

Abstract
FR901464 is a potent antitumor natural product that binds to the SF3b complex and inhibits pre-mRNA splicing. Its analogue, meayamycin, is two orders of magnitude more potent as an antiproliferative agent against human breast cancer MCF-7 cells. Here, we report the picomolar antiproliferative activity of meayamycin against various cancer cell lines and multidrug resistant cells. Time-dependence studies implied that meayamycin may form a covalent bond with its target protein(s). Meayamycin inhibited pre-mRNA splicing in HEK-293 cells but not alternative splicing in a neuronal system. Meayamycin exhibited specificity toward human lung cancer cells compared to non-tumorigenic human lung fibroblasts and retained picomolar growth inhibitory activity against multi-drug resistant cells. These data suggest that meayamycin is a useful chemical probe to study pre-mRNA splicing in live cells and is a promising lead as an anticancer agent.
Keywords: FR901464, meayamycin, pre-mRNA splicing, alternative splicing, multidrug resistance, reversibility, p53, apoptosis, high content cellular analysis
Introduction
FR901464 (Figure 1) is a natural product isolated from the culture broth of the bacterium Pseudomonas sp.2663 (1). FR901464 was discovered as an activator of a stably transfected reporter gene driven by an SV40 DNA tumor virus promoter in human breast adenocarcinoma MCF-7 cells (1). FR901464 exhibited antiproliferative activity against MCF-7, human lung adenocarcinoma A549, colon cancer HCT116 and SW480, and murine leukemia P388 cell lines with IC50 values of 1.8, 1.3, 0.61, 1.0, and 3.3 nM, respectively. The IC50 against mouse bone marrow cells was 9.9 nM, indicating specificity toward tumor cells relative to bone marrow cells (1). FR901464 was effective against P388, A549, murine colon 38 and Meth A cell xenograft models in mice (2). To date, three total chemical syntheses of FR901464 have been reported (3-10). In addition to FR901464, the Jacobsen group synthesized 2, which was slightly more potent than FR901464 against Jurkat cells (5). The Kitahara group reported 3 to be more active than FR901464, although numerical data were not presented (11). We synthesized meayamycin (1) and showed this analog to be two orders of magnitude more potent than FR901464 against MCF-7 cells (9). More recently, the Yoshida group reported that a target of FR901464 was the splicing factor 3b (SF3b) complex (12). Together with pladienolides (13), these two classes of natural products represent the first inhibitors of pre-mRNA splicing via SF3b binding and represent promise for the development of new chemotherapeutic approaches through splicing inhibition (14). A pladienolide analog, E7107, is now in Phase I clinical trial for various cancers (15). In contrast, the development of a compound derived from FR901464 lags behind, although the Webb group recently reported analogues of FR901464 and showed their biological activity (16). Herein we wish to report further studies of meayamycin in a cell-free system and in living cells.
Figure 1.

Structures of FR901464, pladienolide D and their synthetic analogs.
Materials and Methods
Stability of Meayamycin in Buffers and Cell Culture Medium
For the stability of meayamycin in buffers, a Corning tube (15 mL) was filled with phosphate buffer (10 mL), PhCO2H (10 mM in DMSO, 20 μL), and DMSO (70 μL) under an open atmosphere and warmed to 37 °C. To the reaction mixture was added meayamycin (10 mM in DMSO, 10 μL) and the resulting mixture was sealed with a cap, vortexed for 15 s, and then placed in a 37 °C incubator. The decomposition was monitored by HPLC at the indicated times. The resulting data were normalized by dividing the ratios of meayamycin/PhCO2H by the ratio of the first data point.
For the stability of meayamycin in cell culture media, a dram vial was filled with 10% fetal bovine serum (FBS) in RPMI 1640 (1 mL), rhodamine (10 mM in DMSO, 1 μL), and DMSO (7 μL) under open atmosphere and warmed to 37 °C. Meayamycin (10 mM in DMSO, 1 μL) was added to the reaction mixture, and the resulting mixture was sealed with a cap, vortexed for 15 s, and then placed in a 37 °C incubator. The decomposition was monitored by HPLC at the indicated times. The resulting data were normalized by dividing the ratios of meayamycin/rhodamine by the ratio of the first data point.
The HPLC analysis for the stability of meayamycin in buffers was performed on a Varian Pursuit XDR C18 column, 75 × 4.6 mm. The elution conditions — flow: 1.0 mL/min, eluant: 10% MeCN/H2O (containing 1% HCO2H) to 100% MeCN linear gradient elution from 0.5 to 9.5 min. Meayamycin and benzoic acid were monitored at 230 nm. Retention time for meayamycin = 6.7 min. Retention time for benzoic acid = 4.3 min. The HPLC analysis for stability of meayamycin in cell culture media was performed on a Varian Pursuit XRs 5 C18 column, 250 × 10.0 mm. The elution conditions — flow: 2.5 mL/min, eluant: 30% MeOH/H2O (containing 0.1% HCO2H) to 100% MeOH linear gradient elution from 0.5 to 30.5 min. Meayamycin was monitored at 232 nm. Rhodamine was monitored at 550 nm. Retention time for meayamycin = 4.3 min. Retention time for rhodamine = 18.4 min. The data were analyzed by Graphpad Prism 5.0. The calculation of meayamycin degradation in 10% FBS in RPMI 1640 was performed based on a one-phase exponential decay equation in the software.
Cell Culture
The cells used for Table 1 and Figure 2 were grown at 37 °C in an atmosphere containing 5% CO2 in Corning cell culture flasks (25 cm2) in RPMI 1640 cell culture medium containing 10% FBS, 1% penicillin/streptomycin, and 1% l-glutamine. Cells used for Figure 4 were HEK-293 cells with a stably integrated splicing reporter consisting of a human triose phosphate isomerase gene exon 6 – intron 6 – exon 7 cassette upstream of luc2 (17). A549 human lung cancer cells and IMR-90 normal diploid lung fibroblasts were from ATCC (Manassas, VA). DC3F transformed Chinese hamster cells and their multi-drug resistant VCRd5L counterparts were a gift from Prof. John Lazo (University of Pittsburgh). These cells were grown at 37 °C in an atmosphere containing 5% CO2 in DMEM cell culture medium containing 10% FBS. Neurons used for Figure 5 were dissociated from E18 rat cortical tissue and grown at 37 °C with 6% CO2 in Neurobasal medium with B27 supplement.
Table 1.
Fifty percent growth inhibitory (GI50) values (pM) of meayamycin against various human cancer cell lines.a
| MCF-7 b | MDA-MB231 c | HCT116 d | PC-3 e | H1299 f | A549 g | DU-145 h | HeLa i |
|---|---|---|---|---|---|---|---|
| 20±8.9 | 71±55 | 157±33 | 196±42 | 841±271 | 258±162 | 1234±697 | 306±175 |
Data are the averages ± SD of at least three independent experiments (5-day assays for MCF-7 cells, and 3-day assays for other cells), each with quadruplicate determinations.
Breast cancer cells, estrogen receptor positive.
Breast cancer cells, estrogen receptor negative.
Colon carcinoma cells, wild type p53.
