

1,3-Dipolar cycloadditions provide convenient access to carbocyclic and heterocyclic 5-membered rings.[1] In some cases, 1,3-dipoles react with dipolarophiles without activation of either component. For example ozonolysis of olefins and addition of nitrile oxides to alkynes often proceeds without added promoters.[2] With many substrate combinations, however, no cycloadduct is formed when dipole and dipoarophile are simply mixed. To accelerate these types of reactions, two complementary strategies focus on modulating the reactivity of the dipolarophile (Scheme 1). In the first, Lewis acids are included to lower the LUMO of the dipolarophile.[3] Under such conditions the dipole reacts through its HOMO to generate the cycloaddition product.[ 4 ] Alternatively, additives that increase the electron density of the dipolarophile can accelerate cycloaddition via an HOMO(dipolarophile)-LUMO(dipole) interaction. This approach is less common than the Lewis acid-based methods,[5] but is most notably operative in the copper-catalyzed synthesis of triazoles from alkynes and azides.[6] Here we report a new example of inverse electron-demand dipolar cycloadditions: the synthesis of pyrazoles via cycloaddition of copper acetylides with diazocarbonyl compounds (eq 1).
Scheme 1.

Activation of alkynes and alkenes by metals for 1,3-dipolar cycloaddition.
The pyrazole substructure appears in small molecules possessing a wide range of biological activities, and accordingly, represents a valuable target for organic synthesis.[7] A common tactic for the preparation of pyrazoles (1) relies on the condensation of β-diketones with hydrazines.[8] Alternatively, cyclization of diazo-alkenes[9] or unsaturated hydrazines[10] provides improved control of regiochemistry relative to the dicarbonyl/hydrazine condensation, although the precursors are more challenging to prepare. A direct approach to pyrazoles involves the cycloaddition of diazo compounds with alkynes. In this regard, electron-rich diazo compounds (e.g. diazomethane) react under mild conditions with electron-deficient alkynes.[11] In contrast, Lewis acids are required to promote the cycloaddition between electron deficient alkynes and diazocarbonyl compounds.[12] However, simple alkyl- or arylacetylenes generally fail to react with diazocarbonyl compounds under thermal conditions or in the presence of Lewis acids. Seeking an alternative mode of activation, we evaluated metal acetylides in cycloadditions with diazocarbonyl compounds.
When lithium phenylacetylide was treated successively with various Cu(I) sources and benzyl diazoacetate (2), pyrazole 1a was formed along with benzyl alcohol (BnOH, Table 1). In all cases conversion of diazoester was complete, and, with one exception, >90% of the mass balance was accounted for by 1a, BnOH and recovered alkyne. Fumarate and maleate side products were observed in <5% yield, allowing the use of equimolar quantities of reagents. An alkali metal acetylide was required, and in the absence of copper, no pyrazole was formed. Systematic variation of copper source, base, and additive led to the discovery that CuCN·6LiCl[13] minimized BnOH formation and maximized the yield of pyrazole 1a (entry 8).[ 14 ] The relationship between the components of the reaction mixture and the product distribution is complex. For example, while the inclusion of LiCl does not improve reactions involving CuI (entry 1 vs. 3), small but reproducible enhancements in the 1a:BnOH ratio were observed when up to 6 equiv LiCl were added to reaction mixtures containing CuCN (entries 5–9). Similarly, whereas 1:1 molar ratios of CuI and alkyne produced complex mixtures, optimal results were obtained using the lower order cuprate derived from CuCN·6LiCl (entry 1 vs. 2; 4 vs. 5).
Table 1.
Influence of Cu source and LiCl on the cycloaddition of lithium phenylacetylide with benzyl diazoacetate.a
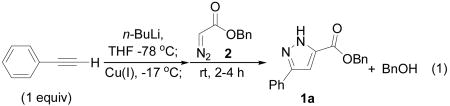
| |||
|---|---|---|---|
| Entry | Cu(I) | Equiv Cu(I) | 1a:BnOHb |
| 1 | CuI | 0.5 | 1.5:1 |
| 2 | CuI | 1.0 | c |
| 3 | CuI·2LiCl | 0.5 | 0.9:1 |
| 4 | CuCN | 0.5 | 2.0:1 |
| 5 | CuCN | 1.0 | 4.0:1 |
| 6 | CuCN·2LiCl | 1.0 | 5.4:1 |
| 7 | CuCN·4LiCl | 1.0 | 5.9:1 |
| 8 | CuCN·6LiCl | 1.0 | 6.0:1 |
| 9 | CuCN·8LiCl | 1.0 | 5.4:1 |
Complete conversion of diazoester in all cases. Mass balances were >90% as determined by 1H NMR relative to an internal standard except in entry 2.
Determined by 1H NMR analysis of the crude reaction mixture.
Complex mixture.
The cycloaddition displays substantial generality with respect to both the alkyne and diazocarbonyl component, and in all cases the product was formed as a single regioisomer (Figure 1). Electron poor and electron rich aryl acetylenes perform well in the cycloaddition. Alkyl-substituted terminal alkynes react cleanly, and halides, tertiary amino groups, esters and nitriles are tolerated. The reaction is not limited to benzyl diazoacetate as the ethyl- and tert-butyl diazoesters and Weinreb’s amides perform similarly in the cycloaddition. In many cases, the pyrazoles can be isolated in >90% purity by tituration with hexanes; in all cases analytically pure product can be isolated following column chromatography on silica gel.
