

Abstract
The catalytic enantioselective formation of tetrasubstituted α-alkoxycarbonyl compounds is an ongoing challenge to synthetic chemists.[1] Fully-substituted α-hydroxyesters and acids comprise essential components of, and building blocks for, many bioactive natural products. These include quinic acid (1), cytotoxic leiodolide A (2),[2] and the anti-cancer agents in the harringtonine series (3a–f), whose activities depend dramatically on the presence and composition of an α-hydroxyester side-chain.[3] While many approaches to these important moieties exist,[4,5] we envisioned applying our recently developed palladium-catalyzed methods for the formation of enantioenriched all-carbon quaternary stereocenters in cyclic alkanones[6] to a general synthesis of C(α)-tetrasubstituted hydroxy carbonyl compounds.[7]
Keywords: Alcohol, Allylic compounds, Asymmetric catalysis, Enantioselectivity, Palladium
For this application, we chose to incorporate oxygenation into a cyclic motif, the 2,2-dimethyldioxanone framework,[8] and employ this platform for the enantioselective synthesis of α-dialkyl ketones (e.g., 4). Dioxanones are challenging alkylation substrates because standard conditions do not permit alkylation, but instead facilitate ketone reduction (e.g., LDA, −78 °C), or self-condensation (e.g., LHMDS), accompanied by decomposition.[9] A technology that avoids this undesirable reactivity would represent a marked advance in dioxanone chemistry. For this purpose, Enders and co-workers have developed a diastereoselective α-alkylation that relies on chirality imparted by (+)-S-1-amino-2-methoxymethylpyrrolidine hydrazones, which can be cleaved in a subsequent step (Figure 2, eq 1). We believed our catalytic palladium technology would be an ideal platform to provide access to valuable tetrasubstituted α-hydroxyketones, esters, and acids (Figure 2, eq 2).
Figure 2.
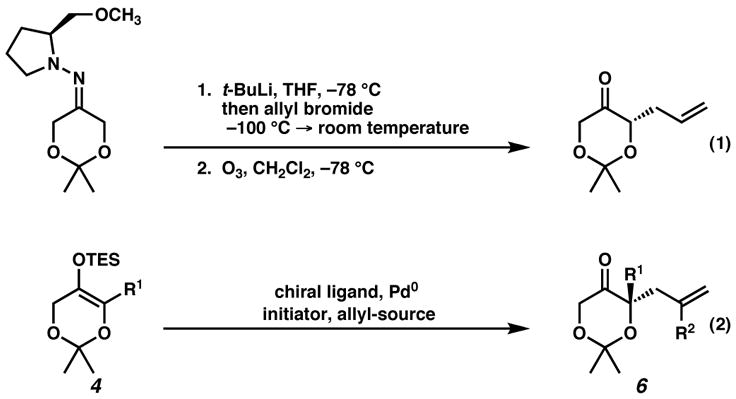
Alkylation technologies using the dioxanone framework.
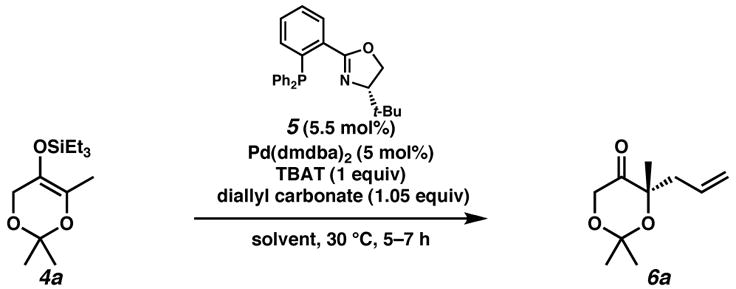
We examined the conversion of silyl enol ether 4 a[10] to enantioenriched tetrasubstituted ether 6a under a variety of palladium-catalyzed conditions, beginning with the standard conditions developed for the all-carbon system (Table 1). Triethylsilyl derivative 4a was chosen for its stability and ease of synthesis compared to the related trimethylsilyl compound. Treatment of silyl ether 4a with Pd(dmdba)2 (5 mol%), (S)-t-BuPHOX (5, 5.5 mol%),[11] Bu4NPh3SiF2 (TBAT, 1 equiv), and diallyl carbonate (1.05 equiv) in THF at 30 °C (entry 1) produced enantioenriched 6a in 40% yield and 81% ee. Interestingly, the choice of solvent played a significant role in the selectivity of tertiary ether formation. In the optimal case, use of PhCH3 as solvent furnished dioxanone 6a in 74% yield and 89% ee (entry 5).
