

Abstract
This study reports results of an extensive and comprehensive study of genetic diversity in 12 genes of the innate immune system in a population of eastern India. Genomic variation was assayed in 171 individuals by resequencing ~75 kb of DNA comprising these genes in each individual. Almost half of the 548 DNA variants discovered was novel. DNA sequence comparisons with human and chimpanzee reference sequences revealed evolutionary features indicative of natural selection operating among individuals, who are residents of an area with a high load of microbial and other pathogens. Significant differences in allele and haplotype frequencies of the study population were observed with the HapMap populations. Gene and haplotype diversities were observed to be high. The genetic positioning of the study population among the HapMap populations based on data of the innate immunity genes substantially differed from what has been observed for Indian populations based on data of other genes. The reported range of variation in SNP density in the human genome is one SNP per 1.19 kb (chromosome 22) to one SNP per 2.18 kb (chromosome 19). The SNP density in innate immunity genes observed in this study (>3 SNPs kb−1) exceeds the highest density observed for any autosomal chromosome in the human genome. The extensive genomic variation and the distinct haplotype structure of innate immunity genes observed among individuals have possibly resulted from the impact of natural selection.
Keywords: Host, Pathogen, Evolution, DNA resequencing, Single nucleotide polymorphism, Haplotype, Genome diversity
1. Introduction
Evolutionarily, the adaptive or acquired immune system is recent. It was built atop the innate immune system, which is phylogenetically more ancient and developed before the separation of vertebrates and invertebrates. Invertebrates and jawless fish depend solely on the innate immune system. The innate immune system controls and assists the adaptive immune system, without which the adaptive immune response offers weak protection (Kimbrell and Beutler, 2001). Being evolutionary ancient, innate immune system may be considered to have been highly optimized by natural selection and, if true, diversity in the innate immunity genes is expected to be low. We, however, note that it has recently been emphasized (Parham, 2003) that innate immunity has the capacity to prevent primary infections from actually causing disease, “an attractive feature that is out of bounds for adaptive immunity,” and therefore the innate immune system may be plastic and continuing to evolve.
We report here the results of an extensive and comprehensive study on the natural variation in 12 innate immunity genes in a slum area of Kolkata (formerly Calcutta), India. Residents of this area - living in extremely impoverished and unhygienic conditions - are episodically exposed to a high load of microbial pathogens, particularly gastro-intestinal pathogens. Annual outbreaks of typhoid, cholera and other gastro-intestinal tract disorders are documented in this area. Based on data from a surveillance study conducted by the National Institute of Cholera and Enteric Diseases, Kolkata, the annual incidence rates (unpublished) of typhoid and cholera were estimated to be: (a) blood or serology-positive typhoid fever = 5.9/1000 population/year, (b) stool-culture positive cholera = 1.5/1000 population/year, and (c) blood culture-positive typhoid fever = 1.1/1000 population/year. Skin infections are also common. We show that there is extensive variation in the innate immunity genes, including the discovery of 259 novel (previously unreported in dbSNP) variations in these genes. We also present an analysis of the comparable portion of our data with HapMap data and draw inferences regarding the population structure of these genes.
2. Materials and methods
2.1. Study populations and participants
The study participants belonged to two different communities - Muslim and Hindu - who do not intermarry. The Muslims of the study area are recent converts from Hinduism. Both groups speak languages that belong to the Indo-European linguistic family. During the course of our study, we have assessed the extent of social substructuring within the two communities, in terms of marriage practices. Within each community, individuals marry freely without any social restrictions. Unrelated, healthy (ascertained to have not suffered from any major infection during the last six months or were not suffering from any chronic disease) individuals (n = 171; Muslim = 86, Hindu = 85), of both genders and of ages 12 years or older, were recruited into this study and blood samples were collected from them by venipuncture with voluntary, informed and written consent, after obtaining institutional ethical approval. From each blood sample, DNA was isolated using Qiagen columns, using the manufacturer's protocol.
2.2. Genes studied and resequencing
We have studied 12 innate immunity genes. These are: cathelicidin antimicrobial peptide (CAMP), defensins (DEFA4, DEFA5, DEFA6 and DEFB1), mannose binding lectin (MBL2), and toll-like receptors (TLR1, TLR2, TLR4, TLR5, TLR6 and TLR9). CAMP is one of the major antimicrobial peptides of the human innate immune system in the intestinal tract (Nizet and Gallo, 2003). The defensins code for small cationic, cysteine-rich peptides. These peptides possess a broad antimicrobial activity. On the basis of the position and bonding of six conserved cysteine residues, defensins in vertebrates are divided into two categories, designated as α-and β-defensins (Ganz and Lehrer, 1994). While α-defensins are produced primarily by intestinal Paneth cells and neutrophils, the β-defensins are primarily produced by epithelial cells. MBL is a calcium-dependent serum protein that plays a role in the innate immune response by binding to carbohydrates on the surface of a wide range of pathogens (Garred et al., 2006). It is also an important component of the complement-activation pathway for activation of macrophages by forming membrane-attack complexes (MACs). The toll-like receptors (TLRs) are a class of single membrane-spanning, non-catalytic receptors that recognize structurally conserved molecules derived from microbes once they have breached physical barriers such as skin or intestinal tract mucosa (Takeda et al., 2003). They are characterized by an extracellular leucine-rich repeat (LRR) domain for ligand recognition and an intracellular tail that bears a homology to the conserved interleukin 1 receptor (TIR) for signal transduction. Ten TLRs (TLR1-TLR10), each with its own ligand specificity, have been identified. We have studied six of these TLR genes.
