

Abstract
Metaplastic tubular complexes (MTC) have been proposed as precursor lesions for pancreatic adenocarcinoma (PDAC). In this study, we investigated the potential role of bone marrow derived progenitor cells (BMPC) in the formation of MTC and PDAC in a rat model. F344 rats defective for CD26 (dipeptidyl peptidase IV, DPPIV) expression were sublethally irradiated and received rescue bone marrow cells from wild type F344 rats that express CD26. After confirming engraftment, recipient animals received dimethylbenzanthracene (DMBA) implantation in their pancreas. Animals were sacrificed monthly from 3 to 7 months. We observed both MTC and tumors in animals that received DMBA. These MTC were ductal complexes since they stained positive for cytokeratin but were negative for chymotrypsin and chromogranin A. Cells that expressed both CD26 and cytokeratin were rarely observed in the MTC. Cells expressing either both CD26 and CD45 or CD26 and smooth muscle actin were also found near the MTC. However, no CD26 signal was detected in the tumors. Within this model, there appeared to be no evidence supporting that BMPC turned into tumor cells directly. BMPC could modulate pancreatic cancer growth through tumor microenvironment.
Keywords: bone marrow, progenitor cell, pancreatic cancer, tumorigenesis, microenvironment
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a highly malignant disease with a bleak prognosis. It ranks 9th in the incidence of solid cancers and 4th for cancer-related death in the United States. The 5-year survival rate is on average less than 4% (1). Most of the PDAC produce no symptoms in their early stage and are, therefore, usually diagnosed at late stage when no effective treatment is available.
Similar to other cancers, PDAC probably begins with small precursor lesions. The nomenclature and classification of such lesions have been made previously (2, 3). It is still controversial regarding the origin of these lesions. In the rat chemical carcinogenesis model, dimethylbenzanthracene (DMBA) has been shown to induce metaplastic tubular complexes (MTC) in pancreas, which are believed to represent the cancer precursor lesions (4). These MTC could be derived from acinar cells by degeneration and atrophy (5, 6) or transdifferentiation (7). Two previous studies suggested that PDAC originates by differentiation of acinar/centroacinar cells or their precursors into ductal-like cells (8, 9). However, a recent study argued that acinar-to-ductal transdifferentiation accounts for only a minority of such metaplastic lesion (10). In addition to transdifferentiation, metaplasia can also occur through other mechanisms such as selective proliferation or loss of a cell type or differentiation of stem cells (11).
The cancer stem cell theory has gained considerable attention the past few years. According to the theory, tissue stem cells are recruited to injury or inflammation to repair damage. Usually this process is transient and tightly regulated. However, in cases of chronic injury or inflammation, such tissue stem cell becomes a perfect target for accumulating genetic mutations because of its long lifespan and instability. These mutated stem cells lose regulatory growth/differentiation controls and eventually lead to cancer (12). Cancer stem-like cells have been identified based on the presence of special markers or high drug efflux capacity in many cancers such as brain cancers (13-15), breast cancer (16), and hepatoma and colorectal cancer (17). Recently, a pancreatic cancer stem cell has been identified based on the presence of a combination of special markers (18, 19). These cancer stem cells all possess the ability of self renewal and can give rise to tumors similar to the original tumor where they were derived from. Unfortunately, the origin of these cancer stem cells remains a mystery. Bone marrow could be a potential source of these cancer stem cells. This idea is supported by an elegant study that showed recipient gastric cancer cells contained donor bone marrow derived marker (20). According to that study, female mice were chronically infected with Helicobacter, a known carcinogen, and received sex-mismatched bone marrow transplantation. The transplanted bone marrow derived cells repopulated the stomach and subsequently progress through metaplasia and dysplasia to intraepithelial cancer. Their findings suggested that epithelial cancers can originate from marrow-derived sources.
In the current study, we investigated the potential role of bone marrow derived progenitor cells (BMPC) in the formation of MTC and pancreatic cancer. We used a rat model for PDAC published previously (4) with some modifications.