Table 1.
Optimization of the enantioselective enol silane allylation.[a]
 | |||
|---|---|---|---|
| Entry | Solvent | Yield [%][b] | ee [%][c] |
| 1 | THF | 40 | 81 |
| 2[d] | Et2O | 59 | 89 |
| 3 | 1,4-dioxane | 65 | 67 |
| 4 | PhH | 67 | 84 |
| 5 | PhCH3 | 74 | 89 |
Reactions were performed using 0.1 mmol of substrate in solvent (0.033 M) at 30 °C over 5–7 h unless stated otherwise.
Isolated yields.
Measured by chiral HPLC following conversion to 6b.[12]
Performed at 35 °C and 0.0167 M. dmdba = bis(3,5-dimethoxybenzylidene)acetone
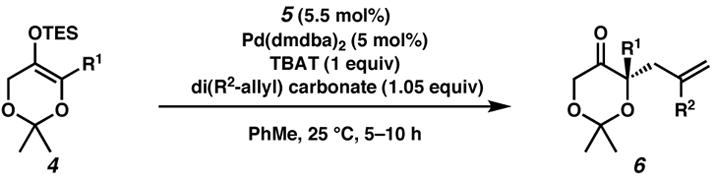
We applied these optimized conditions to silyl enol ethers with diverse α-substituents, in combination with substituted allyl carbonates (Table 2). The asymmetric alkylation tolerates a variety of substituents at the α-position, including alkyl (entries 1 and 2), benzyl (entry 3), and alkenyl (entries 7–9) moieties. Additionally, the reaction proceeds when the allyl group is substituted internally by methyl, chloro, or phenyl (entries 4–7) to afford products in high yields and ee.
Table 2.
Substrate scope for the enantioselective alkylation.[a]
 | ||||
|---|---|---|---|---|
| Entry | R1 | R2 | Yield [%][c] | ee [%][d] |
| 1 | Me | H | 86 | 87 |
| 2 | Et | H | 79 | 93 |
| 3 | PhCH2 | H | 85 | 86 |
| 4[e, f] | Me | Me | 59 | 89 |
| 5[g] | Me | Cl[b] | 59 | 92 |
| 6[h] | Me | Ph | 73 | 94 |
| 7[f] | allyl | Me | 93 | 88 |
| 8 | 2-methallyl | H | 88 | 85 |
| 9 | 1-butenyl | H | 83 | 92 |
Reactions were performed with silyl enol ether (0.5 mmol) and diallyl carbonate (1.05 equiv) in PhMe (0.033 M) unless stated otherwise. When R1 = R2 = H, the reaction proceeds in 13% yield and 82% ee.
Absolute stereochemistry has been assigned by analogy, except in the case of 6c, which was assigned by conversion to (+)-(S)-citramalic acid dimethyl ester.[12]
Isolated yields.
Measured by chiral GC or HPLC.[12]
Performed with trimethylsilyl precursor, 35 mol% TBAT in Et2O (0.0167 M) at 25 °C.
Performed with dimethallyl carbonate (1.05 equiv).
Performed with dichloroallyl carbonate (1.05 equiv) at 35°C.
Performed with diphenylallyl carbonate (1.05 equiv).