To identify and catalog natural variation in these genes, we have carried out double-pass resequencing of the exons, exon-intron boundaries and ~1 kb of each of 5′-upstream and 3′-downstream of each gene, using a capillary sequencer (ABI-3730). The chromosomal location and the length of DNA resequenced for each gene are provided in Supplementary Table 1. A total of ~75 kb of the genome was resequenced for each study participant. In addition to double pass-resequencing, for quality check, negative controls (deionised water) and positive controls (DNA from CEPH samples) were included on each plate. Analyses of sequence chromatograms and genotype calls were carried out using SeqScape ® v2.5 (Applied Biosystems) and PolyPhred (http://droog.mbt.washington.edu/PolyPhred.html) software packages. Coding regions were translated using DNASTAR ® and BioEdit ®packages. Negative controls did not yield any sequences; our genotype calls for the CEPH samples coincided with CEPH data.
2.3. Statistical analysis
Maximum-likelihood estimates of allele frequencies and their standard deviations were estimated using MAXLIK (Reed and Schull, 1968). Tests for equality of genotype frequencies with those expected under Hardy-Weinberg equilibrium were also carried out using MAXLIK. Tests for equality of genotype frequencies between the two study communities, estimation of false discovery rates (Benjamini and Hochberg, 1995; Storey and Tibshirani, 2003), genotype and haplotype diversities (Nei, 1987) were carried out using standard methods implemented in computer programs developed by us. Haplotype identification and estimation of haplotype frequencies were done using PHASE for Windows, version 2.1 (http://www.stat.washington.edu/stephens; Stephens et al., 2001; Stephens and Donnelly, 2001). PHASE was run using the default settings; for the TLR1 tri-allelic SNP, rs4540055, data on individuals possessing the most infrequent allele were not included. We have used the DA distance measure (Nei, 1987) computed on the basis of allele frequencies and DISPAN (http://www.bio.psu.edu/People/Faculty/Nei/Lab/dispan2.htm) to carry out cluster analysis of populations.
3. Results
For each study participant, ~75 kb of DNA spanning 12 innate immunity genes was resequenced. A total of 548 variations were detected (Supplementary Table 2), of which 229 were polymorphic (Table 1). No polymorphic variant was observed in CAMP. Most variants (528; 96%) are single nucleotide changes. However, 20 (4%) changes are single-or multi-nucleotide insertions/deletions (INDELs). One tri-allelic variation (alleles: T, A and G) was also detected in TLR1 (rs4540055). A large number (259 of 548; 47%) of the detected variants were novel, i.e., unreported in dbSNP (build 126) prior to our submission; these have been submitted by us to dbSNP. Of these novel variations, ~10% were polymorphic. From these data it is clear that the observed frequencies of DNA variants and polymorphisms are, respectively, ~7.4 and ~3.1 kb−1. (Further details about the nature of variations are provided in Supplementary Table 3.) Among the 548 variations observed, 92 are in coding regions, of which 50 are non-synonymous (Supplementary Table 2). Of these 92 variants in coding regions, 21 (23%) are polymorphic; 11 (52%) are non-synonymous and 10 (48%) are synonymous (Supplementary Table 2).
Table 1.
Minor allele (allele with lower frequency) frequencies at polymorphica loci in 11 innate immunity genes in the study sample from Kolkata, India
| Gene name | Locus IDb | Minor allele frequency (MAF) | S.D. of MAF |
|---|---|---|---|
| DEFA4 | rs45482601c,d | 0.091 | 0.016 |
| rs2741676 | 0.216 | 0.022 | |
| rs2738098 | 0.157 | 0.020 | |
| DEFA4NV007 | 0.305 | 0.025 | |
| rs2741677 | 0.077 | 0.014 | |
| rs10089687 | 0.320 | 0.025 | |
| rs2239667 | 0.204 | 0.022 | |
| rs2741680 | 0.073 | 0.014 | |
| rs2738100d | 0.463 | 0.028 | |
| rs736227 | 0.300 | 0.025 | |