Colon carcinoma cells, p53 deficient.
Lung carcinoma cells, p53 deficient.
Lung carcinoma cells, wild type p53.
Prostate carcinoma cells.
Cervical cancer cells.
Figure 2.
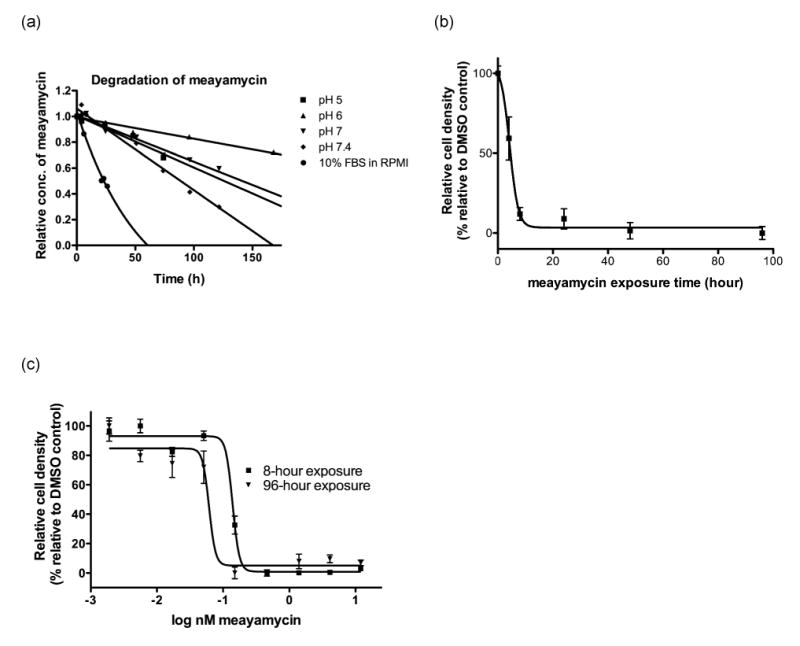
(a) Degradation of meayamycin (10 μM) in phosphate buffers and 10% FBS in RPMI at 37 °C. Benzoic acid and rhodamine were used as an internal standard in phosphate buffers (pH 5, 6, 7 and 7.4) and culture media, respectively. The quantity of meayamycin relative to each of these internal standards was monitored by reversed phase HPLC. ◆: pH 5, R2 = 0.952. ■: pH 6, R2 = 0.9408. σ: pH 7, R2 = 0.968. 5: pH 7.4, R2 = 0.978. Culture media: R2 = 0.989. (b) Exposure time-dependent growth inhibition of A549 cells in the presence of meayamycin at 2 nM for the indicated periods of time. Cell density was measured on day 4. (c) Growth inhibition of A549 cells treated with meayamycin for 96 hours (triangles), and for 8 hours with meayamycin followed by an 88-hour incubation without meayamycin (squares).
Figure 4. Inhibition of pre-mRNA splicing by meayamycin.
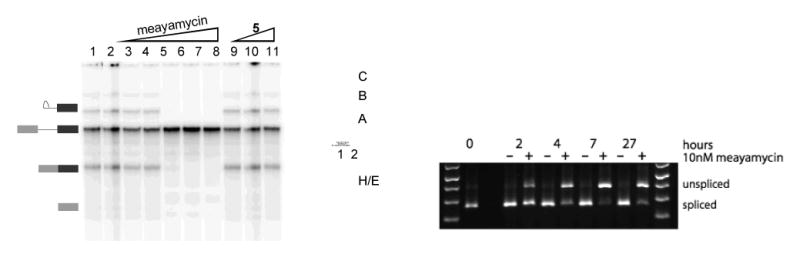
(Left) Denaturing gel of HeLa nuclear extracts. Lane 1: DMSO. Lane 2: [Isoginkgetin] = 50 μM. Lanes 3–8: [1] = 50 pM, 500 pM, 5 nM. 50 nM, 100 nM, 500 nM. Lanes 9–11: [5] = 100 nM, 1 μM, 10 μM. (Middle) Native gel to analyze spliceosomal complex. Lane 1: [1] = 50 nM. Lane 2: DMSO. (Right) RT-PCR analysis of 1 (10 nM)-treated HEK-293 cells.
Figure 5. Analysis of the effects of meayamycin on alternative splicing in cultured neurons.
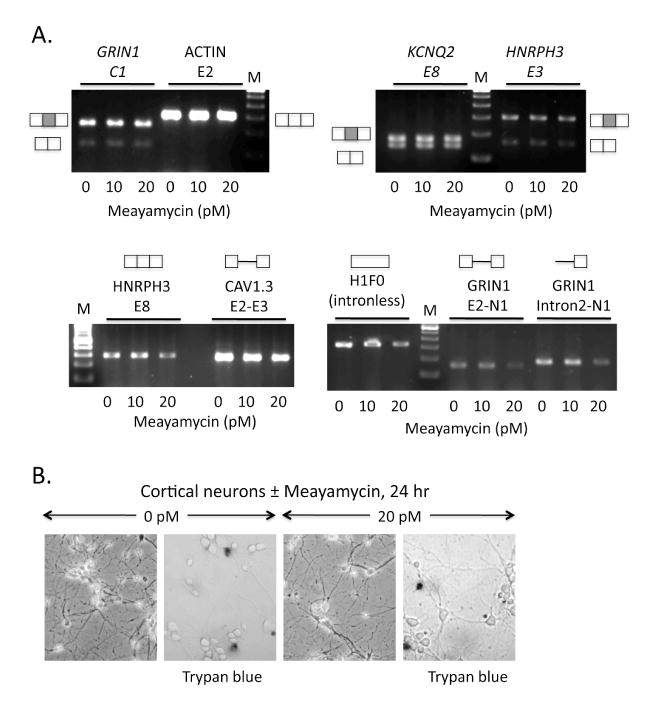
(A) RT-PCR analysis of RNA transcripts after 24-hour treatment with a final concentration of 0, 10, and 20 pM meayamycin in the culture medium. Exons tested include alternative cassette exons from the NMDA R1 receptor (GRIN1, C1 cassette), KCNQ2 potassium channel (KCNQ2, E8), and hnRNP H3 (HNRPH3, E3) transcripts. Constitutively included exons (ACTIN, E2 and HNRPH3, E8) and the intronless histone transcript (H1F0) are included as controls. Assays were negative for the accumulation of intronic regions indicative of splicing inhibition for the calcium channel CAV1.3 and two regions of the GRIN1 transcript (CAV1.3, E2-E3; GRIN1, E2-N1 and intron 2-N1). RT-PCR products were resolved on 1% agarose gels containing ethidium bromide. Schematics represent the structures of the RNA products amplified; boxes, exons; shaded boxes represent alternatively spliced exon; lines, introns. Lanes M, 100 base pair DNA ladder. (B) Neurons were photographed in culture after treatment ± meayamycin; left panel of each pair. Results of trypan blue staining are shown; right panel of each pair. Trypan blue, which stains dead cells, shows similar results for the two samples.