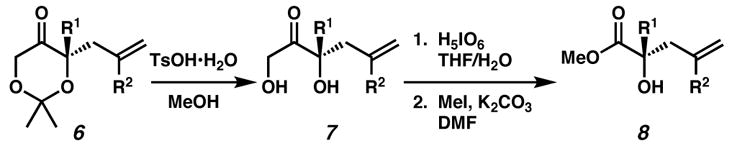
Having established a general route to enantioenriched tetrasubstituted dioxanones 6, we have developed a straightforward sequence to transform these α-alkoxyketones into the corresponding α-hydroxyesters 8 (Table 3). To effect acetonide cleavage, enantioenriched products 6 are treated with catalytic p -toluenesulfonic acid in methanol or ethanol. Dihydroxyketones 7 are oxidatively cleaved in the presence of periodic acid, selectively removing the primary alcohol,[13] to generate α-hydroxyacids. Subsequent methylation furnishes tetrasubstituted enantioenriched α-hydroxyesters 8. These operations are tolerant of a number of substitutents including alkyl (entries 1–4), chloro (entry 5), and aryl (entries 3 and 6) groups. Furthermore, dioxanone derivatives may include enolizable α,β-unsaturated esters (entry 7), or be incorporated into cyclic structures (entries 8 and 9).
Table 3.
Substrate scope for methyl ester formation.[a]
 | ||||
|---|---|---|---|---|
| Entry | Substrate | Yield 7 [%][c, d] | Yield 8 [%][c, d] | |
| R1 | R2 | |||
| 1 | Me | H | 90 | 54 |
| 2 | Et | H | 97 | 60 |
| 3[e] | PhCH2 | H | 91 | 85 |
| 4 | Me | Me | 97 | 84 |
| 5 | Me | Cl[b] | 97 | 76 |
| 6 | Me | Ph[h] | 92 | 77 |
| 7 |
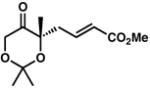
6B |
80 | 51 | |
| 8 |
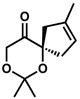
6c |
– | 48[f] | |
| 9[g] |
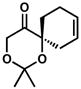
6d |
– | 61[f] | |
Acetonides 6 (1.0 equiv) were cleaved with TsOH•H2O (0.1 equiv) in MeOH (0.1 M) over 2–3 h unless otherwise noted. Diols 7 were oxidized with H5IO6 (1.5 equiv) in 2:1 THF:H2O (0.033 M) unless otherwise noted.
Absolute stereochemistry has been assigned by analogy, except in this case, which was assigned by conversion to (+)-(S)-citramalic acid dimethyl ester.[12]
Isolated yields.
There is no measurable change of ee through this sequence.
Acetonide cleavage was performed in EtOH over 6 h.
Three step yield.
Oxidation performed with H5IO6 (1.0 equiv).
Direct treatment with H5IO6, and methylation provided the ester in 47% yield over two steps.
See the supporting information for a description of the preparation of 6 b from 6a via cross-metathesis. TsOH = p-toluenesulfonic acid
Notably, assembly of acid (S)-10 constitutes a catalytic enantioselective formal synthesis of (−)-quinic acid (1),[14] a useful chiral building block that has been employed in numerous syntheses,[15] including our recent synthesis of dragmacidin F.[16] Toward this end, we recognize that enantioenriched α,ω-dienes can be transformed into cycloalkenes with a stereocenter remote to the olefin (Scheme 1).[17] Chiral diene 6e undergoes ring closing metathesis to generate 3-methylcyclopentene 6c in 95% yield and 85% ee. Likewise, enantioenriched α,ω-diene 6f furnishes cyclohexene 6d in 90% yield and 92% ee. Cyclohexene 6d readily undergoes acetonide cleavage and periodic acid oxidation to provide pure, isolable acid (S)-10, completing the formal synthesis of (−)-quinic acid (1).
Scheme 1.