Materials and methods
Animals
CD26 negative (CD26-) Fisher344 male rats (age 8−10 weeks) were in house bred and maintained on standard laboratory chow and daily cycles, alternating 12 hours of light and dark. Wild-type Fisher344 male rats (age 8−10 weeks) were purchased from Charles River Laboratories (Wilmington, MA). All procedures were performed with the approval of the University of Florida Institutional Animal Care and Usage Committee.
Bone marrow isolation and transplantation
Prior to bone marrow transplantation, CD26- rats (recipients) were exposed to total body gamma irradiation (137Cs, JL Shepherd Mark-I; J. L. Shepherd and Associates, San Fernando, CA), administered in 2 doses (450 rads per dose), 3 hours apart. Whole bone marrow cells were collected from tibias and fibulas of the wild-type rats (donors). Cells were passed through a 130-μm cell strainer, centrifuged at 220 × g, and resuspended in Iscove's modified Dulbecco's medium (IMDM) (GIBCO, Grand Island, NY). These cells were transplanted into the recipient rats via tail vein injection after irradiation (1 × 106 cells/rat). Four weeks later, the donor contribution to bone marrow reconstitution was assessed through analysis of the presence of CD26 in peripheral blood cells.
DMBA implantation
Surgical procedures were performed according to the protocol published previously (4) with modification. Briefly, rats were subjected to general anesthesia and underwent a midline laparotomy with exposure of the pancreatic body. A 5-mm incision was performed on the surface of the pancreas. A pocket in the pancreatic parenchyma was developed at the incision site, where 5 mg DMBA crystals were implanted. The incision was secured with a 6−0 prolene pursestring suture. Control animals underwent the same surgical procedures except no carcinogen implantation. Animals were sacrificed monthly from 3 to 7 months after surgery, or earlier if moribund. Each group contained 3 animals. Five animals served as control group. During necropsy, the previous chemical implantation site was carefully inspected and palpated for nodules/tumors.
Immunohistochemistry
For morphology studies, 5-μm paraffin embedded tissue sections were stained with hematoxylin and eosin (H&E). Immunohistochemistry and immunofluorescence were performed on either paraffin embedded tissue sections, frozen tissue sections or cultured cells using standard protocols. Immunophenotyping of samples used 1:100 mouse anti-CD26 and 1:100 mouse anti-CD45 (common leukocyte antigen) (both from BD Biosciences, San Jose, CA); 1:100 goat anti-amylase, 1:100 goat ant-CD31, 1:100 goat anti-CD34 (all from Santa Cruz Biotechnology, Santa Cruz, CA); 1:1600 mouse anti-smooth muscle actin (Calbiochem, San Diego, CA); 1:100 rabbit anti-chromogranin A (Chemicon, Temecula, CA); 1:400 mouse anti-chymotrypsin (Biogenesis Inc., Kinston, NH); 1:100 rabbit anti-von Willebrand factor, 1:1000 rabbit anti-cytokeratin, 1:100 mouse anti-CD90 (Thy1), and 1:100 mouse anti-desmin (all from Dakocytomation, Carpinteria, CA). We used total serum gamma immunoglobulins (IgGs) (Vector Laboratories, Burlingame, CA) from the same species where the corresponding primary antibodies were generated from as negative controls. Vector ABC-kit (Vector Laboratories, Burlingame, CA) and 3,3’-diaminobenzidine tetrahydrochloride (DAB) reagent (Dakocytomation, Carpinteria, CA) were used in the immunoperoxidase detection procedure. For immunofluorescence staining, Vectastain kit with DAPI, Texas-red, and fluorescein-conjugated secondary antibodies (Vector Laboratories, Burlingame, CA) were used. The enzymatic DPPIV staining procedure was performed as described previously (21). The sections were photographed using an Olympus microscope and Optronics digital camera (Olympus, Melville, NY).