Addition of compounds in Growth Inhibition Assays
The compounds were dissolved in DMSO as 10 mM stocks and stored at -20 °C. For the experiments, aliquots were thawed at room temperature and diluted solutions were prepared in RPMI 1640 medium containing 2% DMSO at 2× the desired concentration and were warmed to 37 °C prior to addition to the cells. Control cultures received pre-warmed medium containing 2% DMSO.
Growth Inhibition Assays
Two independent methods were used to assess growth inhibitory activity. For Table 1, cells were plated in 96-well plates at an initial density of 2,000 cells per well in 100 μL of medium and were incubated for 24 hours prior to compound addition. Serial two-fold dilutions were used from 100 nM to 0.191 pM. The compound was added to the cells at 2× the desired concentration in 100 μL cell culture medium. The compound solutions were pre-warmed to 37 °C. The cells were then incubated for 3 to 5 days. Cell proliferation was measured using a commercial MTS solution (20 μL per well). The absorbance (at 490 nm minus that at 630 nm) was measured by a Spectromax M2 plate reader (Molecular Devices). Test agent concentration points within each experiment were performed in quadruplicate, and experiments were repeated at least three times to ensure reproducibility. The final GI50 values were averaged.
For DC3F and VCRd-5L Chinese hamster cells, we used our previously described image-based analysis procedure (18). Briefly, human cancer cell lines were plated at low density (500 – 2000 cells/well depending on cell line) in 384-well microplates. Cells were allowed to attach overnight and treated with compounds. After three days of continuous exposure to compounds, cells were labeled with Hoechst 33342 and nuclei enumerated on the ArrayScanII. Acquisition parameters were set such that the instrument acquired a minimum of 5,000 cells in each well. Cell densities were calculated as objects per imaging field and normalized to vehicle control density at the end of the study.
Reversibility Tests
Cells were plated in 96 well plates at an initial density of 2,000 cells per well in 100 μL of medium and were incubated for 24 hours prior to compound addition. One concentration was used in each experiment for all times examined. The compound was added to the cells at 2× the desired concentration in 100 μL cell culture medium. At the desired time intervals, the media containing the drug was removed, the wells were washed 5 times with new media and 200 μL of new media containing 1% DMSO was added. At the last time interval, after washing and replacing the media, cell proliferation was measured using a commercial MTS solution (20 μL per well). The absorbance (at 490 nm minus that at 630 nm) was measured by a Spectromax M2 plate reader (Molecular Devices). Each concentration treatment was performed in quadruplicate and the final numbers were averaged.
Pre-mRNA Splicing Inhibition
Uniformly 32P-labeled AdML pre-mRNA splicing substrate was generated by T7 runoff transcription methods and gel purified. HeLa cell nuclear extract was prepared as previously described (19-21). Splicing reactions containing 1 or 5, approximately 20 nM pre-mRNA and 30% nuclear extract, in 40 mM Tris-HCl, pH 8, containing 20 mM KCl, 2.5 mM MgCl2, 10 mM creatine phosphate, 0.5 mM dithiothreitol (DTT), 0.4U/μL RNasin, 0.5 mM ATP and 1% (v/v) DMSO were incubated for 60 minutes at 30 °C. For analysis of splicing, reactions were stopped with 9 volumes of stop buffer (20 mM Tris, pH 7.5, containing 300 mM NaOAc, 0.5% sodium dodecylsulfate (SDS), 100 mM NaCl, 5 mM EDTA and 25 ng/μL tRNA), extracted twice with phenol and precipitated with ethanol. RNAs were separated on 15% denaturing polyacrylamide gels and visualized with a phosphorimager. Native splicing complexes were analyzed as described previously (21, 22). Briefly, heparin was added to a final concentration of 0.5 mg/mL, reactions were incubated for 5 minutes at 30 °C and placed on ice. Reactions were centrifuged at 14,000 rpm in an Eppendorf microcentrifuge and immediately loaded onto a 4% acrylamide (80:1 acrylamide:bis), 50 mM Tris-glycine native gel. Gels were visualized with a phosphorimager.
RT-PCR
RT-PCR was carried out using the SuperScript III One-Step RT-PCR kit (Invitrogen). Total RNA (60 ng) that was DNase-treated was used per 25 μL reaction, run on 1% agarose gels, and stained with ethidium bromide. Primers used for amplification for the experiment of Figure 4 were 5′- TAA ACT TAA GCT TCA GCG CCT CGG – 3′ and 5′- TAG CGC TTC ATG GCT TTG TGC AG-3′. For the experiments shown in Figure 5, 1 μg of total RNA was reverse transcribed with MMLV reverse transcriptase (Invitrogen) with random primers. PCR reactions (10 μL) were performed for 21 to 24 cycles with 1 μL of the reverse transcribed sample. DNA products were separated on 1.2% agarose gels and visualized with ethidium bromide. Forward (F) and reverse (R) primer sequences used for the experiments of Figure 5 are indicated as follows (5′ to 3′). GRIN1 C1 cassette: (F) ATGCCCGTAGGAAGCAGATGC, (R) CGTCGCGGCAGCACTGTGTC; Actin E2: (F) AGCTTCTTTGCAGCTCCTTC, (R) TCTTCTCCATGTCGTCCCAG; KCNQ2 exon 8: (F) TCTATGCTACTAACCTCTCACGCAC, (R) GTGAGTCCAGATTTGCTCTTGAGATTC; HNRPH3 exon 3: (F) GGGACAGTGCGACTTCGTGG, (R) CTGTCCCAGCAATCTTCTTGGTGG; HNRPH3 exon 8: (F) GCTATGGAGGCTATGATG, (R) ACCAGTTACTCTGCCATC; CAV1.3 exon 2-exon 3: (F) TCCTATCTTGGCAAGCTGCAATCG, (R) AGCTAAGGCCACACAATTGGCA; H1F0 (intronless): (F) ATGACCGAGAACTCCACCTCC, (R) TTCTTCTTGCTGGCTCTCTTGG; GRIN1 exon 2-exon N1: (F) AGATGATGCGCGTCTACAACTGGAAC, (R) CATAGGACAGTTGGTCGAGGT; GRIN1 intron2 – exon N1: (F) GGTGTTTGCGAGCTCCAGG, (R) CATAGGACAGTTGGTCGAGGT.
Automated High Content Cellular Analyses
Multiparametric cellular profiling studies followed procedures described previously (23, 24). A549 or IMR-90 cells (10,000 per well) were plated in collagen-1 coated 384-well microplates and treated the next day with vehicle (DMSO) or ten two-fold concentration gradients of test agents. Cells were incubated for 24 h at 37 °C, fixed with formaldehyde, labeled with 10 μg/mL Hoechst 33342 and incubated with a primary antibody cocktail containing rabbit monoclonal anti-cleaved caspase-3 (1:500) and sheep polyclonal anti-p53 (1:500), followed by a mixture of Cy3-labeled donkey anti-rabbit IgG (1:250) and Cy5-labeled donkey anti-sheep IgG (1:250, both from Jackson ImmunoResearch). Microplates were analyzed with an ArrayScanII (Cellomics, Pittsburgh, PA) using the Compartmental Analysis Bioapplication. 1,000 individual cells in each well were imaged at excitation/emission wavelengths of 350/461 nm (Hoechst), 556/573 nm (Cy3), and 649/670 nm (Cy5). A nuclear mask was generated from Hoechst 33342-stained nuclei. Fluorescence intensities in the Cy3 and Cy5 channels were then measured in an area defined by the nuclear mask. The following parameters were extracted for data analysis: Average nuclear intensity (a measure of nuclear condensation), nuclei per field (cell density), object area (nuclear size) and average Cy3 and Cy5 intensities (to determine levels of cleaved caspase and p53, respectively). To calculate the percentages of cells with condensed nuclei, p53 nuclear accumulation, and cleaved caspase, thresholds for Hoechst, Cy3, and Cy5 intensities were defined as the average intensity plus one standard deviation from 28 vehicle-treated wells placed in the center of the microplate. Cells were classified as positive if their average Hoechst, Cy3, or Cy5 intensity exceeded this threshold.