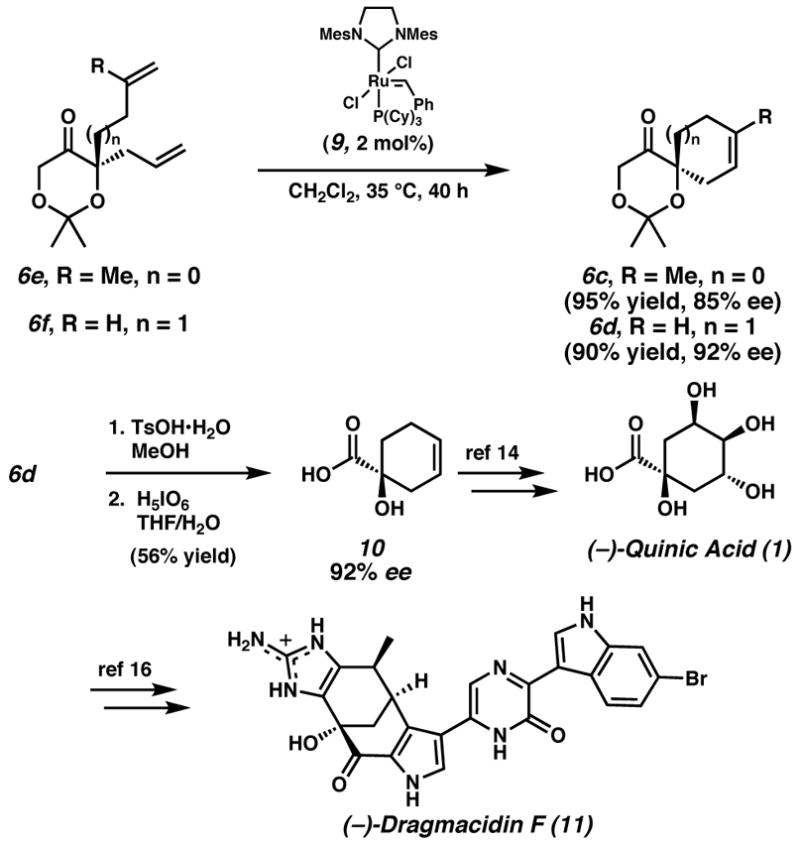
Formal synthesis of (−)-quinic acid (1) and (−)-dragmacidin F (11).
In summary, we have developed a palladium-catalyzed asymmetric alkylation of simple dioxanone derivatives, and transformed the enantioenriched products into α-hydroxyketones, acids, and esters. This mild, straightforward sequence proceeds in high yield and enantioselectivity. This procedure has also enabled a catalytic enantioselective formal synthesis of (−)-quinic acid. Research is underway to extend this chemistry to other α-heteroatom-containing carbonyl derivatives and to employ these methods in the total synthesis of bioactive natural products.
Supplementary Material
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Figure 1.
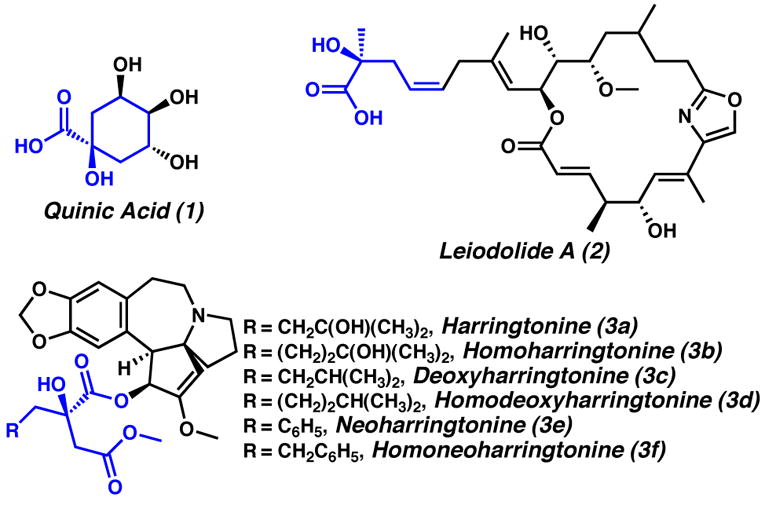
Natural products containing α-hydroxyesters and acids.
Footnotes
The authors wish to thank Takeda Pharmaceutical Company Limited of Japan (postdoctoral fellowship to M.S.), the California Tobacco-Related Disease Research Program of the University of California, Grant Number 14DT-0004 (predoctoral fellowship to J.L.R.), the NIH-NIGMS (R01 GM 080269-01), and Caltech for financial support, Materia, Inc. for their generous donation of catalyst 9 used in these studies, and Dr. S. Virgil, A. Silberstein, and Y. Segawa for experimental assistance.