Figure 1.

Copper-promoted reaction of lithium acetylides with diazo carbonyl compounds.
[a] Representatve tautomer shown
Comparisons between the present cycloaddition and two known reactions are striking. First, a report from the Fu group described a Cu(I)-catalyzed alkynylation of diazoesters (eq 2).[15] Under these neutral conditions, no pyrazole is formed. Likewise, reaction of ethyl dizaoacetate with PhCCLi/CuCN·6LiCl generates no observable alkynyl ester. The critical difference between the two systems is likely the use of alkynyl anions in the cycloaddition versus the neutral alkyne in the Fu reaction. The amount of copper used in the reaction does not appear significant as even catalytic CuI promotes the cycloaddition of lithium phenylacetylene and benzyl diazoacetate.
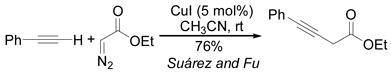 |
(2) |
The copper-mediated cycloaddition of alkynyl anions with diazoesters is also reminiscent of the copper-catalyzed cycloaddition of terminal alkynes with azides.[16] Accordingly, we suspect a similar reaction mechanism is operative in both cases. Copper may serve as an electron-donating group and raise the energy of the alkyne HOMO. Cycloaddition involving the LUMO of the diazocarbonyl compound generates a (pyrazolyl)Cu intermediate 3 which can tautomerize under the reaction conditions. This proposal accounts for several important observations. First, the fact that the reaction rate is similar in THF, ether and toluene is consistent with a concerted cycloaddition but not with stepwise formation of charged intermediates. Second, the observed regioselectivity is consistent with theoretical predictions regarding cycloaddition featuring HOMO(dipolarophile)-LUMO(dipole) interactions.[4] Third, electron withdrawing groups on the alkyne qualitatively slow the reaction. These groups should lower the HOMO of the alkyne and therefore increase the HOMO-LUMO gap. Fourth, diazomethane does not react with lithiumacetylides under the reaction conditions, presumably a reflection of its high-energy LUMO relative to diazoesters. Finally, deuterium labelling experiments support the proposed tautomerization: Copper-mediated cycloaddition with α-D-benzyl diazoacetate (2-d) yields 1a–d with substantial deuterium incorporation on the pyrazole ring (eq 3). Additionally, recovered phenyl acetylene was partially deuterated, likely reflecting deprotonation of the initial cycloadduct 3 by alkynyl anion.[17]
The origin of the benzyl alcohol side product is not clear at present, although several obvious mechanisms for its formation can be ruled out. Generation of BnOH requires a general base and not acetylides or copper salts specifically: benzyl diazoacetate reacts completely with butyllithium, Li2Cu(n-C4H9)2CN, lithium phenylacetylide or LDA to yield BnOH as the major product under mild conditions. This data, and the fact that the pyrazole is stable under the reaction conditions, implicates benzyl diazoacetate as the source of BnOH. In principle, direct E2 elimination of alkoxide from benzyl diazoacetate could account for BnOH formation. If so, reducing the kinetic acidity of 2a might increase the 1a:BnOH ratio.[18] In practice, however, reactions with 2 or 2-d yield nearly identical 1a:BnOH ratios (cf. Table 1, entry 8 and eq 3). Additionally, recovered phenylacetylene was only partially deuterated, so deprotonation could not account for all of the BnOH formation. Direct addition to the carbonyl could release BnOH, yet such a pathway is not supported by the data. In particular, unreacted phenylacetylene is recovered after the reaction, and no addition products are observed in the crude reaction mixtures. Furthermore, the similar yields of pyrazole obtained with small (ethyl) and large (tert-butyl) diazoesters argues against a competing mechanism involving addition to the carbonyl. Finally, simple ester hydrolysis appears unlikely. Specifically, reactions performed with varying sub-stoichiometric amounts of water yield similar amounts of BnOH while excess water inhibits the reaction. Further, the production of BnOH is inversely related to the amount of LiCl (a possible source of water) included in the reaction mixture.
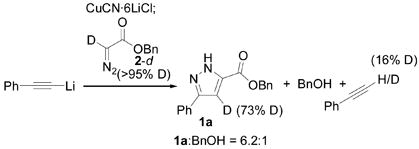 |
(3) |
The copper-promoted cycloaddition of acetylides with diazocarbonyl compounds offers a direct and efficient approach to the synthesis of pyrazoles. The method is operationally simple and tolerates substantial variation in the two reacting partners. Furthermore, as a rare example of an inverse-electron-demand cycloaddition, it represents a conceptually novel approach to this important class of heterocycles. Ongoing studies seek clarification of the mechanisms responsible for pyrazole and side-product formation with an aim to identify reaction parameters which favor the former over the latter.
