

Abstract
The E-cadherin transcriptional repressor Snail is a prognostic marker for metastatic breast carcinoma, as well as a critical determinant of tumor growth and recurrence. We define a non-angiogenic, autocrine function for the vascular endothelial growth factor-A (VEGF-A) in regulating Snail expression in breast tumor cells. The transfection of well-differentiated breast tumor cells with VEGF-A increases Snail mRNA and protein levels, resulting in reduced E-cadherin expression. Conversely, reducing endogenous VEGF-A expression in poorly-differentiated breast tumor cells by siRNA transfection decreases Snail levels. Our studies demonstrate that VEGF and the VEGF receptor Neuropilin-1 increase Snail expression by suppressing the Glycogen Synthase Kinase-3 (GSK-3), an established inhibitor of Snail transcription and protein stability. The VEGF-A neutralizing antibody Avastin® was recently approved by the FDA for the treatment of metastatic breast cancer. We present the provocative finding that beyond its anti-angiogenic activity, Avastin® can reduce Snail expression in breast tumor cells. Collectively, this work describes a novel autocrine function for VEGF in breast tumor cells in driving the expression of Snail, a breast tumor progression factor. Based on our demonstration that Avastin® reduces Snail expression in breast tumor cells, we propose that the treatment of early stage breast cancer patients with Avastin® may impede tumor progression.
Keywords: VEGF, autocrine, Neuropilin, Snail, Avastin
Introduction
The transition of a “normal” epithelial cell to a metastatic tumor cell requires that this cell evolve the ability to survive in foreign environments, to migrate and to invade tissue. The E-cadherin transcriptional repressor Snail is unique in its ability to impart on a cell a number of these activities associated with metastatic tumor progression. Strikingly, Snail is a prognostic marker for metastatic breast carcinoma, being expressed in node-positive, but not in low grade, node-negative specimens[1]. Early studies of Snail focused on its ability to induce an epithelial-mesenchymal transition (EMT) by acting as an E-cadherin transcriptional repressor [2, 3]. However, recent studies indicate that Snail also maintains cell survival and promotes cell migration[4]. Furthermore, in mouse models of cancer, Snail is critical for tumor growth and recurrence[5, 6]. Based on its established importance for breast tumor progression, studies identifying the factors that regulate Snail expression in metastatic breast carcinoma are of utmost importance.
We and others established previously that breast tumor cells express the VEGF receptor Neuropilin-1 (NP-1)[7–9], and that an autocrine VEGF/NP-1 pathway stimulates constitutive PI3-kinase activity in breast tumor cells[7]. Similar to the expression pattern of Snail in breast tumors[1], VEGF-A expression is significantly increased in metastatic compared to non-metastatic breast tumors[10]. Furthermore, recombinant VEGF can stimulate the expression of Snail family members in pancreatic tumor cells [11]. Based on these findings, in the current work, we address the hypothesis that autocrine VEGF-A signaling in breast tumor cells stimulates Snail expression and activity. We also investigate a novel activity for a VEGF-A-directed therapeutic in suppressing tumor cell expression of Snail.
Materials and Methods
Cell lines
T47D and MDA-MB-435 cells were obtained from Duke University Comprehensive Cancer Center’s cell culture facility, and were cultured in RPMI/5%FBS and DMEM/5% FBS, respectively. SUM159 cells were kindly provided by Dr. Stephen Ethier, and cultured in DMEM/F12(1:1)/5% FBS. MCF-7 cells were obtained from Dr. Joseph Geradts (Duke University Medical Center), and were cultured in DMEM/5% FBS.
Antibodies
The following antibodies were used in these studies: Neuropilin-1-neutralizing antibody (R&D Systems-MAB566), mouse IgG2B, (R&D Systems), Avastin® (Genentech)(kindly provided by Dr. Andrew Nixon, Duke University Medical Center), human IgG1κ (Sigma), mouse anti E-cadherin (R&D Systems MAB1838), mouse anti Snail [12], mouse anti β-actin (Sigma), rabbit anti phospho-GSK-3β(Ser9) (Cell Signaling), mouse anti GSK-3 (Upstate), IRDye™700 Donkey anti Mouse IgG and IRDye™800 Goat anti Rabbit IgG (Rockland).