References
- 1.Coppola GM, Schuster HF. α-Hydroxy Acids in Enantioselective Syntheses. WILEY-VCH; Weinheim: 1997. [Google Scholar]
- 2.Sandler JS, Colin PL, Kelly M, Fenical W. J Org Chem. 2006;71:7245–7251. doi: 10.1021/jo060958y. [DOI] [PubMed] [Google Scholar]
- 3.a) Takano I, Yasuda I, Nishijima M, Hitotsuyanagi Y, Takeya K, Itokawa H. Bioorg Med Chem Lett. 1996;6:1689–1690. [Google Scholar]; b) Takano I, Yasuda I, Nishijima M, Yanagi Y, Takeya K, Itokawa H. Phytochemistry. 1997;44:735–738. doi: 10.1016/s0031-9422(96)00574-2. [DOI] [PubMed] [Google Scholar]; c) Morita H, Arisaka M, Yoshida N, Kobayashi J. Tetrahedron. 2000;56:2929–2934. [Google Scholar]; d) Liu Q, Ferreira EM, Stoltz BM. J Org Chem. 2007;72:7352–7358. doi: 10.1021/jo0710883. [DOI] [PubMed] [Google Scholar]
- 4.For reviews, see: Corey EJ, Guzmán-Pérez A. Angew Chem. 1998;110:402–415. doi: 10.1002/(SICI)1521-3773(19980302)37:4<388::AID-ANIE388>3.0.CO;2-V.Angew Chem Int Ed. 1998;37:388–401. doi: 10.1002/(SICI)1521-3773(19980302)37:4<388::AID-ANIE388>3.0.CO;2-V.Ramón DJ, Yus M. Angew Chem. 2004;116:286, 289. doi: 10.1002/anie.200301696.Angew Chem Int Ed. 2004;43:284–287.Ramón DJ, Yus M. Curr Org Chem. 2004;8:149–183.Garcia C, Martin VS. Curr Org Chem. 2006;10:1849–1889.Ley SV, Sheppard TD, Myers RM, Chorghade MS. Bull Chem Soc Jpn. 2007;80:1451–1472.
- 5.For select recent examples, see: Trost BM, Dogra K, Franzini M. J Am Chem Soc. 2004;126:1944–1945. doi: 10.1021/ja031539a.Trost BM, Xu J, Reichle M. J Am Chem Soc. 2006;129:282–283. doi: 10.1021/ja067342a.Li H, Wang B, Deng L. J Am Chem Soc. 2006;128:732–733. doi: 10.1021/ja057237l.Ooi T, Fukumoto K, Maruoka K. Angew Chem. 2006;118:3923–3926. doi: 10.1002/anie.200600470.Angew Chem Int Ed. 2006;45:3839–3842. doi: 10.1002/anie.200600470.Kim SS, Lee SH, Kwak JM. Tetrahedron: Asymmetry. 2006;17:1165–1169.Kim SS. Pure Appl Chem. 2006;78:977–983.Tian SK, Deng L. Tetrahedron. 2006;62:11320–11330.Mikami K, Kawakami Y, Akiyama K, Aikawa K. J Am Chem Soc. 2007;129:12950–12951. doi: 10.1021/ja076539f.Zuend SJ, Jacobsen EN. J Am Chem Soc. 2007;129:15872–15883. doi: 10.1021/ja0735352.Zheng K, Qin B, Liu X, Feng X. J Org Chem. 2007;72:8478–8483. doi: 10.1021/jo701491r.Qin B, Xiao X, Liu X, Huang J, Wen Y, Feng X. J Org Chem. 2007;72:9323–9328. doi: 10.1021/jo701898r.Takada K, Takemura N, Cho K, Sohtome Y, Nagasawa K. Tetrahedron Lett. 2008;49:1623–1626.