Results
Characterization of MTC and tumor
Four weeks after allogenic bone marrow transplantation, engraftment of the donor BMPC in the recipient animals were examined. We found that around 50−70% of the recipient animals’ peripheral blood nucleated cells expressed the donor marker of CD26. After confirming successful bone marrow engraftment, DMBA was surgically implanted into the recipient animals’ pancreatic tail. In the rat pancreas, DMBA has been shown to induce MTC, which is believed to be precursor lesions for PDAC (4). Between 3 and 7 months after the chemical implantation, we observed similar lesions in the pancreas (Fig. 1E). These MTC have wide lumen lined by a monolayer of flat or cuboidal cells. Around the lesions, there were significant amount of inflammation and desmoplastic reaction. These MTC stained positive for pan-cytokeratin (CK) (Fig. 1F), but were negative for chymotrypsin (Fig. 1H). Positive staining to chromogranin was seen at around but not in the MTC (Fig. 1G). Similar staining patterns were also observed previously (22). Among different recipient animals, the extent of such lesions varied. In contrast to the carcinogen implanted animals, no such lesion was seen in the control animals. Hematoxylin & eosin (H&E) (Fig. 1A), CK (Fig. 1B), chromogranin (Fig. 1C), and chymotrypsin (Fig. 1D) staining revealed morphology, ductal cells, islets, and acinar cells of normal rat pancreas, respectively. In addition to these microscopic lesions, 3 of the 15 experimental animals grew large tumor (Fig. 1I) and presenting with hemorrhagic ascites. The tumor sections had spindle swirl morphology under low power microscopic view similar to the findings seen in sarcoma. However, some of the tumor cells stained positive for CK (Fig. 1J) but were negative for chromogranin and chymotrypsin staining (data not shown).
Figure 1.
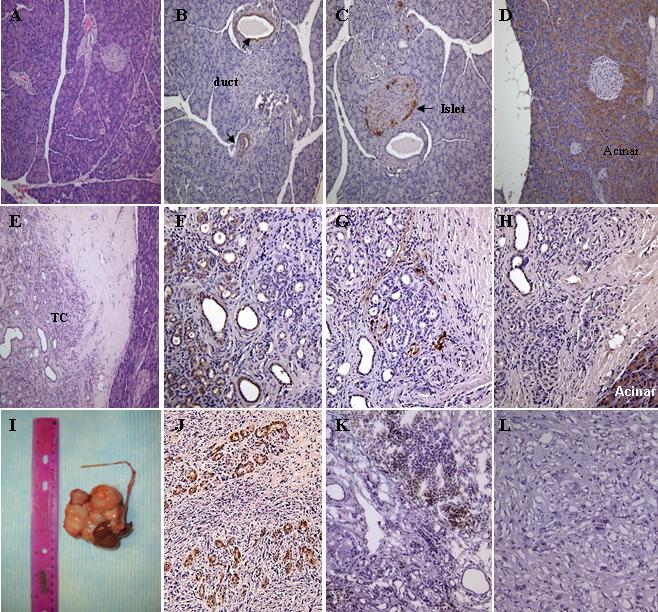
Characterization of MTC and tumor in the pancreas of animals received DMBA. (A) hematoxylin & eosin, (B) CK (arrows indicate pancreatic ducts), (C) chromogranin (arrow indicates pancreatic islet), (D) chymotrypsin staining of paraffin section of normal pancreas (Magnification 200x). (E) hematoxylin & eosin, (F) CK, (G) chromogranin, (H) chymotrypsin staining of paraffin section of pancreatic nodular lesion (Magnification 200x). (I) large tumor found near the DMBA implantation site. (J) CK staining of paraffin section of the tumor (Magnification 200x). (K) CD26 staining of frozen section of the pancreatic nodular lesion (Magnification 200x). (L) CD26 staining of frozen section of the tumor (Magnification 200x). All of the stainings except (A) and (E) were stained with immunoperoxidase conjugated antibodies. TC, tubular complexes.
