

Abstract
Site directed mutagenesis of whole plasmids is a simple way to create slightly different variations of an original plasmid. With this method the cloned target gene can be altered by substitution, deletion or insertion of a few bases directly into a plasmid. It works by simply amplifying the whole plasmid, in a non PCR-based thermocycling reaction. During the reaction mutagenic primers, carrying the desired mutation, are integrated into the newly synthesized plasmid. In this video tutorial we demonstrate an easy and cost effective way to introduce base substitutions into a plasmid. The protocol works with standard reagents and is independent from commercial kits, which often are very expensive. Applying this protocol can reduce the total cost of a reaction to an eighth of what it costs using some of the commercial kits. In this video we also comment on critical steps during the process and give detailed instructions on how to design the mutagenic primers.
Keywords: Basic Protocols, Issue 27, Site directed Mutagenesis, Mutagenesis, Mutation, Plasmid, Thermocycling, PCR, Pfu-Polymerase, Dpn1, cost saving
Protocol
Principle of Method:
The site directed mutagenesis of whole plasmids explained in this video is a mutagenesis method which allows you to alter a cloned target gene by substitution, deletion or insertion of a few bases directly into a plasmid. It works by amplifying the whole plasmid, in a non PCR-based thermocycling reaction. During the reaction mutagenic primers, carrying the desired mutation in form of mismatches to the original plasmid, are integrated into the newly synthesized plasmid. After removal of the original plasmid from the reaction the mutated plasmid gets transformed into E. coli. The following steps are for screening purposes only, because the mutation efficiency of this method is not 100%. The whole procedure takes three days, but the main part can be done within one day.
1.0 Day one:
1.1 Thermocycling reaction:
Original Plasmid: This site directed mutagenesis protocol works best with plasmids up to 10kb. Larger Plasmids are a bit difficult to mutate with this method and may take some patience and adjustment of the thermocycling conditions and/or competent cells. Furthermore the plasmid which you work with must be isolated from a dam+ bacteria strain. For the thermocycling reaction you will need 10 to 60 ngs of the plasmid you want to mutate. Primers: Before you can set up your thermocycling reaction you have to have your primers at hand. You need about 150 ng of every mutagenic primer. It is ok if you take 1.5 µl of a 1:10 diluted 100 pM stock. There are a few simple guidelines you must consider when designing your mutagenic primers:
The primers should be complementary to each other
The primers should be between 25 and 45 nucleotides in length
The mutations in form of mismatches to the original plasmid must be contained in both primers
The mismatches should be centered in the primer and flanked by at least 8 nucleotides on each side
The primers should have a GC-Content of at least 40%
The primers should end 5 prime and 3 prime with one or more Gs or Cs
The primers need not to be phosphorylated, nor do they have to be FPLC or PAGE purified, merely desalted they should be.
For calculation of Tm you don’t need any formula but Tm on the shipping certificate should be higher than 60°C.
For screening purposes it is practical to insert or delete a restriction site with your mutation. Because of the degeneration of the genetic code there are many possibilities to insert a restriction site with your desired mutation. The website of the New England Biolabs provides a tool to find such an appropriate restriction site. Simply enter your primer sequence coding for the amino acid sequence you desire, using the ambiguity code for DNA. The enzyme finder tool will tell you which restriction sites can be introduced parallel to your desired mutation. If you can’t find any possible or practical restriction site by this way, you may insert any new restriction site without concerning the genetic code at the site of mutation. Thereafter you can use this plasmid as template for a series of mutations in which this restriction site will be deleted by insertion of the new mutagenic primers. This approach may be very convenient if you’re planning to do a series of mutations at the same site of the plasmid.
DNA-Polymerase: For this method you need a thermostable DNA-Polymerase which exhibits 3’–5’ Exonuclease activity and creates blunt ends. We always use recombinant Pfu-Polymerase from Fermentas. If you’ve got problems with the amplification steps you may try a higher quality polymerase, but for most purposes the standard Pfu will be enough. Complete the reaction with dNTPs, polymerase buffer and water.
1.2 Thermocycling conditions
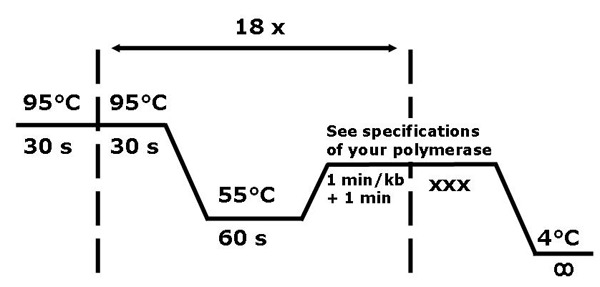
The thermocycler should be set up in the following way. The initial and recurring denaturation phase is set to 30sec at 95°C.