- 6.a) Mohr JT, Stoltz BM. Chem–Asian J. 2007;2:1476–1491. doi: 10.1002/asia.200700183. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mohr JT, Behenna DC, Harned AM, Stoltz BM. Angew Chem. 2005;117:7084–7087. doi: 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:6924–6927. doi: 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]; c) Behenna DC, Stoltz BM. J Am Chem Soc. 2004;126:15044–15045. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]
- 7.For an alternative approach to asymmetric enolate alkylation, see: Braun M, Laicher F, Meier T. Angew Chem. 2000;112:3637–3640. doi: 10.1002/1521-3773(20001002)39:19<3494::aid-anie3494>3.0.co;2-4.Angew Chem Int Ed. 2000;39:3494–3497. doi: 10.1002/1521-3773(20001002)39:19<3494::aid-anie3494>3.0.co;2-4.Braun M, Meier T. Angew Chem. 2006;118:7106–7109.Angew Chem Int Ed. 2006;45:6952–6955. doi: 10.1002/anie.200602169.Braun M, Meier T, Laicher F, Meletis P, Fidan M. Adv Synth Catal. 2008;350:303–314.
- 8.For the use of dioxanones as a template for the synthesis of stereochemically rich substrances, see: Enders D, Bettray W. In: Asymmetric Synthesis with Chemical and Biological Methods. Enders D, Jaeger K-E, editors. WILEY-VCH; Weinheim: 2007. pp. 38–75.Enders D, Breuer I, Drosdow E. Synthesis. 2005:3239–3244.Enders D, Grondal C. Angew Chem. 2005;117:1235–1238. doi: 10.1002/anie.200462428.Angew Chem Int Ed. 2005;44:1210–1212. doi: 10.1002/anie.200462428.Enders D, Bockstiegel B. Synthesis. 1989:493–496.
- 9.Majewski M, Gleave DM, Nowak P. Can J Chem. 1995;73:1616–1626. [Google Scholar]
- 10.Silyl enol ethers were formed from alkylated ketones. The alkylated ketone was treated with triethylsilyl chloride (1.3 equiv), triethylamine (1.6 equiv) and sodium iodide (1.3 equiv) in acetonitrile (0.63 M) and purified by silica gel chromatography in 46–78% yield, with 6–15% yield of the isomeric enol ether. For details, see the supporting information.
- 11.a) Helmchen G, Pfaltz A. Acc Chem Res. 2000;33:336–345. doi: 10.1021/ar9900865. [DOI] [PubMed] [Google Scholar]; b) Williams JMJ. Synlett. 1996:705–710. and references therein. [Google Scholar]
- 12.See supporting information for details.
- 13.Phillips GH, Bailey EJ, Bain BM, Borella RA, Buckton JB, Clark JC, Doherty AE, English AF, Fazakerley H, Laing SB, Lane-Allman E, Robinson JD, Sandford PE, Sharratt PJ, Steeples IP, Stonehouse RD, Williamson C. J Med Chem. 1994;37:3717–3729. doi: 10.1021/jm00048a008. [DOI] [PubMed] [Google Scholar]
- 14.a) Rapado LP, Bulugahapitiya V, Renauld P. Helv Chim Acta. 2000;83:1625–1623. [Google Scholar]; b) Wolinsky J, Novak R, Vasilev R. J Org Chem. 1964;29:3596–3598. [Google Scholar]
- 15.For a review, see: Barco A, Benetti S, De Risi C, Marchetti P, Pollini GP, Zanirato V. Tetrahedron: Asymmetry. 1997;8:3515–3545.
- 16.a) Garg NK, Caspi DD, Stoltz BM. J Am Chem Soc. 2004;126:9552–9553. doi: 10.1021/ja046695b. [DOI] [PubMed] [Google Scholar]; b) Garg NK, Caspi DD, Stoltz BM. J Am Chem Soc. 2005;127:5970–5978. doi: 10.1021/ja050586v. [DOI] [PubMed] [Google Scholar]; c) Garg NK, Stoltz BM. Chem Commun. 2006:3769–3779. doi: 10.1039/b605929e. [DOI] [PubMed] [Google Scholar]; d) Garg NK, Caspi DD, Stoltz BM. Synlett. 2006:3081–3087. [Google Scholar]
- 17.Scholl M, Ding S, Lee CW, Grubbs RH. Org Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.