RNA and protein extraction
Total RNA was extracted from the indicated cells using the RNeasy kit® (Qiagen) according to the manufacturer’s protocol. Proteins were extracted from cells using RIPA lysis buffer [150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris (pH 8.0), 2 mM phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 5 μg/ml pepstatin, 50 μg/ml leupeptin, and 1 mM sodium orthovanadate].
Immunoblotting
Equivalent amounts of total cellular protein extracted from the cells of interest were exposed to SDS-PAGE, transferred to nitrocellulose and probed with the indicated antibodies, followed by an IRDye700 (Rockland Inc.)-conjugated secondary antibody of the appropriate species. Protein bands were visualized using Odyssey Infrared Imaging System (LI-COR Biosciences). Images were cropped and optimized for clarity using Adobe Photoshop.
pCDNA3.1-VEGF-A transfections
Cells at 70% confluence were transfected transiently with either a control vector (PCDNA3.1®, Invitrogen) or VEGF-A165-expressing PCDNA3.1 vector (kindly provided by Dr. Donald Senger, Beth Israel Deaconess Medical Center)[13] using Lipofectamine2000® (Invitrogen).
Incubation of tumor cells with recombinant human VEGF165
Cells at 70% confluence were cultured in reduced serum medium (0.5% FBS) containing 0.5ng/ml recombinant human VEGF165 (R&D Systems).
VEGF-A siRNA transfections
Cells at 70% confluence were transfected using Lipofectamine2000® with either 100nM control siRNA (Santa Cruz) or VEGF siRNA (Dharmacon-{NN-N19}-NNACGCGUAACGCGGGAAUUU).
VEGF-A ELISA
VEGF-A protein levels were measured in cell lysates using a human VEGF-A ELISA kit ( R&D Systems) according to the manufacturer’s protocol. All samples were analyzed in triplicate.
Incubation of breast tumor cells with VEGF-neutralizing antibody
Cells were incubated for 4 hours with 5μg/ml Avastin® or an isotype control antibody (human IgG1κ, Sigma) in reduced serum conditions (DMEM-0.5% FBS). Avastin® (Genentech) was kindly provided by Dr. Andrew Nixon (Duke University Medical Center).
Quantifying mRNA
Reverse Transcription PCR (RT-PCR) for Snail, VEGF and β-Actin was performed using Superscript™ One-Step RT-PCR with Platinum®Taq (Invitrogen) with 0.6μM primers and either 0.5μg (for Snail amplification) or 0.1μg (for VEGF and β-Actin amplifaction) template RNA. Reverse transcription was performed at 55°C for 30min, followed by reverse transcriptase inactivation at 95°C for 15min. The following PCR conditions (35 cycles) were then implemented: 1 min at 94°C, 1 min at 56°C, 1 min at 72°C, 1min, and a final extension at 72°C for 10 min. Primers were obtained from Invitrogen.
Snail-forward 5′-GGGCAGGTATGGAGAGGAAGA-3′.
Snail-reverse 5′-TTCTTCTGCGCTACTGCTGCG-3′.
VEGF-forward 5′-CGAAGTGGTGAAGTTCATGGATG-3′.
VEGF-reverse 5′-TTCTGTATCAGTCTTTCCTGGT-3′.
β-Actin-forward, 5′-AAATCTGGCACCACACCTTC-3′.
β-Actin-reverse, 5′-GGGGTGTTGAAAGGTCTCAAA-3′.
For E-cadherin amplification, 1.2μM E-cadherin primers and100ng template RNA were used. Reverse transcription was performed at 55°C for 30min followed by transcriptase inactivation at 95°C for 15min. PCR (25 cycles) conditions were as follows: 1 min at 94°C, 1 min at 56°C, 1 min at 72°C and a final extension at 72°C for 10 min.