Results
Stability of Meayamycin
FR901464 was not very stable in pH 7.4 phosphate buffer (t1/2 = 4 hours at 37 °C) (9). Although we previously reported the half-life of the right fragment of meayamycin (9), it was not obvious whether the result could be applied to the entire molecule. Thus, meayamycin was incubated at 10 μM in pH 7.4 phosphate buffer at 37 °C, and its decomposition was monitored by HPLC analysis. Under these conditions, the half-life was 80 hours (Figure 2a). When the pH of the solution in which stability was gauged was decreased to 7.4, 7, and 6, meayamycin exhibited progressively longer half-lives. At pH 5, the stability of meayamycin was similar to that at pH 7. A DMSO solution of meayamycin kept at 24 °C for 4 weeks then examined by HPLC showed no evidence of the decomposition (data not shown).
More relevant data in terms of a drug's biological effect on cells would be their stability in tissue culture media. Yoshida and co-workers showed that the half-life of FR901464 in RPMI culture medium containing 10% FBS was only 45 minutes (12). We found the half-life of meayamycin in RPMI containing 10% FBS to be 37 hours (Figure 2). Therefore, meayamycin is 50 times more stable than its parent compound and during cell culture experiments, more than 50% of the initial dose of meayamycin would be presumed to be preserved during the first 24 hours of compound treatment.
Because we have not yet determined the mechanisms by which meayamycin is degraded, initial-rate kinetics of meayamycin was analyzed under pseudo-first order conditions (-d[meayamycin]/dt = k[meayamycin]). The rate constants were found to be 1.1 × 10-6, 0.44 × 10-6, 1.0 × 10-6, 1.8 × 10-6, and 5.2 × 10-6 s-1 in pH 5 buffer, pH 6 buffer, pH 7 buffer, pH 7.4 buffer, and culture media, respectively.
Potency of Meayamycin Against Human Cancer Cell Lines
We examined antiproliferative properties of meayamycin against various tumor cell lines. The average 50% growth inhibitory concentrations (GI50) are reported in Table 1. Human breast cancer MCF-7 (gift from Professor Marc Lippman, Georgetown University) and MDA-MB231 cells were particularly responsive to the inhibitory effects of meayamycin, against which low picomolar GI50's were observed. Human colon cancer HCT-116 and human prostate cancer PC-3 cells were also quite responsive. Human non-small cell lung cancer H1299 and human lung cancer A549 cells showed very respectable mid-picomolar responses toward meayamycin. Human prostate cancer DU-145 cells were slightly less responsive.
Reversibility of Meayamycin-Induced Cell Growth Inhibition
The reversibility of growth inhibition by meayamycin was examined to determine whether the epoxide might form a covalent bond with target biomolecule(s). A549 cells were treated with meayamycin at 2 nM, and after the indicated times (Figure 2b), the compound was removed from some of the wells. The assay was continued with and without meayamycin for total of 4 days, at which point cell growth was assessed by the MTS assay. An 8-hour exposure of cells to 2 nM meayamycin was sufficient to irreversibly inhibit growth even after drug removal. We observed similar results with MCF-7 cells and HeLa cells (data not shown)
The data in Figure 2b suggest that meayamycin exerted its effect on cells within 8 hours. We therefore examined drug concentration-dependent cell proliferation after 8 hours of exposure to meayamycin followed by its removal from the culture medium. If meayamycin covalently binds to intracellular target protein(s) within 8 hours, the growth inhibition curve of the cells treated with meayamycin for 8 hours should be similar to that of cells treated with meayamycin for the entire assay period (25). As Figure 2c shows, when A549 cells were exposed to meayamycin for 8 hours and incubated for an additional 88 hours in its absence, the concentration-dependent growth inhibition was similar to that observed with meayamycin for 96 hours.
Activity Against Multi-Drug Resistant Cells
A major challenge in cancer therapy is the development of small molecules that are active against cells that have acquired multi-drug resistance (MDR), a phenotype that is typically mediated by expression of xenobiotic efflux pumps such as P-glycoprotein (26). We used the DC-3F (drug sensitive) / VCRd-5L (multi-drug resistant) cell line pair that has been extensively used and is well described in the literature (27). VCRd-5L cells were rendered resistant to vincristine and show cross-resistance to a variety of agents due to expression of efflux pumps (27). Figure 3 shows that VCRd5L cells were resistant to vincristine, paclitaxel, and to a lesser extent, FR901464, but retained complete sensitivity to picomolar concentrations of meayamycin.
Figure 3. Meayamycin's activity against multi-drug resistant VCRd-5L cells.
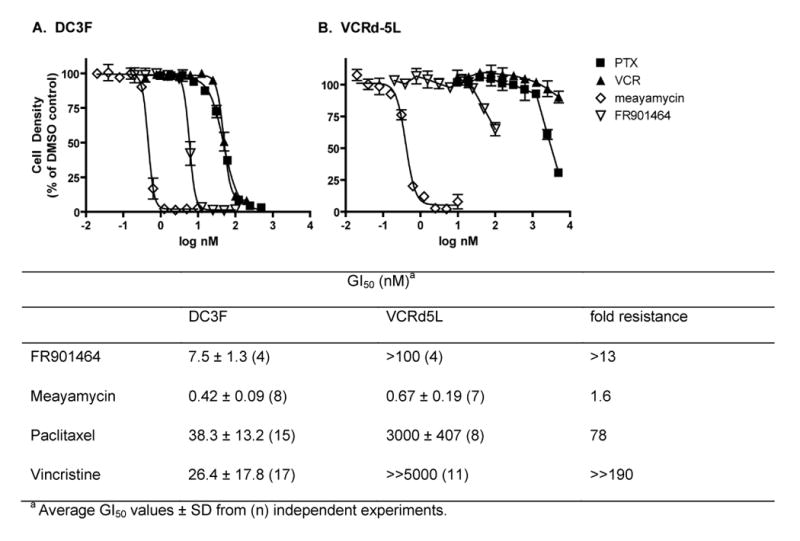
DC3F (A) or VCRd5L (B) cells were seeded into 384-well plates and treated for 72 hours with the indicated concentrations of paclitaxel (PTX, closed squares), vincristine (VCR, closed triangles), meayamycin (open diamonds) or FR901464 (open triangles). Cells were stained with Hoechst 33342 and nuclei enumerated on an ArrayScan II high-content reader. Nuclei counts were normalized to wells treated with vehicle (0.1% DMSO). Data represent the averages ± SE of quadruplicate determinations from a single representative experiment that has been repeated at least three times.