Characterization of the role of BMPC in the MTC and tumor
To study the role of bone marrow derived cells in the MTC and tumor, tissue sections were first stained for CD26 alone. Positive CD26 staining was found at around and in the MTC (Fig. 1K) but not in the tumor itself (Fig. 1L). To further investigate the role of bone marrow derived cells, double stainings for CD26 in combination with various markers including von Willebrand factor (vWF), CD45, smooth muscle actin (SMA), and CK were performed. There was no area stained positive for both CD26 and vWF in the tissue sections (data not shown), which suggests that BMPC did not contribute to neoangiogenesis in this model. Cells expressing both CD26 and CD45 (Fig. 2G) or CD26 and SMA (Fig. 2C) were found scattered around the MTC, although at a low frequency. Finally, less than 5% of the cells in the MTC expressed both CD26 and CK (Fig. 2K).
Figure 2.
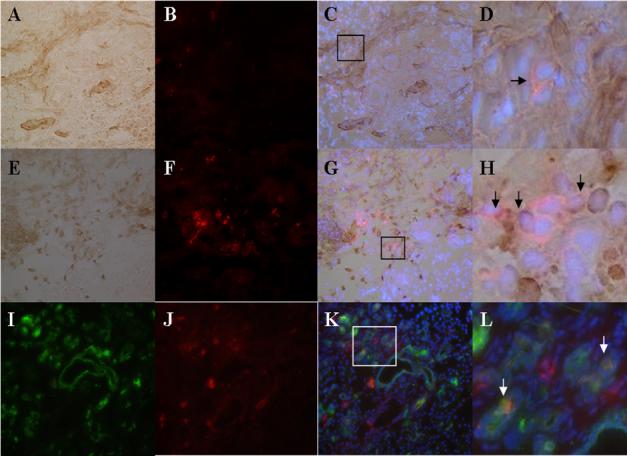
Characterization of the role of bone marrow derived cells in the MTC. (A) immunoperoxidase staining for SMA (Magnification 200x). (E) immunoperoxidase staining for CD45 (Magnification 200x). (I) immunofluorescence staining for CK (Magnification 200x). (B, F, J) immunofluoresence staining of CD26 (Magnification 200x). (C) merge image of (A) and (B). (G) merge image of (E) and (F). (K) merge image of (I) and (J). (D) magnification of the box in (C) (Arrow indicates cell expressing both CD26 and SMA). (H) magnification of the box in (G) (Arrows indicate cells expressing both CD26 and CD45). (L) magnification of the box in (K) (Arrows indicate cells expressing both CD26 and CK). DAPI was used to counter stain nuclei in (C), (G), and (K).
Discussion
Tissue metaplasia occurs through a variety of mechanisms, but not limited to; including selective proliferation or loss of a cell type, transdifferentiation, or differentiation of stem cells (11). The current study was conducted to investigate the role of BMPC in the formation MTC and tumor. By losing acinar components through acinar degeneration and atrophy, MTC could become the predominant findings in acute and chronic pancreatitis (5, 6). However, MTC could also come from acinar-to-ductal transdifferentiation as shown previously (7).
Cells expressed either both CD26 and CD45 or CD26 and SMA were found near the MTC in vivo, albeit in low frequency. There were other cells near the MTC expressing either CD45 or SMA only but no CD26 expression was observed. Since the study animals are chimera in bone marrow components, it is likely that these single labeled cells were derived from the recipient animals’ own BMPC. However, these findings suggest that BMPC could influence MTC and tumor formation though differentiation to lymphocytes and myofibroblasts. Inflammation has been proposed to be a critical component of tumor progression (23). Many cancers arise from sites of infection, chronic irritation and inflammation. Tumor microenvironment, which is largely orchestrated by inflammatory cells, fosters tumor proliferation, survival and migration (23). In a chemically induced pancreatic carcinogenesis study, similar to this study, lymphocyte infiltrates were observed very early after chemical implantation and that infiltration increased with time paralleling pancreatic lesions (24). The current consensus is that PDAC is not characterized by a lack of specific T-cell immunity but by a potent barrier established by complex cancer-stroma interactions that inhibit T-cell activity in situ (25). Several studies have shown that bone marrow could contribute to both the myofibroblast and fibroblast populations within the tumor stroma (26-28). By labeling the donor bone marrow, the frequency of donor derived myofibroblasts varied significantly among different studies (26-28). It is also believed that these myofibroblasts are responsible for the desmoplastic reaction commonly seen in PDAC (29).