The primer sequences for E-cadherin were as follows:
E-cadherin-forward, 5′-CAGCACGTACACAGCCCTAA-3′;
E-cadherin-reverse, 5′-GCTGGCTCAAGTCAAAGTCC-3′.
PCR products was separated by elecrophorisis on a 1.5% agarose gel containing 1μg/ml ethidium bromide, and DNA bands were visualized using a UV light source.
Results/Discussion
We investigated the impact of introducing VEGF-A to two well-differentiated breast tumor cell lines in vitro. MCF-7 and T47D cells were transfected transiently with a VEGF-A-expressing or control vector. After 24 hours, a 5-fold increase in VEGF-A expression levels was observed in VEGF-A transfectants compared to control transfectants (Figures 1A and 1B). Control transfectants grew as islands of clustered cells maintaining strong cell-cell adhesion, reflecting the fact that these cells express E-cadherin. In contrast, in the VEGF-A transfectants, we observed single cells with membrane ruffles budding off from islands of clustered cells (Fig. 1A). This VEGF-A induced morphology was suggestive of cells undergoing an epithelial-mesencyhmal transition (EMT). To examine the effects of increased VEGF-A expression in breast tumor cells on EMT markers, we measured Snail mRNA levels in VEGF-A and control transfectants by performing reverse-transcription PCR. As shown in Fig. 1B, VEGF-A transfectants expressed significantly higher levels of Snail mRNA than control transfectants. Moreover, Snail protein levels were increased in VEGF-A transfectants as compared to control transfectants, as assessed by immunoblotting (Fig. 1C). As evidence that VEGF-A increases Snail activity, we observed that the mRNA (Fig. 1B) and protein (Fig. 1C) levels of E-cadherin were significantly reduced in VEGF-A-transfected tumor cells relative to control transfectants. The incubation of breast tumor cells with recombinant VEGF-A also increased Snail and decreased E-cadherin expression levels (Fig. 1D). These studies demonstrate that VEGF-A increases Snail mRNA and protein levels in breast tumor cells. Surprisingly, VEGF-A did not increase the activity of a reporter gene regulated by a Snail promoter composed of base pairs −869/+59 (data not shown). This result indicates that either: 1) VEGF-A increases Snail mRNA stability or 2) VEGF-A stimulates Snail transcription by increasing the expression of factors that bind to regions of the Snail promoter not included in our Snail promoter construct.
Figure 1. VEGF-A increases Snail and reduces E-cadherin expression levels in breast tumor cells.

A–C. Well-differentiated breast tumor cells (MCF-7 and T47D) were transfected transiently with a control plasmid (pcDNA) or a VEGF-A-expressing plasmid (pcDNA-VEGF-A). A. After 24 hours, cell morphology was examined by phase contrast microscopy. VEGF-A expression levels were measured by ELISA, using equivalent amounts of total cellular protein extracted from the indicated cells. Arrows indicate the budding of single cells from islands of cells maintaining strong cell-cell contacts. B. After 24 hours, mRNA was harvested from the indicated cells, and reverse-transcription PCR reactions for VEGF-A and β-actin were performed. Alternatively, using a VEGF-A ELISA kit, VEGF-A protein was quantified from equivalent amounts of total cellular protein extracted from these cells. B. mRNA was extracted from cells 24 hours post-transfection, and reverse transcription PCR reactions for Snail, E-cadherin, and β-actin were performed. C. Equivalent amounts of total cellular proteins from MCF-7 transfectants (24 hours post-transfection) were run on SDS polyacrylamide gels and immunoblotted with Snail, E-cadherin and β-actin antibodies. D. T47D cells were incubated with BSA or recombinant human VEGF-A (rhVEGF). After 30 min., RNA was extracted from these cells. Snail, E-cadherin and β-actin levels were measured by reverse transcription PCR. E. SUM159 breast tumor cells were transfected with 100nM of control or VEGF-A siRNA. After 4 hours, Snail, VEGF-A, and β-actin levels were assessed by reverse-transcription PCR. For A–E, similar results were obtained in 3 independent trials.