Pre-mRNA Splicing Inhibition
Although FR901464 inhibits pre-mRNA splicing in live cells (12), it was not obvious whether meayamycin, a far more stable analog of FR901464 (9), would exhibit similar biological activity. As such, HeLa nuclear extracts were treated with meayamycin and 5 at various concentrations and with isoginkgetin, a known inhibitor of pre-mRNA splicing (17) as a comparison (Figure 4). Meayamycin inhibited pre-mRNA splicing of the AdML pre-mRNA substrate in a concentration-dependent manner and 50 nM was sufficient for complete inhibition (Figure 4, left panel). Incubation of the same nuclear extract with up to 10 μM of the non-epoxide analog 5 had no effect upon splicing. Furthermore, compound 5 showed no antiproliferative activity even at 10 μM (data not shown). These data imply that the epoxide functionality in both FR910464 and meayamycin is necessary for both antiproliferative activity and pre-mRNA splicing inhibition. Analysis of the RNA-containing complex on a native gel showed that meayamycin blocked assembly of the spliceosomal A complex (Figure 4, middle panel), which is consistent with SF3b inhibition (12).
We then treated HEK-293 cells with meayamycin (10 nM) and monitored splicing inhibition over time (Figure 4, right panel). In 2 hours, unspliced RNA was noticeable, and in 4 to 7 hours meayamycin exhibited its full inhibitory activity. This time-dependence is similar to meayamycin's antiproliferative activity against human cancer cells (Figures 2b and 2c), supporting the notion that the antiproliferative activity of meayamycin is mediated by pre-mRNA splicing inhibition.
Alternative Splicing is Resistant to Meayamycin When Transcript Levels Are Low
To extend this analysis, we measured the effects of meayamycin on alternative splicing. Cortical neurons were chosen as the experimental system, since at high frequency alternative splicing produces modular changes in the expression of ion channels and other protein factors as these cells differentiate in culture (28, 29). Neurons were treated with meayamycin for 24 hours in culture, and total RNA was harvested for analysis of endogenous splicing patterns by RT-PCR. Primers were chosen for the RT-PCR analysis to amplify three-exon regions of these transcripts corresponding to the exons of interest. As additional controls, we tested a region of the intronless histone H1F0 transcript, as well as intron-containing regions of known pre-mRNAs.
A group of three alternatively-spliced cassette exons from the glutamate NMDA R1 receptor, GRIN1 (exon C1), the potassium channel KCNQ2 (exon 8), and the splicing regulatory factor, HNRPH3 (exon 3) showed no change in their splicing patterns upon treatment with meayamycin (Figure 6A). That is, the sizes of the observed exon-included and exon-skipped products were as expected (30), and the relative ratios of these products did not vary with the presence or absence of meayamycin. Constitutive exons from actin (exon 2) and HNRPH3 (exon 8) also showed no variations with the expected (single) mRNA product. We noticed that the mRNA product levels decreased for some transcripts at a final concentration of 20 pM meayamycin. A similar decrease was also observed for the intronless H1F0 transcript and for the pre-mRNA fragments containing introns. Nonetheless, the neurons remained viable to the same extent in the presence or absence of meayamycin as measured by trypan blue staining (Figure 6B). Thus, it is likely that meayamycin induces a decrease in transcription and/or mRNA stability, but does not affect alternative splicing regulation in these cells.
Figure 6. High-content analysis of A549 and IMR-90 cells treated with meayamycin.
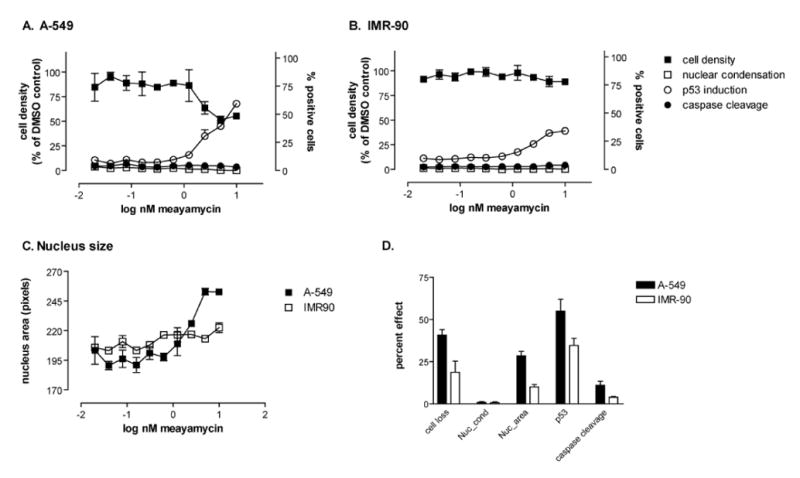
A549 (A) or IMR-90 cells (B) were plated at high density (10,000 cells per well), treated with vehicle or ten two-fold serial dilutions meayamycin for 24 hours and analyzed for cell density, chromatin condensation, nucleus area, p53 induction, and caspase cleavage on the ArrayScan II high-content reader. (A-C) Concentration dependence. Meayamycin caused non-cell loss (closed squares) and induced p53 (open circles) in A549 but not IMR-90 cells. Neither cell line underwent apoptosis as indicated by the absence of nuclear condensation (open squares) or caspase cleavage (close circles). Data are the averages ± SE of quadruplicate wells from a single representative experiment that has been repeated at least twice. D. Magnitude of response at 10 nM meayamycin. Data are the averages ± SE of multiple independent experiments performed in quadruplicate (n=6 for A549 and n=3 for IMR-90) at the highest concentration of meayamycin used (10 nM). Cell loss, percent of cells lost during treatment; nuc_cond, % cells with condensed nuclei; nuc_area, % increase in nuclear size compared with vehicle; p53, % p53 positive cells; caspase cleavage, % of cleaved caspase positive cells.
High-Content Profiling of Meayamycin in Human Lung Cancer Cells Compared with Non-tumorigenic Human Lung Fibroblasts
Since the intracellular functions of SF3b are poorly understood, it was not clear what phenotype should be observable when cells are treated with FR901464-class compounds, nor if inhibitors of SF3b would show selectivity for the transformed phenotype. We therefore probed cancer-relevant activities of meayamycin by high-content multiparametric cellular profiling in A549 human lung cancer cells and IMR-90 non-tumorigenic human lung fibroblasts. We chose the IMR-90 line because it is well characterized and known to be resistant to a variety of cytotoxic and antisignaling agents (31-34). The IMR-90 cells have a finite lifespan of approximately 60 population doublings (35) before entering crisis, which we confirmed in our laboratory (data not shown).