In this study, we also observed that less than 5% of the cells expressed both CD26 and CK in the MTC. These so called MTC have been demonstrated previously deriving from acinar cells by degeneration and atrophy (5, 6) or transdifferentiation (7). Strobel et al. recently published an elegant study showing that although acinar-to-ductal transdifferentiation occurs in adult pancreas in vivo, it accounts for only a minority of MTC lesions (10). Although not a dominant theory, MTC could also possibly come from ductal proliferation or merely represent preexisting ducts that are less sensitive to injury than acinar cells and persist during acinar loss (30). In the current study, our findings support that although BMPC could differentiate to pancreatic ductal cells in vivo, directly or through transdifferentiation, it is not a common event.
Regarding the fate of MTC, it is still controversial although it has been considered as precursor lesions of PDAC by other studies (7, 22). In 3 out of the 15 experimental animals in this study, large tumors were found close to the chemical implantation site. These tumors showed spindle swirl morphology under low power microscopic view similar to the findings seen in sarcoma. However, immunoactivity of CK was also seen in the tumor sections but no CD26 immunoactivity was seen in tumor tissue. Despite the rare CD26 positive cells seen in the MTC, no CD26 positive cells were seen within the tumors. These findings suggest that BMPC did not contribute to tumor formation directly in this model, i.e., they did not act as cancer stem cells. Alternatively, MTC could lose the CD26 donor marker as they progressed to tumors although we can neither support nor rule out this possibility. Similar to our study, a previous study using allogenic bone marrow cells and transgenic animals susceptible to develop islet tumors also failed to show the donor marker(s) in the tumor. However, about 25% of myofibroblasts around the tumor were derived from the donor bone marrow (27). Their findings also suggested that BMPC do not act as cancer stem cells.
There are several limitations in this study. Since we used whole bone marrow cells for in vivo studies, we do not know what type of bone marrow derived progenitor cells differentiate to MTC. A recent study reported preferential homing of systemically administered bone marrow derived mesenchymal stem cells to xenografted pancreatic cancer but not normal pancreatic tissue (31). This raised the possibility that mesenchymal stem cells could be the source of MTC and myofibroblast in our study. Because of the study design, we did not observe acinar-to-ductal transdifferentiation in this study. Nevertheless, we cannot completely rule out this possibility if it happens early in the study as suggested by others (7).
In summary, we report in this study that there appeared to be no evidence supporting that BMPC turned into pancreatic cancer directly within this chemical carcinogenesis model. However, the BMPC could modulate tumor growth through tumor microenvironment by differentiating to inflammatory cells and myofibroblasts. Clearly, more studies are required to fully understand the role of BMPC in tumor formation.
Acknowledgement
This study was supported by NIH grant T32DK60443.