Previous studies indicate that Snail is expressed in high-grade, metastatic breast tumors, but not in low-grade, non-metastatic tumors [1]. Accordingly, we sought to determine if endogenous VEGF-A is a determinant of Snail expression in poorly-differentiated breast tumor cells. SUM159 breast tumor cells (VEGF+, Snail+) were transfected transiently with a VEGF-A or control siRNA. As shown in Fig. 1E, VEGF-A mRNA and protein levels were reduced in VEGF siRNA transfectants compared to control transfectants. Importantly, VEGF siRNA-transfected cells also exhibited significantly reduced Snail expression as compared to control transfectants (Fig. 1E).
Our previous studies indicate that breast tumor cells support autocrine VEGF-A signaling involving the VEGF receptor Neuropilin-1 [7, 14]. Intruigingly, our work also indicated that tyrosine kinase VEGF receptors (KDR, Flt-1) are not important for autocrine VEGF-A signaling in breast tumor cells [7,14]. Based on these findings, we next addressed the hypothesis that the VEGF receptor NP-1 stimulates Snail expression in breast tumor cells. Specifically, we investigated the effect on Snail of incubating breast tumor cells that express endogenous NP-1 and Snail with an antibody that inhibits VEGF/NP-1 interactions. The incubation of MDA-MB-435 breast tumor cells, which express NP-1[14], with this NP-1-neutralizing antibody significantly reduced Snail mRNA levels, as assessed by reverse transcription PCR (Fig. 2A). Of note, we were unable to induce de novo E-cadherin expression by incubating MDA-MB-435 breast tumor cells with this NP-1 neutralizing antibody (data not shown), most likely because the E-cadherin gene is suppressed by promoter methylation in these cells[15]. As evidence that this NP-1-neutralizing antibody reduces Snail activity, the incubation of T47D cells (NP-1-positive, E-cadherin-positive) with this NP-1 antibody reduced Snail mRNA levels while increasing E-cadherin mRNA levels (Fig. 2A). Collectively, these results indicate that an autocrine VEGF-A/NP-1 pathway increases Snail expression in breast tumor cells. Our demonstration that autocrine VEGF-A signaling in breast tumors drives Snail expression does not exclude the possibility that exogenous sources of VEGF, as exist in stromal cells, also stimulate Snail expression in tumor cells. This possibility is supported by our observation that the addition of recombinant VEGF to breast tumor cells increases Snail expression (Fig. 1D). Of interest, previous studies indicate that the introduction of Snail to epithelial cells drives VEGF-A expression [16]. Accordingly, autocrine VEGF-A signaling, which increases Snail expression, may be subject to a positive feedback loop involving Snail stimulation of VEGF-A expression.
Figure 2. Snail expression in breast tumor cells is regulated by the VEGF receptor Neuropilin-1 and Glycogen Synthase Kinase-3.

A. MDA-MB-435 and T47D cells were incubated with a NP-1-neutralizing antibody (Anti-NP-1) or an isotype control antibody (IgG) at a concentration of 10μg/ml. After 4 hours, RNA was extracted, and Snail, E-cadherin and β-actin mRNA levels were measured as described in A. B. SUM159 breast tumor cells were transfected with 100nM of a control or VEGF-A siRNA. Four hours after transfection, equivalent amounts of total cellular proteins extracted from these cells were subjected to SDS-PAGE and immunoblotted with a phospho-GSK-3β or total GSK-3β antibody. C & D. MDA-MB-435 cells were incubated with the VEGF-A-neutralizing antibody Avastin (Anti-VEGF), a NP-1-neutralizing (Anti-NP-1) antibody, or an isotype control antibody (IgG) in the presence of DMSO (1:1000) or a small molecule GSK-3 inhibitor (SB415286, 25 μM). After 4 hours, RNA was extracted, and Snail and β-actin levels were determined by reverse-transcription PCR. Similar results for A–D were observed in 3 trials.