Twenty-four hours after treatment, meayamycin consistently caused a more pronounced cell loss in the A549 lung cancer cells compared with normal cells (Figures 6A and 6B). Nuclear morphology measurements indicated that meayamycin did not induce chromatin condensation (Figure 6A and 6B, open squares), suggesting a non-apoptotic mode of cell death and the absence of mitotic arrest. Consistent with non-apoptotic death, no caspase cleavage was observed (Figure 6A and 6B, closed circles). We occasionally observed a low level of caspase cleavage that appeared to be higher in the A549 cells, which accounts for the slightly higher response seen with A549 cell in Figure 6d but the effect was not seen consistently and its magnitude was low. Similar results were obtained after a 48-hour treatment (data not shown). Both cell lines responded to meayamycin with p53 induction, although the magnitude of response was smaller in the normal cells (Figure 6D). The meayamycin-induced increase in p53 was, however, not responsible for antiproliferative activity because wild type (wt) Tp53+/+ and Tp53−/− HCT116 cell lines (36) were equally sensitive to meayamycin, with GI50 values of 0.66 nM for wt HCT116 and 0.95 nM for p53−/− HCT116 cells.
Meayamycin consistently caused enlarged nuclei (Figure 6C), which may be related to splicing inhibition-induced RNA accumulation that results in speckle enlargement (37, 38). Similar results were obtained after a 48-hour exposure to meayamycin (data not shown). Data were repeatable and consistent over the course of several independent experiments (Figure 6D) and suggested that meayamycin preferentially affected the survival of lung cancer cells compared with non-tumorigenic lung fibroblasts but did not cause apoptosis.
Discussion
Stability of Meayamycin
Meayamycin was very stable in pH 7.4 phosphate buffer and relatively stable in cell culture medium. The enhanced chemical stability relative to FR901464 is meritorious because unlike FR901464, solutions of meayamycin could be stored even at ambient temperature for months without significant decomposition. The half-life of meayamycin in tissue culture media is useful information to design cell-based experiments and interpret data.
Potency of Meayamycin Against Human Cancer Cell Lines
The growth of various tumor cell lines was inhibited by picomolar concentrations of meayamycin. The activity profile of meayamycin is noticeably different from that of pladienolide; for example, although the MCF-7 and HCT-116 cell lines are very sensitive to both pladienolide and meayamycin, the MDA-MB231 cell line is far more affected by meayamycin than by pladienolide (13). Additionally, the A549 and DU-145 cell lines are very sensitive to pladienolide but less so to meayamycin.
Reversibility of Meayamycin-Induced Cell Growth Inhibition
Some epoxide-containing anticancer natural products such as fumagillin form a covalent bond with specific proteins in vivo, while others such as the epothilones do not (39), which is consistent with the low intrinsic electrophilicity of epoxides under biological conditions (39, 40). Therefore, we asked if meayamycin would act as an electrophile to form a covalent bond with target biomacromolecule(s). This is a testable question because covalent bond formation between drugs and biomacromolecules typically results in irreversible growth inhibition within the experimental time frame (3–5 days), which manifests itself as continuous growth inhibition after removing drug from culture media (25). Non-covalent binding usually results in a return to cell proliferation after drug removal (17).
Biochemical studies by the Yoshida group showed that FR901464-SF3b protein binding may be reversible (12). If FR901464 reversibly binds to a splicing factor and this binding is responsible for the antiproliferative activity, removal of meayamycin at an early stage should have resulted in a return to the typical rate of cell growth. However, transient treatment of A549 (Figures 2b and 2c), HeLa, and MCF-7 cells (data not shown) with meayamycin resulted in growth inhibition profiles similar to those seen with cells treated with meayamycin for 4 days. These data suggest that, at least in these cell lines, the binding mode of meayamycin may be irreversible. Alternatively, reversible binding of meayamycin may induce irreversible cellular events that lead to cell growth arrest. Further studies are needed to precisely determine the nature of the interaction of meayamycin with its proposed cellular targets.
Therefore, together with the lack of activity of 4 (5) and 5, there is a line of evidence suggesting that the antiproliferative activity of meayamycin is mediated by biomolecule(s) that attack the epoxide to form a covalent bond.
Activity Against Multi-Drug Resistant Cells
A striking advantage of meayamycin, aside from its improved potency and stability compared to FR901464, was found to be its potency against an MDR cell line. The improved potency (100-fold) of meayamycin relative to that of FR901464 may be explained by its enhanced stability in cell culture media (56-fold); however, the explanation behind its improved potency against cells with the MDR phenotype is less obvious. A common mechanism of MDR is the overexpression of P-glycoprotein (ABCB1), which often exports aromatic ring-containing small molecules (41). However, other factors play a significant role in MDR mechanisms. For example, when the ester moiety of an epothilone was replaced by an amide bond, the resulting analog lost cytotoxicity against an MDR cell line (42). Clearly, the minor chemical modifications represented by the change in FR901464 to meayamycin dramatically improved the potency against an MDR cell line. Currently we do not know what causes the extraordinary ability of meayamycin to inhibit the growth of MDR cells. The interaction of meayamycin with cellular proteins that mediate multi-drug resistance should be a focus of future investigations.
Pre-mRNA Splicing Inhibition
The activity of meayamycin in splicing assays in vitro and in live cells was found to be similar to that of 3 and pladienolide D. In in vitro assays, pre-mRNA substrate is initially coated with a heterogeneous mixture of RNA-binding proteins, referred to as the H complex. The first step in spliceosome assembly (the E complex) is the recognition of the 5′ splice site by the U1 snRNP and recognition of the branchpoint by U2 snRNA and its associated proteins (43). Spliceosomal A complex assembly is the first ATP-requiring step and results in stabilization of the branchpoint-U2 snRNP interaction. Addition of the U4/U5/U6 tri-snRNP, forming the B complex, and subsequent structural rearrangements lead to the catalytically competent C complex spliceosome. Native gel electrophoresis of pre-mRNA transcripts from in vitro splicing reactions shows that a pre-mRNA substrate moves through a progression of complexes as defined by their electrophoretic mobilities. Consistent with interfering with binding and/or function of SF3b, in vitro spliceosome assembly was blocked by meayamycin at its earliest stages. As expected from the SF3b binding, the presence of meayamycin in in vitro splicing reactions prevented the transition from the H complex to the A complex.
Alternative Splicing
The increased complexity in human proteome relative to its genomic counterpart is primarily due to alternative RNA splicing. As such, the regulation of alternative splicing plays pivotal roles in many biological processes and human disease (44, 45). We examined whether meayamycin perturbed alternative splicing in cortical neuron cells. None of the alternative splicing processes that we examined was affected by meayamycin at 20 pM. We observed a decrease in the mRNA level of intronless H1F0, suggesting that meayamycin may induce a decrease in transcription and/or mRNA stability. This observation raises a question about whether the meayamycin-mediated splicing inhibition is less sensitive under in vivo conditions in which splicing is coupled to transcriptional regulation, for which further studies are needed (46).
Although meayamycin does not perturb alternative RNA splicing in the system we examined, this does not exclude the possibility that this compound interferes with other splicing regulatory systems. For example, SAP155, one of several possible targets for FR901464 (12), binds to ceramide-responsive RNA cis-element 1 and regulates the alternative splicing of Bcl-x (47). Therefore, meayamycin may regulate the alternative splicing of Bcl-x.