References
- 1.Zhang J, Dhakal I, Yan H, Phillips M, Kesteloot H. Trends in pancreatic cancer incidence in nine SEER Cancer Registries, 1973−2002. Ann. Oncol. 2007;18:1268–1279. doi: 10.1093/annonc/mdm123. [DOI] [PubMed] [Google Scholar]
- 2.Hruban RH, Adsay NV, Albores-Saavedra J, Compton C, Garrett ES, Goodman SN, Kern SE, Klimstra DS, Kloppel G, Longnecker DS, Luttges J, Offerhaus GJ. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am. J. Surg. Pathol. 2001;25:579–586. doi: 10.1097/00000478-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 3.Kern S, Hruban R, Hollingsworth MA, Brand R, Adrian TE, Jaffee E, Tempero MA. A white paper: the product of a pancreas cancer think tank. Cancer Res. 2001;61:4923–4932. [PubMed] [Google Scholar]
- 4.Rivera JA, Graeme-Cook F, Werner J, Z'Graggen K, Rustgi AK, Rattner DW, Warshaw AL, Fernandez-del Castillo C. A rat model of pancreatic ductal adenocarcinoma: targeting chemical carcinogens. Surgery. 1997;122:82–90. doi: 10.1016/s0039-6060(97)90268-3. [DOI] [PubMed] [Google Scholar]
- 5.Bockman DE, Boydston WR, Anderson MC. Origin of tubular complexes in human chronic pancreatitis. Am. J. Surg. 1982;144:243–249. doi: 10.1016/0002-9610(82)90518-9. [DOI] [PubMed] [Google Scholar]
- 6.Willemer S, Adler G. Histochemical and ultrastructural characteristics of tubular complexes in human acute pancreatitis. Dig. Dis. Sci. 1989;34:46–55. doi: 10.1007/BF01536153. [DOI] [PubMed] [Google Scholar]
- 7.Bockman DE, Guo J, Buchler P, Muller MW, Bergmann F, Friess H. Origin and development of the precursor lesions in experimental pancreatic cancer in rats. Lab. Invest. 2003;83:853–859. doi: 10.1097/01.lab.0000074918.31303.5a. [DOI] [PubMed] [Google Scholar]
- 8.Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, Dubus P, Sandgren EP, Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 9.Esposito I, Seiler C, Bergmann F, Kleeff J, Friess H, Schirmacher P. Hypothetical progression model of pancreatic cancer with origin in the centroacinar-acinar compartment. Pancreas. 2007;35:212–217. doi: 10.1097/mpa.0b013e31805d0190. [DOI] [PubMed] [Google Scholar]
- 10.Strobel O, Dor Y, Alsina J, Stirman A, Lauwers G, Trainor A, Castillo CF, Warshaw AL, Thayer SP. In vivo lineage tracing defines the role of acinar-to-ductal transdifferentiation in inflammatory ductal metaplasia. Gastroenterology. 2007;133:1999–2009. doi: 10.1053/j.gastro.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tosh D, Slack JM. How cells change their phenotype. Nat. Rev. Mol. Cell. Biol. 2002;3:187–194. doi: 10.1038/nrm761. [DOI] [PubMed] [Google Scholar]
- 12.Perryman SV, Sylvester KG. Repair and regeneration: opportunities for carcinogenesis from tissue stem cells. J. Cell. Mol. Med. 2006;10:292–308. doi: 10.1111/j.1582-4934.2006.tb00400.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 14.Kondo T, Setoguchi T, Taga T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc. Natl. Acad. Sci. U S A. 2004;101:781–786. doi: 10.1073/pnas.0307618100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 16.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haraguchi N, Utsunomiya T, Inoue H, Tanaka F, Mimori K, Barnard GF, Mori M. Characterization of a side population of cancer cells from human gastrointestinal system. Stem Cells. 2006;24:506–513. doi: 10.1634/stemcells.2005-0282. [DOI] [PubMed] [Google Scholar]