Considering that NP-1 lacks consensus signaling sites, we are currently investigating the co-factors in these breast tumor cells that support NP-1 in its regulation of Snail expression. Of note, we have previously shown that Plexin A1, a NP-1 co-receptor in neurons, is expressed in breast tumor cells, and is a determinant of NP-1 signaling in these cells [8]. A role for Plexin A1 in the regulation of Snail expression in breast tumor cells is currently being investigated.
We next examined the hypothesis that VEGF-A/NP-1 increases Snail expression by inhibiting the Glycogen Synthase Kinase-3 (GSK-3). This hypothesis is founded on the knowledge that: 1) VEGF-A stimulates PI3-kinase, 2) the PI3-kinase pathway inhibits GSK-3, and 3) GSK-3 inhibits Snail expression and activity[17, 18]. According to our hypothesis that VEGF regulates Snail by inhibiting GSK-3, we predicted that autocrine VEGF-A signaling in SUM159 cells, which express Snail, inhibits GSK-3 activity. As shown in Fig. 2B, the transfection of SUM159 cells with a VEGF-A-specific siRNA decreased the level of phosphorylated GSK-3β, representing the inactive form of this kinase. Next, we investigated the importance of GSK-3 in autocrine VEGF/NP-1 regulation of Snail. The incubation of MDA-MB-435 cells, which express both VEGF-A and Snail, with a VEGF-neutralizing antibody (Fig. 2C) or NP-1-neutralizing antibody (Fig. 2D), but not with an isotype control antibody, significantly decreased Snail expression levels. However, neither the VEGF nor the NP-1-neutralizing antibody decreased Snail expression levels in cells that had been preincubated with a GSK-3 inhibitor (Fig. 2C and 2D). These findings indicate that autocrine VEGF/NP-1 increases Snail expression in breast tumor cells by suppressing GSK-3.
Avastin® (Genentech) is a VEGF-neutralizing antibody that, when used in combination with chemotherapy, prolongs the progression-free survival of breast cancer patients[19, 20], presumably due to its ability to suppress tumor angiogenesis[21]. Based on this activity, Avastin® was recently approved as a therapy for patients with HER2-negative metastatic breast tumors. We examined a novel activity for Avastin® in influencing Snail expression levels in breast tumor cells. Snail mRNA (Fig. 3A) and Snail protein (Fig. 3B) levels were significantly reduced in poorly-differentiated breast tumor cells that had been incubated in vitro with Avastin® as compared to the same cells incubated with an isotype control antibody. The therapeutic effect of Avastin® in breast cancer patients has thus far been attributed exclusively to its anti-angiogenic activities[21]. Our data indicate that Avastin® can also reduce the expression in breast tumor cells of Snail, a tumor progression factor. These findings raise the exciting possibility that beyond its angiogenic effects, Avastin® may inhibit tumor metastatic progression by reducing Snail expression. Our findings underscore the importance of determining if the administration of Avastin to patients with early stage breast cancer impedes tumor metastatic progression by suppressing Snail expression.
Figure 3. Incubation of breast tumor cells with Avastin® reduces Snail expression levels.