High-Content Profiling of Meayamycin in Human Lung Cancer Cells
The toxicity of meayamycin in non-tumorigenic cells was addressed; the result was encouraging in that the compound did not show significant toxicity against IMR-90 human lung fibroblasts. At this point, it is not clear whether such cell-type specificity can be explained by splicing inhibition. Nonetheless, this result should warrant future drug development. Meayamycin had a unique profile of cellular activities. Exposure to meayamycin resulted in non-apoptotic toxicity that was selective for transformed cells as measured by cell loss, chromatin condensation, and caspase cleavage. Consistent with previous reports using FR901464 (1, 2), meayamycin did not induce mitotic arrest (Figure 6 and data not shown). We also examined alterations in the levels of the p53 protein, an indicator for DNA damage. As Figure 6a and b illustrate, meayamycin concentration-dependently increased the level of p53 protein in A549 and, to a lesser extent, in IMR-90 cells. This is interesting because FR901464 has been reported to lower the mRNA level of p53, potentially via splicing inhibition (2). It is possible that p53 protein degradation is inhibited because the p53 mRNA impairs Mdm2 through direct binding, resulting in increased levels of p53 protein (48). The meayamycin-induced increase in p53 was, however, not responsible for antiproliferative activity because wild type (wt) and p53−/− HCT116 cell lines were equally sensitive to meayamycin.
Acknowledgments
We would like to thank Professor Stephen Weber (University of Pittsburgh) for sharing his Spectromax M2 plate reader with us. We thank Professors Bert Vogelstein (Johns Hopkins) and Lin Zhang (University of Pittsburgh) for sharing their p53−/− cell line with us.
Grant support: Graduate Excellence Fellowship from the University of Pittsburgh to B.J.A. The Arnold and Mabel Beckman Scholar Award, the Lilly Summer Research Fellowship, and the Averill Scholarship to N.L.C. The National Cancer Institute (R01 CA120792 to K.K. and CA 78039 to A.V.). The National Institutes of Health (R01 GM35007) to M.J.M. HHMI to M.J.M.
Abbreviations
- DMSO
dimethyl sulfoxide
- SF3b
splicing factor 3b
- MDR
multidrug resistance
Footnotes
Potential Conflicts of Interest. No potential conflicts of interest were listed.
References
- 1.Nakajima H, Sato B, Fujita T, Takase S, Terano H, Okuhara M. New antitumor substances, FR901463, FR901464 and FR901465. 1. Taxonomy, fermentation, isolation, physico-chemical properties and biological activities. J Antibiot. 1996;49:1196–203. doi: 10.7164/antibiotics.49.1196. [DOI] [PubMed] [Google Scholar]
- 2.Nakajima H, Hori Y, Terano H, et al. New antitumor substances, FR901463, FR901464 and FR901465. II. Activities against experimental tumors in mice and mechanism of action. J Antibiot. 1996;49:1204–11. doi: 10.7164/antibiotics.49.1204. [DOI] [PubMed] [Google Scholar]
- 3.Thompson CF, Jamison TF, Jacobsen EN. Total synthesis of FR901464. Convergent assembly of chiral components prepared by asymmetric catalysis. J Am Chem Soc. 2000;122:10482–3. [Google Scholar]
- 4.Horigome M, Motoyoshi H, Watanabe H, Kitahara T. A synthesis of FR901464. Tetrahedron Letters. 2001;42:8207–10. [Google Scholar]
- 5.Thompson CF, Jamison TF, Jacobsen EN. FR901464: total synthesis, proof of structure, and evaluation of synthetic analogues. J Am Chem Soc. 2001;123:9974–83. doi: 10.1021/ja016615t. [DOI] [PubMed] [Google Scholar]
- 6.Albert BJ, Koide K. Synthesis of a C4-epi-C1-C6 fragment of FR901464 using a novel bromolactolization. Org Lett. 2004;6:3655–8. doi: 10.1021/ol049160w. [DOI] [PubMed] [Google Scholar]
- 7.Albert BJ, Sivaramakrishnan A, Naka T, Koide K. Total synthesis of FR901464, an antitumor agent that regulates the transcription of oncogenes and tumor suppressor genes. J Am Chem Soc. 2006;128:2792–3. doi: 10.1021/ja058216u. [DOI] [PubMed] [Google Scholar]
- 8.Motoyoshi H, Horigome M, Watanabe H, Kitahara T. Total synthesis of FR901464: second generation. Tetrahedron. 2006;62:1378–89. [Google Scholar]
- 9.Albert BJ, Sivaramakrishnan A, Naka T, Czaicki NL, Koide K. Total syntheses, fragmentation studies, and antitumor/antiproliferative activities of FR901464 and its low picomolar analogue. J Am Chem Soc. 2007;129:2648–59. doi: 10.1021/ja067870m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koide K, Albert BJ. Review: Total syntheses of FR901464. J Synth Org Chem, Jpn. 2007;65:119–26. [Google Scholar]
- 11.Motoyoshi H, Horigome M, Ishigami K, et al. Structure-activity relationship for FR901464: A versatile method tor the conversion and preparation of biologically active biotinylated probes. Biosci, Biotechnol, Biochem. 2004;68:2178–82. doi: 10.1271/bbb.68.2178. [DOI] [PubMed] [Google Scholar]
- 12.Kaida D, Motoyoshi H, Tashiro E, et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat Chem Biol. 2007;3:576–83. doi: 10.1038/nchembio.2007.18. [DOI] [PubMed] [Google Scholar]
- 13.Mizui Y, Sakai T, Iwata M, et al. Pladienolides, new substances from culture of Streptomyces platensis Mer-11107 III. In vitro and In vivo antitumor activities. J Antibiot. 2004;57:188–96. doi: 10.7164/antibiotics.57.188. [DOI] [PubMed] [Google Scholar]
- 14.Grosso AR, Martins S, Carmo-Fonseca M. The emerging role of splicing factors in cancer. EMBO Reports. 2008;9:1087–93. doi: 10.1038/embor.2008.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanada RM, Itoh D, Nagai M, et al. Total synthesis of the potent antitumor macrolides pladienolide B and D. Angew Chem Int Ed. 2007;46:4350–5. doi: 10.1002/anie.200604997. [DOI] [PubMed] [Google Scholar]
- 16.Lagisetti C, Pourpak A, Jiang Q, et al. Antitumor compounds based on a natural product consensus pharmacophore. J Med Chem. 2008;51:6220–4. doi: 10.1021/jm8006195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Brien K, Matlin AJ, Lowell AM, Moore MJ. The biflavonoid isoginkgetin is a general inhibitor of pre-mRNA splicing. J Biol Chem. 2008;283:33147–54. doi: 10.1074/jbc.M805556200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vogt A, Kalb EN, Lazo JS. Scalable high-content cytotoxicity assay insensitive to changes in mitochondrial metabolic activity. Oncol Res. 2004;14:305–14. doi: 10.3727/096504003773994842. [DOI] [PubMed] [Google Scholar]