| rs2702867 | 0.294 | 0.025 | |
| DEFA5 | rs4610776 | 0.201 | 0.022 |
| rs2272719 | 0.269 | 0.024 | |
| rs45477802 | 0.133 | 0.018 | |
| rs45628240 | 0.133 | 0.018 | |
| DEFA6 | rs3842204 | 0.132 | 0.018 |
| rs3824304 | 0.132 | 0.018 | |
| rs2741689 | 0.171 | 0.020 | |
| rs2741690 | 0.171 | 0.020 | |
| rs12721595 | 0.067 | 0.014 | |
| rs4458901 | 0.453 | 0.027 | |
| rs11784359 | 0.331 | 0.026 | |
| rs45479905 | 0.073 | 0.014 | |
| rs2738120 | 0.314 | 0.030 | |
| rs712276 | 0.157 | 0.020 | |
| DEFB1 | rs2472143 | 0.188 | 0.032 |
| rs5743399 | 0.152 | 0.020 | |
| rs2978862 | 0.288 | 0.025 | |
| rs5743401 | 0.160 | 0.026 | |
| rs5743402 | 0.243 | 0.023 | |
| rs2741137 | 0.227 | 0.023 | |
| rs2741136 | 0.138 | 0.019 | |
| rs5743404 | 0.399 | 0.027 | |
| rs5743407 | 0.494 | 0.028 | |
| rs2741135 | 0.298 | 0.025 | |
| rs2978863 | 0.227 | 0.023 | |
| rs2741134 | 0.365 | 0.027 | |
| rs2702877 | 0.162 | 0.021 | |
| rs2741133 | 0.359 | 0.027 | |
| rs5743415 | 0.082 | 0.015 | |
| rs5743416 | 0.082 | 0.015 | |
| rs2702876 | 0.365 | 0.027 | |
| rs2741132 | 0.025 | 1.000 | |
| rs2738182 | 0.027 | 0.265 | |
| rs1799946 | 0.026 | 0.148 | |
| rs1800972 | 0.019 | 1.000 | |
| rs11362 | 0.027 | 1.000 | |
| rs45613938 | 0.012 | 0.354 | |
| rs2293959 | 0.019 | 1.000 | |
| rs2293960 | 0.019 | 1.000 | |
| rs2702945 | 0.023 | 1.000 | |
| rs5743428 | 0.013 | 0.018 | |
| rs7003198 | 0.020 | 0.228 | |
| rs5743432 | 0.013 | 0.419 | |
| rs5743433 | 0.013 | 0.419 | |
| rs5743435 | 0.013 | 0.071 | |
| rs2741130 | 0.028 | 0.115 | |
| rs5743437 | 0.019 | 0.747 | |
| rs5743439 | 0.019 | 0.037 | |
| rs5743440 | 0.020 | 0.751 | |
| rs2978864 | 0.019 | 1.000 | |
| rs45588335 | 0.014 | 0.568 | |
| rs45499493 | 0.015 | 0.277 | |
| rs10543366 | 0.019 | 0.742 | |
| rs2741129 | 0.025 | 0.597 | |
| rs2977772 | 0.019 | 1.000 | |
| rs5743450 | 0.013 | 0.417 | |
| rs2980923 | 0.019 | 1.000 | |
| rs5743454 | 0.013 | 0.452 | |
| rs2951854 | 0.020 | 0.000 | |
| rs2741126 | 0.027 | 0.009 | |
| rs5743470 | 0.027 | 1.000 | |
| rs2978871 | 0.019 | 1.000 | |
| rs45474591 | 0.015 | 0.311 | |
| rs5743476 | 0.020 | 0.362 | |
| rs2980926 | 0.019 | 0.743 | |
| rs2977778 | 0.019 | 0.742 | |
| rs2977779 | 0.023 | 0.219 | |
| rs2980927 | 0.018 | 0.742 | |
| rs2980928 | 0.019 | 0.742 | |
| rs2977780 | 0.019 | 0.742 | |
| rs5743480 | 0.013 | 0.450 | |
| rs2977781 | 0.023 | 1.000 | |
| rs2978873 | 0.019 | 0.743 | |
| rs5743482 | 0.027 | 0.876 | |
| rs2978874 | 0.023 | 0.219 | |
| rs2978875 | 0.019 | 0.743 | |
| rs767423 | 0.023 | 1.000 | |
| rs1344197 | 0.019 | 1.000 | |
| rs2741124 | 0.023 | 0.297 | |
| rs2702885 | 0.024 | 0.293 | |
| rs1047031 | 0.024 | 0.053 | |
| rs1800971 | 0.016 | 0.642 | |
| rs45442801 | 0.016 | 0.365 | |
| MBL2 | rs11003125 | 0.333 | 0.025 |
| rs11003124 | 0.284 | 0.024 | |
| rs7084554 | 0.284 | 0.024 | |
| rs36014597 | 0.284 | 0.024 | |
| rs10556764 | 0.284 | 0.024 | |
| rs7096206 | 0.240 | 0.023 | |
| rs11003123 | 0.284 | 0.024 | |
| rs45602536 | 0.067 | 0.014 | |
| rs7095891 | 0.282 | 0.024 | |
| rs1800450 | 0.146 | 0.019 | |
| rs1800451 | 0.067 | 0.014 | |
| rs4647964 | 0.284 | 0.024 | |
| rs1982266 | 0.474 | 0.027 | |
| rs1838066 | 0.318 | 0.026 | |
| rs1838065 | 0.316 | 0.025 | |
| rs930509 | 0.225 | 0.023 | |
| rs930508 | 0.234 | 0.023 | |
| rs930507 | 0.225 | 0.023 | |
| rs10082466 | 0.244 | 0.023 | |
| rs10824792d | 0.471 | 0.027 | |
| rs2120132 | 0.243 | 0.023 | |
| rs2120131 | 0.243 | 0.023 | |
| rs2165813 | 0.243 | 0.023 | |
| rs2099903 | 0.243 | 0.023 | |
| rs2099902 | 0.249 | 0.023 | |
| rs2083771 | 0.249 | 0.023 | |
| rs2506 | 0.247 | 0.023 | |
| TLR1 | rs5743551d | 0.363 | 0.032 |
| TLR1NV002 | 0.175 | 0.022 | |
| TLR1NV003 | 0.164 | 0.021 | |
| rs45588337 | 0.160 | 0.020 | |
| rs1873196 | 0.053 | 0.012 | |
| rs5743553 | 0.074 | 0.014 | |
| rs5743556 | 0.161 | 0.020 | |
| rs5743557 | 0.162 | 0.020 | |
| rs5743558 | 0.166 | 0.020 | |
| rs4072548 | 0.129 | 0.018 | |
