Most favored | 90.1 |
| Allowed | 9.4 |
Rmerge = ΣhΣi|Ihi − <Ih>|/ΣhΣiIhi, where Ihi is the observation of the reflection h, and <Ih> is the mean intensity of reflection h.
Rfactor = Σ‖Fo| − |Fc‖/|Fo|. Rfree was calculated with a set of randomly selected reflections (5%).
pT26-6p has an elongated shape that consists of three contiguous domains from N to C terminus: two β-sandwich domains (regions comprising residues 148–368 and residues 369–529) and a bundle of five α helices (region 530–670) [Fig. 2(a,c)]. The sheets of the two β-sandwich domains are lying in the same plane and the longitudinal axis of the helix bundle is perpendicular to it. Both β-sandwich domains share the same fold, composed of two sheets made of four antiparallel β strands. Despite the low amino acid sequence identity (11%) between these β-sandwich domains, their structures superpose very well (rms deviation of 2.18Å for 101 Cα positions aligned) [Fig. 2(b)]. The β-sheets of the second sandwich domain pack against four helices, two are originating from the N-terminal and two are inserted between β6 and β7 from the first β-sandwich domain [Fig. 2(a,c)]. The helical bundle domain contains five antiparallel helices (α5–α9). In the global structure of pT26-6p the N- and C-termini, where the putative membrane spanning segments were truncated, are in close proximity. This suggests that both N and C terminal segments may form a single membrane spanning domain.
Figure 2.
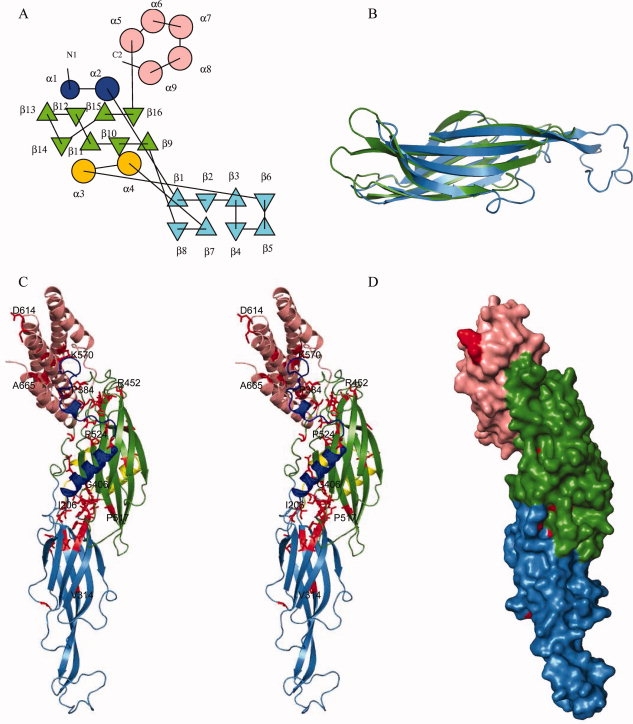
Structure of pT26-2-6: (a) Topology diagram of pT26-6p. The repeated β-sandwich domains are colored blue and green and the helical bundle pink. The N-terminal helical peptide is in deep blue and the helical insertion in the fist sandwich domain in yellow. (b) Superposition of the two β-sandwich domains. (c) Cartoon stereo view representation using the same color scheme as (a) conserved residues are colored in red, some of them are labeled. (d) Surface representation of conserved residues (in red).
The elongated structure of pT26-6p is stabilized by extensive inter domain interactions. The second central β-domain extensively interacts both with the N-terminal β-domain and with the helical bundle. The interaction between the two β-domains involves a hydrophobic patch surrounded by polar interactions: 37 hydrogen bonds and seven salt bridges, burying 20% of the N-terminal and 31% of the central domain.
Dimer formation
Two copies of the protein are present in the asymmetric unit related by a two-fold symmetry axis, suggesting that the protein exists in a dimeric form. The dimerization occurs via antiparallel alignment of the central β-domain of each monomer (not shown). Dimerization is stabilized by eight hydrogen bonds. The β-strand pairing creates an extended intersubunit β sandwich. The interacting surface of the two monomers represents only 3.2% of the total solvent-accessible surface of the protein. These values are rather low when compared with surface areas identified in well-characterized homodimers.10 Analytical gel filtration experiments showed that pT26-6p is present as a monomer in solution (not shown). We, therefore, suggest that the dimer observed in the structure is induced by the crystallization conditions.
pT26-6p homologues
Using the MSD-SSM server (http://www.ebi.ac.uk/msd-srv/ssm/), we were unable to identify close structural analogues for any of the domains. In absence of clear structural analogues, amino acid conservation features may give some hints on broad biochemical function (nucleic acid binding, membrane association, active sites, etc.). We therefore analyzed sequence conservation mapped on the 3D-structure of pT26-6p. pT26-6p represents one of the best conserved ORFs from the integrated elements in genomes of Thermococcales and Methanococcales, sharing 22% sequence identity on average with its homologues encoded by the mobile elements previously described. Figure S1 (Supporting Information) shows the sequence alignment of pT26-6p homologues with the superposed secondary structure elements retrieved from the pT26-6p crystal structure. The majority of the well-conserved residues is hydrophobic and is important for maintaining the protein fold. A considerable amount of conserved hydrophobic and hydrophilic residues are found at the interfaces between the three domains [Fig. 2(c,d)]. This suggests that residue conservation of pT26-6p has endeavored the maintenance of an elongated shape.