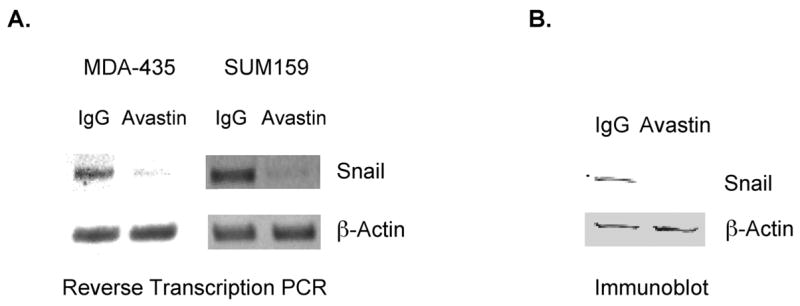
MDA-MB-435 and SUM159 cells were incubated with Avastin®; or an isotype control antibody (IgG) at a concentration of 5 μg/mL. A. After 4 hours, RNA was extracted from these cells, and Snail and β-actin levels were assessed by reverse-transcription PCR. B. Total cellular proteins were extracted from SUM159 cells incubated for 4 hours with the indicated antibodies. Equivalent amounts of protein from these cels were subjected to SDS-PAGE and immunoblotted with Snail or β-actin antibodies. Similar results were obtained in 3 independent trials.
The concept that autocrine factors may drive Snail expression in breast tumor cells is an attractive idea. By expressing autocrine factors that regulate Snail, tumor cells could direct their own metastatic progression by increasing the expression of an autocrine factor and/or its receptor. VEGF-A expression has been linked to breast tumor metastatic progression[10], and it is assumed that this correlation relates to the angiogenic activity of this growth factor[22]. Our data suggest that VEGF-A also contributes to progression by stimulating autocrine signaling in tumor cells that induces the expression of Snail, a breast tumor progression factor. Based on our findings, we propose that anti-VEGF or anti-VEGF receptor-based therapies may impede tumor progression in part by reducing Snail expression in tumor cells.
Acknowledgments
We thank Dr. Valentina Folgiero for her assistance with this study. In addition, we thank Dr. Arthur Mercurio for his valuable advice on this work. This work was supported by NIH grant CA093855 (R.E.B.) and a pilot award (R.E.B.) from the Duke Comprehensive Cancer Center‘s Breast and Ovarian Oncology Research Program.
References
- 1.Blanco MJ, Moreno-Bueno G, Sarrio D, Locascio A, Cano A, Palacios J, Nieto MA. Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene. 2002;21:3241–3246. doi: 10.1038/sj.onc.1205416. [DOI] [PubMed] [Google Scholar]
- 2.Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia De Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 3.Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 4.Barrallo-Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development (Cambridge, England) 2005;132:3151–3161. doi: 10.1242/dev.01907. [DOI] [PubMed] [Google Scholar]
- 5.Olmeda D, Jorda M, Peinado H, Fabra A, Cano A. Snail silencing effectively suppresses tumour growth and invasiveness. Oncogene. 2007;26:1862–1874. doi: 10.1038/sj.onc.1209997. [DOI] [PubMed] [Google Scholar]
- 6.Moody SE, Perez D, Pan TC, Sarkisian CJ, Portocarrero CP, Sterner CJ, Notorfrancesco KL, Cardiff RD, Chodosh LA. The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell. 2005;8:197–209. doi: 10.1016/j.ccr.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 7.Bachelder RE, Crago A, Chung J, Wendt MA, Shaw LM, Robinson G, Mercurio AM. Vascular endothelial growth factor is an autocrine survival factor for neuropilin-expressing breast carcinoma cells. Cancer Res. 2001;61:5736–5740. [PubMed] [Google Scholar]
- 8.Bachelder RE, Lipscomb EA, Lin X, Wendt MA, Chadborn NH, Eickholt BJ, Mercurio AM. Competing autocrine pathways involving alternative neuropilin-1 ligands regulate chemotaxis of carcinoma cells. Cancer Res. 2003;63:5230–5233. [PubMed] [Google Scholar]
- 9.Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 1998;92:735–745. doi: 10.1016/s0092-8674(00)81402-6. [DOI] [PubMed] [Google Scholar]