| rs5743560 | 0.164 | 0.020 | |
| rs5743562 | 0.164 | 0.020 | |
| rs5743563 | 0.164 | 0.020 | |
| rs5743564 | 0.134 | 0.019 | |
| rs5743565 | 0.164 | 0.020 | |
| rs5743566 | 0.164 | 0.020 | |
| rs5743567 | 0.131 | 0.018 | |
| rs5743571 | 0.164 | 0.020 | |
| rs5743572 | 0.064 | 0.013 | |
| rs5743574 | 0.052 | 0.012 | |
| rs45482391 | 0.111 | 0.017 | |
| rs5743578 | 0.125 | 0.018 | |
| rs5743580 | 0.168 | 0.020 | |
| rs5743584 | 0.169 | 0.021 | |
| rs5743585 | 0.164 | 0.020 | |
| rs4540055e | 0.355 | 0.032 | |
| rs5743592 | 0.166 | 0.020 | |
| rs5743593 | 0.161 | 0.020 | |
| rs5743595 | 0.164 | 0.020 | |
| rs5743596 | 0.155 | 0.020 | |
| rs5743599c | 0.131 | 0.019 | |
| rs45592140 | 0.065 | 0.014 | |
| rs5743604 | 0.429 | 0.027 | |
| rs45610931 | 0.065 | 0.013 | |
| rs4833095d | 0.500 | 0.027 | |
| rs5743614d | 0.500 | 0.028 | |
| rs5743618 | 0.058 | 0.013 | |
| TLR2 | rs4696480 | 0.320 | 0.026 |
| rs5743688 | 0.081 | 0.015 | |
| rs1898830 | 0.409 | 0.027 | |
| rs1816702 | 0.093 | 0.019 | |
| rs3804099 | 0.269 | 0.024 | |
| rs3804100 | 0.107 | 0.017 | |
| TLR4 | rs45478793 | 0.054 | 0.013 |
| rs10983755 | 0.096 | 0.016 | |
| rs1927914 | 0.395 | 0.027 | |
| rs10759932 | 0.120 | 0.018 | |
| rs1927911 | 0.261 | 0.024 | |
| rs10759933 | 0.121 | 0.018 | |
| rs4986790 | 0.114 | 0.021 | |
| rs4986791 | 0.126 | 0.018 | |
| rs7869402 | 0.087 | 0.018 | |
| rs11536889 | 0.162 | 0.025 | |
| rs7873784 | 0.113 | 0.017 | |
| rs11536891 | 0.102 | 0.019 | |
| rs11536896 | 0.118 | 0.018 | |
| rs1927906 | 0.209 | 0.022 | |
| rs11536898 | 0.100 | 0.016 | |
| TLR5 | rs1773766 | 0.054 | 0.012 |
| rs3044336 | 0.058 | 0.013 | |
| rs45487102 | 0.053 | 0.012 | |
| rs851181 | 0.429 | 0.027 | |
| rs851183 | 0.429 | 0.027 | |
| rs851184 | 0.429 | 0.027 | |
| rs45506101 | 0.064 | 0.014 | |
| rs45528236 | 0.092 | 0.016 | |
| rs5744168 | 0.091 | 0.016 | |
| rs2072493 | 0.160 | 0.020 | |
| rs5744174 | 0.426 | 0.027 | |
| rs1861172 | 0.419 | 0.029 | |
| rs45508499 | 0.429 | 0.027 | |
| rs5744190 | 0.429 | 0.027 | |
| rs3839019 or rs5744195f | 0.435 | 0.027 | |
| rs3795743 | 0.430 | 0.027 | |
| rs45578733 | 0.059 | 0.013 | |
| rs3795742 | 0.430 | 0.027 | |
| rs4661282 | 0.426 | 0.027 | |
| rs4661281 | 0.426 | 0.027 | |
| rs4661280 | 0.426 | 0.027 | |
| TLR6 | rs5743788 | 0.433 | 0.027 |
| rs5743789 | 0.188 | 0.022 | |
| rs5743790 | 0.379 | 0.027 | |
| rs5743792 | 0.224 | 0.023 | |
| rs5743794 | 0.188 | 0.021 | |
| rs5743795 | 0.184 | 0.021 | |
| rs5743798 | 0.064 | 0.013 | |
| rs5743802 | 0.376 | 0.026 | |
| rs5743805 | 0.222 | 0.022 | |
| rs5743806 | 0.376 | 0.026 | |
| rs1039559 | 0.432 | 0.027 | |
| rs1039560 | 0.433 | 0.027 | |
| rs3821985 | 0.383 | 0.026 | |
| rs3775073 | 0.383 | 0.026 | |
| rs5743818 | 0.065 | 0.013 | |
| rs5743826 | 0.060 | 0.013 | |
| rs3043438 | 0.433 | 0.027 | |
| rs45609936 | 0.433 | 0.027 | |
| rs2381288 | 0.433 | 0.027 | |
| rs5743827 | 0.161 | 0.020 | |
| rs5743828 | 0.106 | 0.017 | |
| rs5743831 | 0.158 | 0.020 | |
| rs2890664 | 0.347 | 0.026 | |
| rs2381289 | 0.347 | 0.026 | |
| rs2381290 | 0.312 | 0.025 | |
| TLR9 | rs187084 | 0.350 | 0.026 |
| rs5743836 | 0.112 | 0.018 | |
| rs352139 | 0.441 | 0.027 | |
| rs352140 | 0.435 | 0.027 |
Significance of p-values for tests of Hardy-Weinberg equilibrium and allele frequency differences were assessed by two False Discovery Rate methods (Benjamini and Hochberg, 1995; Storey and Tibshirani, 2003).
A locus is scored as polymorphic if the MAF at this locus exceeds 0.05 in pooled (Muslim and Hindu) population.
Locus ID in italics indicates that the major allele in the study populations is not the RefSeq allele.
Significantly (p< 0.005) deviated from Hardy-Weinberg equilibrium (see footnote e).
Minor allele frequencies differ significantly (p < 0.005) between Muslim and Hindu (see footnote e).
Triallelic (T > A > G) locus. The frequency of the G allele among Muslim and Hindu are, respectively, 0.185 and 0.189.