The annealing temperature of the primers must not be calculated with complicated formulae. We usually set the annealing temperature to 55°C and the annealing time to one minute. For primers of 25 to 30 nucleotides which you will be using mostly, this works for us 95% of the time. Anyhow if this should not work, simply try varying the annealing temperature between 50- and 60°C.
The elongation temperature depends on the polymerase you use. In this demonstration we use the Pfu-Polymerase from Fermentas, which calls for a elongation temperature of 72°C. The elongation time varies according to the size of the plasmid. We always calculate 1 min per kb, and add one extra minute to that time. For example, for a 9kb plasmid we would choose 10min as elongation time.
18 cycles are sufficient to create enough mutated plasmid for the further use. Keeping the number of cycles low, also saves you time.
1.3 Gelcheck after Thermocycling reaction
The successful amplification should be checked by performing an electrophoresis. Directly after the cycling has finished, load 5 µl of the reaction onto a 1% TAE agarose gel. If the amplification was successful, you should see a distinct band. However if the newly synthesized DNA is not clearly visible on the gel, you may try to precipitate the whole reaction and use it for transformation in the next step. For us this has seldom worked but it is worth to give a try. The best thing to do if the reaction does not work is to adjust the annealing temperature.
1.4 DpnI digestion:
Before transformation the original plasmid which served as a template must be removed from the reaction to prevent strong background. This is done by restriction digestion with DpnI. This restriction endonuclease cuts only methylated plasmid. Its recognition and restriction site is the sequence GATC whereas A has to be methylated. When digesting with DpnI only the original unmutated plasmid which was isolated from a dam+ strain gets cut, the newly synthesized mutated plasmid which is not methylated is not affected by DpnI. Simply add 1-2 µl of DpnI to the reaction and incubate it at least one hour at 37°. When using the Fast digest DpnI the incubation time can be decreased to about 15 minutes. The quality of your DpnI and the incubation time of this restriction digestion greatly determines how strong your background with unmutated plasmid will be.
1.5 Transformation:
After DpnI digestion the plasmid is ready for transformation into the competent E. coli cells. Simply add 5 µl from the DpnI digestion reaction into the competent cells and perform the transformation as recommended in your lab. In our case we use the heat shock procedure by incubating the bacteria-plasmid mixture on ice for 30 min and then heat shock it at 42°C for 90 sec. After adding 200 µl SOC-solution, the bacteria are incubated, vigorously shaking, at 37°C for 1 h. After 1 hour the bacteria are plated on selecting agar media.
2.0 Day two
2.1 Screening of clones part 1: Selection of clones
Because the mutation efficiency isn’t 100% you need to screen for your mutants. We do this by restriction digestion in which we check for presence or absence of the restriction site which we added or deleted through primer integration. For this purpose we usually pick up eight colonies and grow them over night for plasmid preparation on the next day.
3.0 Day three
3.1 Screening of clones part 2: Restriction digestion with marker enzyme
On the third day you perform a Mini Prep and subsequent restriction digestion with your marker enzyme. On the gel you then can see which one of your clones carry the desired mutation and which ones not.
The screening method using restriction digestion works very well. Nevertheless, you should confirm your successful mutation by sequencing.
Discussion
Site directed mutagenesis is a mutagenesis-method which provides a fast way to mutate a gene carried by a plasmid. The whole reaction can be done in only one day. With this method, it will need only a pair of complimentary primers carrying the desired mutations and a proofreading polymerase such as Pfu-Polymerase. The newly synthesized plasmid can be separated from the parental plasmid by digesting the reaction with the restriction enzyme DpnI. This enzyme digests only the methylated DNA. Therefore only the newly synthesized plasmid can be transformed. In this tutorial we demonstrated performing a site directed mutagenesis by using a homemade mutagenesis kit. The benefit of this kit is the cost saving while the effectiveness remains. The critical points of this method are the primer design and the annealing temperature. In case the newly synthesized DNA can not be visualized after electrophoresis, which may indicate the failure of the method, one can try to precipitate the whole reaction and use it for transformation in bacteria. However the best way to solve this problem is trying to optimize the annealing temperature or increase the length of the constant region in the primers. Further, using a high quality polymerase or restriction enzyme may also improve the sensitivity of the reaction. The competent cells also are a key factor which effects the successfulness of this method. Therefore middle (107 cfu/µg DNA) or highly (109 cfu/µg DNA) competent cells are preferable.
Although in this tutorial we did not demonstrate the deletion or insertion by using the homemade kit, we were successful to do these in our laboratory. We were able to successfully substitute, insert or delete at least 3 bases at the same time.
Disclosures
The authors have nothing to disclose.
References
- Papworth C, Bauer JC, Braman J, Wright DA. QuikChange site-directed mutagenesis. Strategies. 1996;9:3–4. [Google Scholar]