- 10.Brown LF, Guidi AJ, Schnitt SJ, Van De Water L, Iruela-Arispe ML, Yeo TK, Tognazzi K, Dvorak HF. Vascular stroma formation in carcinoma in situ, invasive carcinoma, and metastatic carcinoma of the breast. Clin Cancer Res. 1999;5:1041–1056. [PubMed] [Google Scholar]
- 11.Yang AD, Camp ER, Fan F, Shen L, Gray MJ, Liu W, Somcio R, Bauer TW, Wu Y, Hicklin DJ, Ellis LM. Vascular endothelial growth factor receptor-1 activation mediates epithelial to mesenchymal transition in human pancreatic carcinoma cells. Cancer Res. 2006;66:46–51. doi: 10.1158/0008-5472.CAN-05-3086. [DOI] [PubMed] [Google Scholar]
- 12.Franci C, Takkunen M, Dave N, Alameda F, Gomez S, Rodriguez R, Escriva M, Montserrat-Sentis B, Baro T, Garrido M, Bonilla F, Virtanen I, Garcia de Herreros A. Expression of Snail protein in tumor-stroma interface. Oncogene. 2006;25:5134–5144. doi: 10.1038/sj.onc.1209519. [DOI] [PubMed] [Google Scholar]
- 13.Hoang MV, Senger DR. In vivo and in vitro models of Mammalian angiogenesis. Methods Mol Biol. 2005;294:269–285. doi: 10.1385/1-59259-860-9:269. [DOI] [PubMed] [Google Scholar]
- 14.Bachelder RE, Wendt MA, Mercurio AM. Vascular endothelial growth factor promotes breast carcinoma invasion in an autocrine manner by regulating the chemokine receptor CXCR4. Cancer Res. 2002;62:7203–7206. [PubMed] [Google Scholar]
- 15.Graff JR, Herman JG, Lapidus RG, Chopra H, Xu R, Jarrard DF, Isaacs WB, Pitha PM, Davidson NE, Baylin SB. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res. 1995;55:5195–5199. [PubMed] [Google Scholar]
- 16.Peinado H, Marin F, Cubillo E, Stark HJ, Fusenig N, Nieto MA, Cano A. Snail and E47 repressors of E-cadherin induce distinct invasive and angiogenic properties in vivo. Journal of cell science. 2004;117:2827–2839. doi: 10.1242/jcs.01145. [DOI] [PubMed] [Google Scholar]
- 17.Bachelder RE, Yoon SO, Franci C, de Herreros AG, Mercurio AM. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: implications for the epithelial-mesenchymal transition. J Cell Biol. 2005;168:29–33. doi: 10.1083/jcb.200409067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6:931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- 19.Ramaswamy B, Elias AD, Kelbick NT, Dodley A, Morrow M, Hauger M, Allen J, Rhoades C, Kendra K, Chen HX, Eckhardt SG, Shapiro CL. Phase II trial of bevacizumab in combination with weekly docetaxel in metastatic breast cancer patients. Clin Cancer Res. 2006;12:3124–3129. doi: 10.1158/1078-0432.CCR-05-2603. [DOI] [PubMed] [Google Scholar]
- 20.Wedam SB, Low JA, Yang SX, Chow CK, Choyke P, Danforth D, Hewitt SM, Berman A, Steinberg SM, Liewehr DJ, Plehn J, Doshi A, Thomasson D, McCarthy N, Koeppen H, Sherman M, Zujewski J, Camphausen K, Chen H, Swain SM. Antiangiogenic and antitumor effects of bevacizumab in patients with inflammatory and locally advanced breast cancer. J Clin Oncol. 2006;24:769–777. doi: 10.1200/JCO.2005.03.4645. [DOI] [PubMed] [Google Scholar]
- 21.Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, Ferrara N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 1993;362:841–844. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- 22.Connolly DT, Heuvelman DM, Nelson R, Olander JV, Eppley BL, Delfino JJ, Siegel NR, Leimgruber RM, Feder J. Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J Clin Invest. 1989;84:1470–1478. doi: 10.1172/JCI114322. [DOI] [PMC free article] [PubMed] [Google Scholar]