There is a “CGTTTTCTT” or “GTTTTCTTC” deletion. From the sequence data, it was not possible to decide the exact sequence of the inserted nucleotides.
Nearly all SNPs are in Hardy-Weinberg equilibrium. The frequencies of genotypes in the two communities are also not significantly different at 210 of 216 comparable loci (Table 1), possibly because the Muslims are recent religious converts. Prior to religious conversion, the two groups were possibly one large intra-marrying group. Therefore, data on the two communities were pooled for most analyses, and the pooled group has been referred to as the study population. The observed average heterozygosities are variable across genes; CAMP exhibits a very low heterozygosity, while average heterozygosity values are relatively higher for MBL2, TLR5, TLR6 (Fig. 1(a)).
Fig. 1.

(a) Average heterozygosity and (b) haplotype diversity values at the 12 innate immunity gene loci in the pooled (Muslim and Hindu combined) population.
Since population genetic theory suggests that the most frequent allele is the oldest (Watterson and Guess, 1977) and therefore likely to be the ancestral allele, we compared our data at the variable positions with the corresponding data from the chimpanzee reference sequence (http://www.genome.ucsc.edu/; March 2006 assembly). While the major allele at the majority (412 of 544; 76%) of variable sites in our data coincided with the ancestral (chimpanzee reference sequence) allele, at the remaining 132 (24%) sites the major allele is not the ancestral allele. Such observations have been made earlier (Hacia et al., 1999). It is not unlikely to observe non-ancestral alleles to be major alleles in humans, when these pertain to loci that are influenced by natural selection. Indirectly, our observation lends support to natural selection acting on the innate immunity genes.
Haplotypes were reconstructed using genotype data at the polymorphic loci and their frequencies were estimated (Supplementary Table 4). For most genes, three or four haplotypes are in high frequencies (Fig. 2), and haplotype frequencies are similar between Muslim and Hindu (data not shown). For most genes, the human reference sequence haplotype (build 36, http://www.ncbi.nlm.nih.gov/) is not the most common haplotype in the study population and for some genes (e.g., DEFA4) the reference sequence haplotype was not even observed (Supplementary Table 4). The haplotype diversity values are very high (from 62% for TLR9 to 88% for DEFA6) for most genes (Fig. 1(b)). The ratio of SNPs to haplotypes varies from 0.41 (DEFA6) to 2.08 (TLR6).
Fig. 2.

Frequencies of haplotypes I10% in the study population (haplotypes with frequencies <10% have been pooled into a single category, ■.).
Haplotypes based on some specific loci in MBL2 are known to be associated with MBL serum concentration variability (Garred et al., 2006). MBL serum concentration influences susceptibility to and clinical course of many infectious and chronic diseases (Nuytinck and Shapiro, 2004). The relevant details about these haplotypes and their frequencies observed in our study population are presented in Table 2.
Table 2.
Frequencies of individuals in the study population who express MBL2 at different levels, and the underlying haplotypes
| Level of MBL expression |
Haplotype (using traditional allelic nomenclature) |
Frequency in study population (%) |
Detailed definition of haplotype |
|||||
|---|---|---|---|---|---|---|---|---|
| rs# (location) | ||||||||
| Nucleotide (traditional allelic nomenclature) |
||||||||
| rs11003125 (promoter) |
rs7096206 (promoter) |
rs7095891 (5′ UTR) |
rs5030737a,b (exon 1) |
rs1800450a (exon 1) |
rs1800451a (exon 1) |
|||
| High | HYPA | 53.22 | G (H) | G (Y) | C (P) | C (A) | G/A | G/A |
| LYQA | C (L) | G (Y) | T (Q) | C (A) | G/A | G/A | ||
| Intermediate | LYPA | 1.46 | C (L) | G (Y) | C (P) | C (A) | G/A | G/A |
| Low | LXPA | 23.98 | C (L) | C (X) | C (P) | C (A) | G/A | G/A |
| None | HYPD | 19.59 | G (H) | G (Y) | C (P) | T (D) | G/A | G/A |
| LYQC | C (L) | G (Y) | T (Q) | C/T | G/A | A (C) | ||
| LYPB | C (L) | G (Y) | C (P) | C/T | A (B) | G/A | ||
Non-synonymous change.
Not polymorphic in the study population.
Recently (Ferwerda et al., 2007), it has been shown that there is a strong continental structuring of haplotype frequencies at two non-synonymous sites (rs4986790, Asp > Gly and rs4986791, Thr > Ile) in TLR4. This structuring was inferred to be due to the interaction between our innate immune system and the infectious pressures in particular environments during the out-of-Africa migration of modern humans. The observed percent frequencies of the Asp/Thr, Asp/Ile, Gly/Thr and Gly/Ile haplotypes in our data were, respectively, 86.52, 2.33, 1.16 and 9.94.
For the variants in these innate immunity genes that were also assayed in the HapMap project (10 genes), we compared our data with the HapMap data (http://www.hapmap.org; HapMap Data Release 21a/phaseII 07 January, on NCBI B35 assembly, dbSNP build 125). We found that there are three nucleotide positions (1 in DEFB1, rs2978873; 1 in TLR1, rs1873196; and, 1 in TLR2, rs4696480) that are monomorphic in all four HapMap populations, but are polymorphic in our study population. None of these variants is in a coding region, however. On the other hand, there are 26 positions that are polymorphic in at least one of the four HapMap populations, but are monomorphic in our study population. There is also a tremendous variation in allele frequencies at highly polymorphic loci (i.e., allele frequencies ranging between 0.2 and 0.8 in at least one population) among the HapMap populations and our study population (Supplementary Figure 1). This is also reflected in the pattern of clustering of the populations (Fig. 3) based on allele frequency data on 177 common SNPs from the innate immunity genes.