- 18.Olempska M, Eisenach PA, Ammerpohl O, Ungefroren H, Fandrich F, Kalthoff H. Detection of tumor stem cell markers in pancreatic carcinoma cell lines. Hepatobiliary Pancreat. Dis. Int. 2007;6:92–97. [PubMed] [Google Scholar]
- 19.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 20.Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, Li H, Cai X, Fox JG, Goldenring JR, Wang TC. Gastric cancer originating from bone marrow-derived cells. Science. 2004;306:1568–1571. doi: 10.1126/science.1099513. [DOI] [PubMed] [Google Scholar]
- 21.Dabeva MD, Hwang SG, Vasa SR, Hurston E, Novikoff PM, Hixson DC, Gupta S, Shafritz DA. Differentiation of pancreatic epithelial progenitor cells into hepatocytes following transplantation into rat liver. Proc. Natl. Acad. Sci. U S A. 1997;94:7356–7361. doi: 10.1073/pnas.94.14.7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jimenez RE, Z'Graggen K, Hartwig W, Graeme-Cook F, Warshaw AL, Fernandez-del Castillo C. Immunohistochemical characterization of pancreatic tumors induced by dimethylbenzanthracene in rats. Am. J. Pathol. 1999;154:1223–1229. doi: 10.1016/S0002-9440(10)65374-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ubirajara-Garcia I, Escribano MJ. Immunosurveillance by T-lymphocytes in pretumoral stages of chemically induced pancreatic carcinogenesis. Cancer Lett. 1992;67:79–86. doi: 10.1016/0304-3835(92)90011-j. [DOI] [PubMed] [Google Scholar]
- 25.Kleeff J, Beckhove P, Esposito I, Herzig S, Huber PE, Lohr JM, Friess H. Pancreatic cancer microenvironment. Int. J. Cancer. 2007;121:699–705. doi: 10.1002/ijc.22871. [DOI] [PubMed] [Google Scholar]
- 26.Ishii G, Sangai T, Oda T, Aoyagi Y, Hasebe T, Kanomata N, Endoh Y, Okumura C, Okuhara Y, Magae J, Emura M, Ochiya T, Ochiai A. Bone-marrow-derived myofibroblasts contribute to the cancer-induced stromal reaction. Biochem. Biophys. Res. Commun. 2003;309:232–240. doi: 10.1016/s0006-291x(03)01544-4. [DOI] [PubMed] [Google Scholar]
- 27.Direkze NC, Hodivala-Dilke K, Jeffery R, Hunt T, Poulsom R, Oukrif D, Alison MR, Wright NA. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. 2004;64:8492–8495. doi: 10.1158/0008-5472.CAN-04-1708. [DOI] [PubMed] [Google Scholar]
- 28.Sangai T, Ishii G, Kodama K, Miyamoto S, Aoyagi Y, Ito T, Magae J, Sasaki H, Nagashima T, Miyazaki M, Ochiai A. Effect of differences in cancer cells and tumor growth sites on recruiting bone marrow-derived endothelial cells and myofibroblasts in cancer-induced stroma. Int. J. Cancer. 2005;115:885–892. doi: 10.1002/ijc.20969. [DOI] [PubMed] [Google Scholar]
- 29.Yen TW, Aardal NP, Bronner MP, Thorning DR, Savard CE, Lee SP, Bell RH., Jr. Myofibroblasts are responsible for the desmoplastic reaction surrounding human pancreatic carcinomas. Surgery. 2002;131:129–134. doi: 10.1067/msy.2002.119192. [DOI] [PubMed] [Google Scholar]
- 30.Reid LE, Walker NI. Acinar cell apoptosis and the origin of tubular complexes in caerulein-induced pancreatitis. Int. J. Exp. Pathol. 1999;80:205–215. doi: 10.1046/j.1365-2613.1999.00116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kallifatidis G, Beckermann BM, Groth A, Schubert M, Apel A, Khamidjanov A, Ryschich E, Wenger T, Wagner W, Diehlmann A, Saffrich R, Krause U, Eckstein V, Mattern J, Chai M, Schutz G, Ho AD, Gebhard MM, Buchler MW, Friess H, Buchler P, Herr I. Improved lentiviral transduction of human mesenchymal stem cells for therapeutic intervention in pancreatic cancer. Cancer Gene Ther. 2008;15:231–240. doi: 10.1038/sj.cgt.7701097. [DOI] [PubMed] [Google Scholar]