Materials and Methods
Cloning, expression, and purification
The coding sequence of pT26-6p deleted by 146 residues at the N- and 78 residues at the C-terminal (ΔN147–ΔC671) was amplified by PCR from cDNA. The cDNA was cloned in a pDEST30a plasmid. Expression was done at 37°C using the E. coli Rosetta (DE3) pLysS strain and the 2xYT medium (BIO 101 Inc.). When the cell culture reached an OD600 nm of 0.8, induction at 37°C was performed during 3 h with 0.5 mM IPTG (Sigma). Cells were harvested by centrifugation and resuspended in buffer A (20 mM tris Tris-HCl pH 7.5, 200 mM NaCl, 5 mM β-mercaptoethanol). Cell lysis was completed by sonication and the lysate was heated at 20 min at 70°C before centrifugation at 20,000 rpm for 20 min. The soluble fraction was loaded on a NiNTA column (Qiagen Inc.) equilibrated with buffer A. The protein was eluted with imidazole and subsequently loaded on a Superdex200 column (Amersham Pharmacia Biotech) equilibrated against buffer A supplemented with 10 mM β-mercaptoethanol. Selenomethionine substituted protein was produced and purified as the native protein. The homogeneity of the protein samples was checked by SDS-PAGE.
Structure resolution
Crystals of SeMet substituted pT26-6p were grown from a 1:1 μL mixture of protein (14 mg/mL) with 1.5–2M (NH4)2SO4, 0.025M KH2PO4, 8% PEG 8000, 10–16% MPD, using the hanging drop vapor diffusion method at 23°C. Crystals were soaked in a mixture of mother liquor and 20% MPD before flash freezing at 100 K. X-ray diffraction data were collected from a crystal of the SeMet substituted pT26-2-6 on beamline ID23-1 (ESRF) at the wavelength of the Se K-edge. The crystals belong to the P65 space group with two copies per asymmetric unit, corresponding to a 65.6% solvent content. Data were collected at a resolution of 2.6Å and processed with the program XDS11 and SCALA12 for merging and scaling. The structure was solved using the SAD method using diffraction data collected at 2.6Å resolution from SeMet-substituted crystals. Twenty two Selenium atom sites were found with the program SHELXD13 in the 20–3.5Å resolution range. These sites were used for phasing with the program SOLVE14. After solvent flattening with the program RESOLVE,15 the quality of the electron density map allowed automated construction of 70% of the model. The missing residues were built by hand using O16 molecular graphics program and the model was refined with REFMAC.17
Conclusions
The crystal structure of the pT26-6p protein encoded by a Thermococcus plasmid shows that it consists of three domains, with unexpected fold duplication for the two N-terminal domains. The pT26-6p protein has an elongated shape, stabilized by inter domain interactions. None of the three domains has structural analogues in the data base and therefore their 3D-structure does not suggest a biochemical function. pT26-6p is frequently juxtaposed to pT26-5p encoding a large putative membrane associated protein of unknown function. Experiments are underway to determine if pT26-5p and pT26-6p physically interact.
The proteins pT26-5p and pT26-6p are systematically present in a new family of mobile elements (plasmids and/or viruses) detected in two closely related archaeal orders (Thermococcales and Methanococcales). They have no sequence similarities with other proteins and can, therefore, be considered as typical plasmid/viral specific. In this work, we failed also to detect cellular homologues to pT26-6p by structural similarity. This supports the hypothesis that viruses and plasmids might represent a reservoir of new folds for structural genomic studies. Of course, one cannot conclude from a single example, but previous structural and phylogenetic analyses of other viral specific proteins suggests that these proteins indeed contain also unique folds that are not present in cellular proteins (reviewed in Refs. 1,3). It would be important now to accumulate more examples to generalize (or not) these preliminary observations.
Coordinates
Coordinates and structure factors have been deposited in the Protein Data Bank with the accession code 2WB7.
Acknowledgments
The authors thank Evelyne Marguet who isolated the pTN2 plasmid and helped in its characterization.
References
- 1.Forterre P. The origin of viruses and their possible roles in major evolutionary transitions. Virus Res. 2006;117:5–16. doi: 10.1016/j.virusres.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 2.Prangishvili D, Garrett RA, Koonin EV. Evolutionary genomics of archaeal viruses: unique viral genomes in the third domain of life. Virus Res. 2006;117:52–67. doi: 10.1016/j.virusres.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 3.Koonin EV, Senkevich TG, Dolja VV. The ancient Virus World and evolution of cells. Biol Direct. 2006;1:29. doi: 10.1186/1745-6150-1-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chandonia JM, Brenner SE. The impact of structural genomics: expectations and outcomes. Science. 2006;311:347–351. doi: 10.1126/science.1121018. [DOI] [PubMed] [Google Scholar]
- 5.Lepage E, Marguet E, Geslin C, Matte-Tailliez O, Zillig W, Forterre P, Tailliez P. Molecular diversity of new Thermococcales isolates from a single area of hydrothermal deep-sea vents as revealed by randomly amplified polymorphic DNA fingerprinting and 16S rRNA gene sequence analysis. Appl Environ Microbiol. 2004;70:1277–1286. doi: 10.1128/AEM.70.3.1277-1286.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fukui T, Atomi H, Kanai T, Matsumi R, Fujiwara S, Imanaka T. Complete genome sequence of the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1 and comparison with Pyrococcus genomes. Genome Res. 2005;15:352–363. doi: 10.1101/gr.3003105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 9.Brochier C, Forterre P, Gribaldo S. An emerging phylogenetic core of Archaea: phylogenies of transcription and translation machineries converge following addition of new genome sequences. BMC Evol Biol. 2005;5:36. doi: 10.1186/1471-2148-5-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Janin J, Rodier F. Protein-protein interaction at crystal contacts. Proteins. 1995;23:580–587. doi: 10.1002/prot.340230413. [DOI] [PubMed] [Google Scholar]
- 11.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Cryst. 1993;26:795–800. [Google Scholar]
- 12.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 13.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr D Biol Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 14.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 17.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]