Fig. 3.
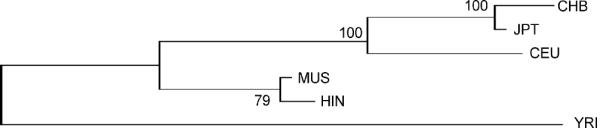
Neighbor-joining tree depicting relationship among the two study communities (Muslim and Hindu) and the four HapMap populations, constructed on the basis of allele frequency data of 177 common polymorphisms in innate immunity genes.
There is also considerable variation in haplotype frequencies (Supplementary Figure 2), calculated on the basis of loci that were common to HapMap and our study populations. (Because of lack of data in HapMap database for TLR5, haplotype frequencies could not be compared for this gene.) Detailed descriptions of the haplotypes and their frequencies are given in Supplementary Table 5.
4. Discussion
Large-scale resequencing studies have primarily been conducted on DNA samples collected from individuals residing in regions of the world that are relatively free of microbial pathogens and infectious diseases. To the best of our knowledge, this is the first extensive and comprehensive population-based study of natural variation in a large number of innate immunity genes assayed by DNA resequencing carried out on individuals residing in an area with a high load of microbial pathogens. Almost half of the variation discovered was novel. Further, among variable sites at which the alleles present in the human and chimpanzee (ancestral) reference sequences matched, at 6% of these sites the major allele in our study population was different. At a further 38% of variable sites at which there was mismatch between alleles present in human and chimpanzee reference sequences, the allele found in our study was different from the human reference sequence. Thus, in the study population different alleles in the innate immunity genes may have been selectively favored.
Most of the non-synonymous SNPs detected in this study have been reported earlier and some have been found to be associated with various diseases. The single nucleotide change resulting in a stop codon in TLR5 (rs5744168) has been shown to be associated with susceptibility to Legionnaire's disease and to flagellated bacterial infections (Hawn et al., 2003; Hawn et al., 2005). The observed non-synonymous SNPs in TLR4 (rs4986790 and rs4986791) have been reported to be associated with impaired LPS signalling and resultantly to Gram-negative bacterial infections (Arbour et al., 2000). The (GT)n repeat polymorphism in intron-2 of TLR2 which has been reported to be associated with many mycobacterial diseases (Yim et al., 2006) was also detected in our study, but the genotypes based on the numbers of repeats in the two chromosomes could not be scored by us on the basis of sequence data. However, the Arg > Gln change (rs5743708) in TLR2 which is polymorphic in many populations and has been shown to be associated with several diseases (Lorenz et al., 2000; Ogus et al., 2004) was found to be non-polymorphic in our study population. For the MBL2 gene, while the frequencies of haplotypes in our study population associated with high (53%) and deficient (20%) levels of MBL serum concentration are comparable to those observed worldwide (49% and 22%), the frequencies of intermediate (1.5%) and low (24%) haplotypes are significantly different from those observed worldwide (15% and 13%) (Verdu et al., 2006). The significantly higher frequency of the low-secretor haplotype (and, consequently, a lower frequency of intermediate-secretor haplotypes) is noteworthy. While low MBL concentration is known to be clinically disadvantageous (Nuytinck and Shapiro, 2004), the high frequency of the haplotype associated with low MBL concentration in our study population resident in an area with a high load of pathogens may be indicative of selective advantage being conferred on the carriers of this haplotype (Seyfarth et al., 2005), although a recent study has concluded that the pattern of genomic variation in MBL2 is compatible with neutral evolution, as opposed to evolution under the effect of natural selection (Verdu et al., 2006). In any case, it is evident that the population included in this study show many similarities with other populations, but manifest many independent features possibly resulting from the action of natural selection.
With respect to two non-synonymous changes [rs4986790 (Asp > Gly) and rs4986791 (Thr > Ile)] in TLR4 that are 300 nucleotides apart and show strong geographical clustering of haplotypes, we noted that the frequency (10%) of the Gly/Ile haplotype in our study population is the highest reported so far (Ferwerda et al., 2007). Interestingly, this haplotype is absent (or nearly so) in Africa and also among the Han Chinese, but is present in European populations with frequencies ranging between 5 and 9% (Ferwerda et al., 2007). These haplotypes have been shown (Arbour et al., 2000) to be associated with responsiveness to lipopolysaccharides (which are present in many microbes, including typhoid fever causing Salmonella). Based on limited data available from Asian populations, Ferwerda et al. (2007) concluded that the Gly allele is lost from Asian populations and has speculated on possible causes for this loss. However, this allele is present in our study population with frequencies exceeding 10%. The geographic distribution of these TLR4 haplotypes in populations varies according to susceptibility to infection and is indicative of our innate immune system having been modified by infectious pressures (Ferwerda et al., 2007).