Experimental Section
BuLi (1.0 mmol) was added to a solution of alkyne (1.0 mmol) in THF (4 mL) at −78 °C. After 1 hour this solution was transferred to a solution of CuCN·6LiCl (1.0 mmol) in THF (6 mL). The reaction mixture was warmed to −17 °C (dry ice/brine) and stirred for 1 h. A solution of diazo compound (1.0 mmol) in THF (4 mL) was added. After stirring for 2–4 h at rt, NH4Cl(aq) and ether were added. The aqueous layer was extracted with ether and the combined organic phases were concentrated and purified by flash chromatography. Full characterization data is supplied as supporting information.
Supplementary Material
Scheme 2.

Proposed cycloaddition of copper acetylide with diazo carbonyl compounds.
Footnotes
Financial support from the NIGMS (GM074822) and UT Southwestern (J.M.R is a Southwestern Medical Foundation Scholar in Biomedical Research).
References
- 1.Padwa A, Pearson WH, editors. Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products. John Wiley & Sons, Inc; Hoboken: 2003. [Google Scholar]
- 2.Melo TMVDP. Curr Org Chem. 2005;9:925–958. [Google Scholar]
- 3.Gothelf KV, Jorgensen KA. Chem Rev. 1998;98:863–910. doi: 10.1021/cr970324e. [DOI] [PubMed] [Google Scholar]
- 4.a) Sustmann R. Tetrahedron Lett. 1971;29:2717–2720. [Google Scholar]; b) Bastide J, Henri-Rousseau NCO. Tetrahedron Lett. 1972;41:4225–4228. [Google Scholar]; c) Houk KN. Accts Chem Res. 1975;8:361–369. [Google Scholar]
- 5.a) Barluenga J, Valdes C, Beltran G, Escribano M, Aznar F. Angew Chem Int Ed. 2006;45:6893–6896. doi: 10.1002/anie.200601045. [DOI] [PubMed] [Google Scholar]; b) Barluenga J, Valdes C, Beltran G, Escribano M, Aznar F. Angew Chem. 2006;118:7047–7050. doi: 10.1002/anie.200601045. [DOI] [PubMed] [Google Scholar]
- 6.a) Himo F, Lovell T, Hilgraf R, Rostovtsev VV, Noodleman L, Sharpless KB, Fokin VV. J Am Chem Soc. 2005;127:210–216. doi: 10.1021/ja0471525. [DOI] [PubMed] [Google Scholar]; b) Bock VD, Hiemstra H, van Maarseveen JH. Eur J Org Chem. 2006:51–68. [Google Scholar]
- 7.For examples of biologically active pyrazoles, see the supporting information.
- 8.For lead references: Heller ST, Natarajan SR. Org Lett. 2006;8:2675–2678. doi: 10.1021/ol060570p.
- 9.Padwa A, Kulkarni YS, Zhang Z. J Org Chem. 1990;55:4144–4153. [Google Scholar]
- 10.a) Martin R, Rivero MR, Buchwald SL. Angew Chem Int Ed. 2006;45:7079–7082. doi: 10.1002/anie.200602917. [DOI] [PubMed] [Google Scholar]; b) Martin R, Rivero MR, Buchwald SL. Angew Chem. 2006;118:7237–7240. [Google Scholar]
- 11.(a) Sauer DR, Schneller SW. J Org Chem. 1990;55:5535–5538. [Google Scholar]; (b) Aggarwal VK, de Vicente J, Bonnert RB. J Org Chem. 2003;68:5381–5383. doi: 10.1021/jo0268409. [DOI] [PubMed] [Google Scholar]; c) Maas G. pp. 623–680. in ref [1] [Google Scholar]
- 12.Jiang N, Li CJ. Chem Commun. 2004:394–395. doi: 10.1039/b311763d.Intramolecular versions: Kende AS, Journet M. Tetrahedron Lett. 1995;36:3087.Maas G, Gettwert V. Tetrahedron. 2000;56:4139–4147.
- 13.CuCN·2LiCl: Knochel P, Yeh MCP, Berk SC, Talbert J. J Org Chem. 1988;53:2392–2394.
- 14.For selected optimization studies, see the supporting information.
- 15.a) Suarez A, Fu G. Angew Chem Int Ed. 2004;43:3580–3582. doi: 10.1002/anie.200454070. [DOI] [PubMed] [Google Scholar]; b) Suarez A, Fu G. Angew Chem. 2004;116:3664–3666. [Google Scholar]
- 16.(a) Himo F, Lovell T, Hilgraf R, Rostovtsev VV, Noodleman L, Sharpless KB, Fokin VV. J Am Chem Soc. 2005;127:210–216. doi: 10.1021/ja0471525. [DOI] [PubMed] [Google Scholar]; (b) Bock VD, Hiemstra H, van Maarseveen JH. Eur J Org Chem. 2006:51–68. [Google Scholar]
- 17.Quenching with DCl yielded >90% deuterated recovered alkyne and 79% deuterated 1a–d.
- 18.a) Bestmann HJ, Soliman FM. Angew Chem Int Ed. 1979;18:947–948. [Google Scholar]; b) Bestmann HJ, Soliman FM. Angew Chem Int Ed. 1979;91:1012–1013. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.