- 19.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–89. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abmayr SM, Workman JL, Roeder RG. The pseudorabies immediate early protein stimulates in vitro transcription by facilitating TFIID: promoter interactions. Genes Dev. 1988;2:542–53. doi: 10.1101/gad.2.5.542. [DOI] [PubMed] [Google Scholar]
- 21.Reichert V, Moore MJ. Better conditions for mammalian in vitro splicing provided by acetate and glutamate as potassium counterions. Nucleic Acids Res. 2000;28:416–23. doi: 10.1093/nar/28.2.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Konarska MM, Sharp PP. Interactions between small nuclear ribonucleoprotein particles in formation of spliceosomes. Cell. 1987;49:763–74. doi: 10.1016/0092-8674(87)90614-3. [DOI] [PubMed] [Google Scholar]
- 23.Wipf P, Graham TH, Vogt A, Sikorski RP, Ducruet AP, Lazo JS. Cellular analysis of disorazole C-1 and structure-activity relationship of analogs of the natural product. Chem Biol Drug Des. 2006;67:66–73. doi: 10.1111/j.1747-0285.2005.00313.x. [DOI] [PubMed] [Google Scholar]
- 24.Giuliano KA. High-content profiling of drug-drug interactions: Cellular targets involved in the modulation of microtubule drug action by the antifungal ketoconazole. J Biomol Screen. 2003;8:125–35. doi: 10.1177/1087057103252616. [DOI] [PubMed] [Google Scholar]
- 25.Leonce S, Perez V, Lambel S, et al. Induction of cyclin E and inhibition of DNA synthesis by the novel acronycine derivative S23906-1 precede the irreversible arrest of tumor cells in S phase leading to apoptosis. Mol Pharmacol. 2001;60:1383–91. doi: 10.1124/mol.60.6.1383. [DOI] [PubMed] [Google Scholar]
- 26.Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Disc. 2006;5:219–34. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 27.Scotto KW, Biedler JL, Melera PW. Amplification and expression of genes associated with multidrug resistance in mammalian cells. Science. 1986;232:751–5. doi: 10.1126/science.2421411. [DOI] [PubMed] [Google Scholar]
- 28.Xing Y, Lee CJ. Protein modularity of alternatively spliced exons is associated with tissue-specific regulation of alternative splicing. PLoS Genet. 2005;1:e34. doi: 10.1371/journal.pgen.0010034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Q, Lee JA, Black DL. Neuronal regulation of alternative pre-mRNA splicing. Nat Rev Neurosci. 2007;8:819–31. doi: 10.1038/nrn2237. [DOI] [PubMed] [Google Scholar]
- 30.An P, Grabowski PJ. Exon silencing by UAGG motifs in response to neuronal excitation. PLoS Biol. 2007;5:263–80. doi: 10.1371/journal.pbio.0050036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Erickson LC, Bradley MO, Ducore JM, Ewig RA, Kohn KW. DNA crosslinking and cytotoxicity in normal and transformed human cells treated with antitumor nitrosoureas. Proc Natl Acad Sci U S A. 1980;77:467–71. doi: 10.1073/pnas.77.1.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dusre L, Covey JM, Collins C, Sinha BK. DNA damage, cytotoxicity and free radical formation by mitomycin C in human cells. Chem Biol Interact. 1989;71:63–78. doi: 10.1016/0009-2797(89)90090-2. [DOI] [PubMed] [Google Scholar]
- 33.Hsieh TJ, Liu TZ, Chern CL, et al. Liriodenine inhibits the proliferation of human hepatoma cell lines by blocking cell cycle progression and nitric oxide-mediated activation of p53 expression. Food Chem Toxicol. 2005;43:1117–26. doi: 10.1016/j.fct.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 34.Suzuki H, Aoshiba K, Yokohori N, Nagai A. Epidermal growth factor receptor tyrosine kinase inhibition augments a murine model of pulmonary fibrosis. Cancer Res. 2003;63:5054–9. [PubMed] [Google Scholar]
- 35.Nichols WW, Murphy DG, Cristofalo VJ, Toji LH, Greene AE, Dwight SA. Characterization of a new human diploid cell strain, IMR-90. Science. 1977;196:60–3. doi: 10.1126/science.841339. [DOI] [PubMed] [Google Scholar]
- 36.Bunz F, Dutriaux A, Lengauer C, et al. Requirement for p53 and p21 to sustain G(2) arrest after DNA damage. Science. 1998;282:1497–501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 37.Harris H. The use of cell fusion in the analysis of gene action. Proc Royal Soc Lond B. 1970;176:315–7. doi: 10.1098/rspb.1970.0051. [DOI] [PubMed] [Google Scholar]
- 38.O'Keefe RT, Mayeda A, Sadowski CL, Krainer AR, Spector DL. Disruption of premessenger RNA splicing in-vivo results in reorganization of splicing factors. J Cell Biol. 1994;124:249–60. doi: 10.1083/jcb.124.3.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Albert BI, Koide K. How rapidly do epoxides nonspecifically form covalent bonds with thiols in water? ChemBioChem. 2007;8:1912–5. doi: 10.1002/cbic.200700365. [DOI] [PubMed] [Google Scholar]
- 40.Weerapana E, Simon GM, Cravatt BF. Disparate proteome reactivity profiles of carbon electrophiles. Nat Chem Biol. 2008;4:405–7. doi: 10.1038/nchembio.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yeh GC, Lopaczynska J, Poore CM, Phang JM. A new functional-role for P-glycoprotein - efflux pump for benzo(a)pyrene in human breast-cancer MCF-7 cells. Cancer Res. 1992;52:6692–5. [PubMed] [Google Scholar]
- 42.Stachel SJ, Chappell MD, Lee CB, et al. On the total synthesis and preliminary biological evaluations of 15(R) and 15(S) aza-dEpoB: A Mitsunobu inversion at C15 in pre-epothilone fragments. Org Lett. 2000;2:1637–9. doi: 10.1021/ol005932m. [DOI] [PubMed] [Google Scholar]
- 43.Will CL, Lührmann R. The RNA World. 3rd. Cold Spring Harbor Laboratory Press; 2006. [Google Scholar]
- 44.Licatalosi DD, Darnell RB. Splicing regulation in neurologic disease. Neuron. 2006;52:93–101. doi: 10.1016/j.neuron.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 45.Orengo JP, Cooper TA. Alternative splicing in the postgenomic era. 2007 [Google Scholar]
- 46.Bentley DL. Rules of engagement: co-transcriptional recruitment of pre-mRNA processing factors. Curr Opin Cell Biol. 2005;17:251–6. doi: 10.1016/j.ceb.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 47.Massiello A, Roesser JR, Chalfant CE. SAP155 binds to ceramide-responsive RNA cis-element 1 and regulates the alternative 5 ′ splice site selection of Bcl-x pre-mRNA. FASEB J. 2006;20:1680–2. doi: 10.1096/fj.05-5021fje. [DOI] [PubMed] [Google Scholar]
- 48.Candeias MM, Malbert-Colas L, Powell DJ, et al. p53 mRNA controls p53 activity by managing Mdm2 functions. Nat Cell Biol. 2008;10:1098–105. doi: 10.1038/ncb1770. [DOI] [PubMed] [Google Scholar]