The extensive variation in the innate immunity genes observed among our study participants is possibly an outcome of their inhabiting in an area that is endemic to a variety of infectious diseases. Compared to the average of one SNP occurring in 1.91 kb (Sachidanandam et al., 2001) or one variant per 1.3 kb (Miller et al., 2005) in the autosomal genome, the densities of SNPs (>3 SNPs kb−1) and variants (>7 variants kb−1) in the innate immunity genes in our study population are significantly higher (p < 0.02). Even compared to the more recent estimate (The International HapMap Consortium, 2007) of one SNP per 0.88 kb, the observed SNP density in our study is higher. There is, of course, variation in SNP density across autosomal chromosomes. The range of variation (see Table 1 of Sachidanandam et al., 2001) is one SNP per 1.19 kb (chromosome 22) to one SNP per 2.18 kb (chromosome 19); thus, the SNP density in the innate immunity genes observed in this study exceeds the highest density observed for any autosomal chromosome in the human genome. In fact, at the genome-wide level, there are very few genomic regions where SNP density exceeds 2.5 SNPs kb−1 (see Fig. 1 of The International HapMap Consortium, 2007). Thus, compared with all existing data, the extent of genetic variation in the human innate immunity genes observed in this study is very high. This is consistent with Parham's (2003) proposition that the innate immunity genes are continuing to evolve due to pressures of natural selection in different directions, possibly because of the extent and diversity of pathogens to which humans are exposed.
With respect to the allele frequency profiles of innate immunity genes, the positioning of the study population relative to the HapMap populations (Fig. 3) is interesting. Not unexpectedly (Conrad et al., 2006), the African Yoruba (YRI) forms a separate cluster. What is, however, striking is that the Muslim and Hindu communities of this study form a cluster that is distinct from the remaining HapMap populations. A recent study (The Indian Genome Variation Consortium, 2008) that has used data on 405 SNPs from 55 diverse ethnic groups of India has shown that Indian populations are intermediate between the CEU and {JPT, CHB}. However, with respect to the allele frequencies of innate immunity SNPs, the Muslim and Hindu show very distinct allele frequency profiles compared to the CEU, JPT and CHB (Fig. 3) with a strong bootstrap support. The haplotype frequency profile of the study population is also distinct from those of the HapMap populations (Supplementary Figure 2). For many of the genes, the ratio of SNPs to haplotypes exceeds 1, indicating that there is repression of recombination in these genes or, more likely, that many recombinant haplotypes are selected against. One possible reason is that the allele and haplotype frequencies of polymorphic loci in innate immunity genes in the study population has been reshaped by natural selection, perhaps because of their long and continued exposure to a high load of pathogens. Specific alleles in many human genes are now known to have been strongly selected in recent human history, especially in relation to exposure to infectious pathogens, such as CCR5 (Novembre et al., 2005; Sabeti et al., 2005) and FY (Hamblin et al., 2002). It has also been speculated that genes related to disease resistance are very likely to show evidence of recent positive selection (Wang et al., 2006), including resistance to infections - such as smallpox, malaria, yellow fever, typhus and cholera - that have become important causes of mortality after the origin and spread of agriculture (McNeill, 1976; Wolfe et al., 2007). Since innate immunity genes play a paramount role in eliminating pathogens, it is very likely that these genes have been strongly influenced by natural selection. We are now conducting analyses to formally test this possibility.
Supplementary Material
Acknowledgements
We thank Mr. Manab Mukherjee, Minister-in-Charge of Tourism, Cottage and Small-Scale Industries Departments, Government of West Bengal, India, and Mr. S. Panja for their immense help with the organization of this field study; Mr. Ardhendu Endow for his assistance with fieldwork management; Ms. Jane Hammond and Mr. Christopher McClure of RTI International for their assistance with project management; and, Professor V. Nanjundiah for his comments on an earlier draft of the manuscript. This work was supported by NIAID, National Institutes of Health, U.S.A.; Contract No. HHSN200400067C.
Footnotes
Appendix A. Supplementary data Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.meegid.2008.02.009.
References
- Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, Frees K, Watt JL, Schwartz DA. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat. Genet. 2000;25:187–191. doi: 10.1038/76048. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B. 1995;57:289–300. [Google Scholar]
- Conrad DF, Jakobsson M, Coop G, Wen X, Wall JD, Rosenberg NA, Pritchard JK. A worldwide survey of haplotype variation and linkage disequilibrium in the human genome. Nat. Genet. 2006;38:126–1251. doi: 10.1038/ng1911. [DOI] [PubMed] [Google Scholar]
- Ferwerda B, McCall MBB, Alonso S, Giamarellos-Bourboulise EJ, Mouktaroudi M, Izagirre N, et al. TLR4 polymorphisms, infectious diseases, and evolutionary pressure during migration of modern humans. Proc. Natl. Acad. Sci. U.S.A. 2007;42:16645–16650. doi: 10.1073/pnas.0704828104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganz T, Lehrer RI. Defensins. Curr. Opin. Immunol. 1994;6:584–589. doi: 10.1016/0952-7915(94)90145-7. [DOI] [PubMed] [Google Scholar]
- Garred P, Larsen F, Seyfarth J, Fujita R, Madsen HO. Mannose-binding lectin and its genetic variants. Genes Immun. 2006;7:85–94. doi: 10.1038/sj.gene.6364283. [DOI] [PubMed] [Google Scholar]
- Hacia JG, Fan J-B, Ryder O, Jin L, Edgemon K, Ghandbur G, Mayer RA, Sun B, Hsie L, Robbins CM, Brody LC, Wang D, Lander ES, Lipshutz R, Fodor SPA, Collins FS. Determination of ancestral alleles for human single-nucleotide polymorphisms using high-density oligonucleotide arrays. Nat. Genet. 1999;22:164–167. doi: 10.1038/9674. [DOI] [PubMed] [Google Scholar]
- Hamblin MT, Thompson EE, Di Rienzo A. Complex signatures of natural selection at the Duffy blood group locus. Am. J. Hum. Genet. 2002;70:369–383. doi: 10.1086/338628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawn TR, Verbon A, Lettinga KD, Zhao LP, Li SS, Laws RJ, Skerrett SJ, Beutler B, Schroeder L, Nachman A, Ozinsky A, Smith KD, Aderem A. A common dominant TLR5 stop codon polymorphism abolishes flagellin signaling and is associated with susceptibility to Legionnaires' disease. J. Exp. Med. 2003;198:1563–1572. doi: 10.1084/jem.20031220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawn TR, Wu H, Grossman JM, Hahn BH, Tsao BP, Aderem A. A stop codon polymorphism of Toll-like receptor 5 is associated with resistance to systemic lupus erythematosus. Proc. Natl. Acad. Sci. U.S.A. 2005;102:10593–10597. doi: 10.1073/pnas.0501165102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz E, Mira JP, Cornish KL, Arbour NC, Schwartz DA. A novel polymorphism in the toll-like receptor 2 gene and its potential association with staphylococcal infection. Infect. Immun. 2000;68:6398–6401. doi: 10.1128/iai.68.11.6398-6401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimbrell DA, Beutler B. The evolution and genetics of innate immunity. Nat. Rev. Genet. 2001;2:256–267. doi: 10.1038/35066006. [DOI] [PubMed] [Google Scholar]
- Miller RD, Phillips MS, Jo I, Donaldson MA, Studebaker JF, Addleman N, Steven Alfisi V, Ankener WM, Bhatti HA, Callahan CE, Carey BJ, Conley CL, Cyr JM. High-density single-nucleotide polymorphism maps of the human genome. Genomics. 2005;86:117–126. doi: 10.1016/j.ygeno.2005.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill W. Plagues and Peoples. Doubleday; Garden City, NY: 1976. [Google Scholar]
- Nei M. Molecular Evolutionary Genetics. Columbia University Press; New York: 1987. [Google Scholar]
- Nizet V, Gallo RL. Cathelicidins and innate defense against invasive bacterial infection. Scand. J. Infect. Dis. 2003;35:670–676. doi: 10.1080/00365540310015629. [DOI] [PubMed] [Google Scholar]
- Novembre J, Galvani AP, Slatkin M. The geographic spread of the CCR5 Delta32 HIV-resistance allele. PLoS Biol. 2005;3:e339. doi: 10.1371/journal.pbio.0030339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuytinck L, Shapiro F. Mannose-binding lectin: laying the stepping stones from clinical research to personalized medicine. Personalized Med. 2004;1:35–52. doi: 10.1517/17410541.1.1.35. [DOI] [PubMed] [Google Scholar]
- Ogus AC, Yoldas B, Ozdemir T, Uguz A, Olcen S, Keser I, Coskun M, Cilli A, Yegin O. The Arg753GLn polymorphism of the human toll-like receptor 2 gene in tuberculosis disease. Eur. Respir. J. 2004;23:219–223. doi: 10.1183/09031936.03.00061703. [DOI] [PubMed] [Google Scholar]
- Parham P. The unsung heroes. Nature. 2003;423:20. doi: 10.1038/423020a. [DOI] [PubMed] [Google Scholar]
- Reed TE, Schull WJ. A general maximum likelihood estimation program. Am. J. Hum. Genet. 1968;20:579–580. [PMC free article] [PubMed] [Google Scholar]
- Sabeti PC, Walsh E, Schaffner SF, Varilly P, Fry B, Hutcheson HB, Cullen M, Mikkelsen TS, Roy J, Patterson N, Cooper R, Reich D, Altshuler D, O'Brien S, Lander ES. The case for selection at CCR5-Delta32. PLoS Biol. 2005;3:e378. doi: 10.1371/journal.pbio.0030378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachidanandam R, et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409:928–933. doi: 10.1038/35057149. [DOI] [PubMed] [Google Scholar]
- Seyfarth J, Garred P, Madsen HO. The `involution' of mannose-binding lectin. Hum. Mol. Genet. 2005;14:2859–2869. doi: 10.1093/hmg/ddi318. [DOI] [PubMed] [Google Scholar]
- Stephens M, Donnelly P. A comparison of Bayesian methods for haplotype reconstruction. Am. J. Hum. Genet. 2001;73:1162–1169. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U.S.A. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu. Rev. Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- The Indian Genome Variation Consortium Genetic Landscape of the People of India: a Canvas for Disease Gene Exploration. J. Genet. 2008 doi: 10.1007/s12041-008-0002-x. in press. [DOI] [PubMed] [Google Scholar]
- The International HapMap Consortium A second generation human haplo-type map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdu P, Barreiro LB, Patin E, Gessain A, Cassar O, Kidd JR, et al. Evolutionary insights into the high worldwide prevalence of MBL2 deficiency alleles. Hum. Mol. Genet. 2006;15:2650–2658. doi: 10.1093/hmg/ddl193. [DOI] [PubMed] [Google Scholar]
- Wang ET, Kodama G, Baldi P, Moyzis RK. Global landscape of recent inferred Darwinian selection for Homo sapiens. Proc. Natl. Acad. Sci. U.S.A. 2006;103:135–140. doi: 10.1073/pnas.0509691102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson GA, Guess HA. Is the most frequent allele the oldest? Theor. Popul. Biol. 1977;11:141–160. doi: 10.1016/0040-5809(77)90023-5. [DOI] [PubMed] [Google Scholar]
- Wolfe ND, Dunavan CP, Diamond J. Origins of major human infectious diseases. Nature. 2007;447:279–283. doi: 10.1038/nature05775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim JJ, Lee HW, Lee HS, Kim YW, Han SK, Shim YS, Holland SM. The association between microsatellite polymorphisms in intron II of the human Toll-like receptor 2 gene and tuberculosis among Koreans. Genes Immun. 2006;7:150–155. doi: 10.1038/sj.gene.6364274. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.