

Abstract
The BH3-only BCL-2 family protein BID is activated by caspase-8 cleavage upon engagement of cell surface death receptors. The resulting 15 kDa C-terminal fragment, tBID, translocates to mitochondria triggering the release of cytotoxic molecules and cell death. The pro-apoptotic activity of tBID is regulated by its interactions with pro-survival BCL-XL and pro-death BAX, both in the cytosol and at the mitochondrial membrane. In this study we characterize the molecular interactions between full-length tBID and BCL-XL using NMR spectroscopy and isothermal titration calorimetry (ITC). In aqueous solution, tBID adopts an α-helical but dynamically disordered conformation, however, the three-dimensional conformation is stabilized when tBID engages its BH3 domain in the BH3-binding hydrophobic groove of BCL-XL to form a stable heterodimeric complex. Characterization of the binding thermodynamics by ITC reveals that the interaction between tBID and BCL-XL is driven by enthalpy, but disfavored by the entropy associated with the conformational order induced in tBID upon binding BCL-XL.
The BCL-2 family proteins are the principal regulators of the mitochondrion-dependent pathway for programmed cell death (1-7). Their opposing activities are exerted through a network of interactions between pro- and anti-apoptotic family members, in the cytosol and in intracellular membranes, mediated by four BCL-2 homology (BH1–BH4) domains that are conserved in the amino acid sequences of the proteins. Several BCL-2 proteins also contain a ∼20-residue hydrophobic C-terminus that is important for intracellular membrane targeting. The anti-apoptotic family members (e.g. BCL-2, BCL-XL) possess all four BH domains, while the pro-apoptotic family members have more diverse sequences and can contain multiple BH domains (e.g. BAX, BAK) or just the single BH3 domain (e.g. BID, BIM), which is highly conserved and essential for both the cell killing activity of the pro-apoptotic family members and for mediating their interactions with their pro-survival counterparts. The BH3-only proteins provide a key link between the extracellular and intrinsic mitochondrion pathways to cell death because they are activated when extracellular stress cues, transmitted by death receptors, trigger their expression or post-translational modification, thus enabling their association with anti- or pro-apoptotic BCL-2 proteins in the cytosol and in mitochondrial membranes.
The BH3-only protein BID is activated after cleavage by caspases, calpains, and lysosomal proteases, upon engagement of the Fas or TNFR1 cell surface receptors, which link it to both apoptotic and necrotic cell death pathways in stroke, neuro-degeneration, hepatitis, and other ailments (8). Cleavage of BID by caspase-8 generates the 15 kDa C-terminal fragment, tBID, which has a 10-fold greater binding affinity for BCL-XL and is 100-fold more efficient in inducing cytochrome-c release from mitochondria than its full-length precursor (9, 10). The newly generated tBID N-terminus becomes myristoylated following caspase-8 cleavage (11) and this may assist its association with mitochondrial membranes, which is implicated in the oligomerization and activation of BAX and BAK, mitochondrial membrane remodeling, and the release of mitochondrial cytotoxic molecules, ultimately leading to cell death (9, 10, 12-17).
The cytosolic form of intact BID has a well-defined globular structure that consists of eight α-helices arranged with two central hydrophobic helices in the core of the molecule (18, 19). The third helix (h3) contains the BH3 domain and is connected to the first two helices by a long flexible loop, which includes the caspase-8 cleavage site at Asp60 (Figure 1). Despite the low sequence homology, the structure of BID is strikingly similar to those of BCL-XL, BAX, and other anti- and pro-apoptotic multi-domain BCL-2 family proteins, as well as to those of the pore-forming domains of bacterial toxins (20, 21). In contrast, we previously reported that the three-dimensional conformation of tBID in water is highly disordered, even though the cleaved polypeptide retains the helical secondary structure of its intact precursor (22).
Figure 1.
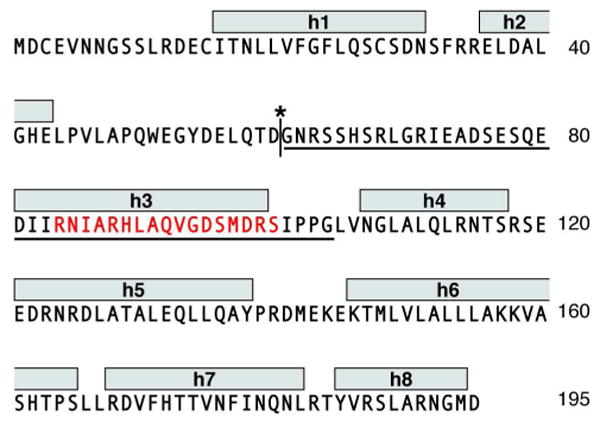
Amino acid sequence of human BID showing: the BH3 domain (red), the helices identified in the solution NMR structure (h1-h8) (18), the caspase-8 cleavage site (asterisk) and start of the tBID sequence (G61). The sequence of the BH3-spanning BID61-104 is underlined.
The existing data indicate that pro-apoptotic BAX and anti-apoptotic BCL-XL act as competing receptors for the common ligand tBID (2). Apoptosis can be promoted when tBID binds and activates BAX to induce membrane permeabilization, or inhibited when tBID binds and is sequestered by BCL-XL acting as a buffer for pro-death signals. These interactions occur both in the cytosol and in mitochondrial membranes where they culminate in decisions of life and death for the cell. Structural studies with short peptides spanning various pro-apoptotic BH3 domain sequences have shown that they all bind a hydrophobic groove on the surface of pro-survival BCL-XL, formed by the BH1, BH2, and BH3 domains of BCL-XL and by residues located in BCL-XL helices h2-h5 and h8 (23-25). The interaction is mediated by conserved Leu and Asp residues in each peptide's BH3 domain and induces a localized structural rearrangement in the BCL-XL binding groove. This specific association of BH3 domains with the hydrophobic groove of their pro-survival target is highly conserved and has also been documented for the anti-apoptotic proteins BCL-W and MCL-1, which are both structurally homologous to BCL-XL (26, 27). In addition, the direct association of a short BIM BH3 peptide with BAX has been described recently (28). This peptide, which was chemically modified to stabilize its α-helical structure, binds BAX at an interaction site that is distinct from the canonical binding groove characterized for anti-apoptotic proteins, providing an explanation for the direct activation of BAX and BAK by the BH3-only proteins.
Although all of the short BH3 peptides are unstructured in solution, they become α-helical upon binding their anti-apoptotic target and binding is favored by their propensity toward helical structure (24, 26, 27, 29). However, while some of the full-length proteins from which the BH3 peptides are derived are intrinsically unstructured in their free state (30), tBID is distinctly α-helical (22, 31). For such pre-structured BH3-only proteins, binding their anti-apoptotic target does not involve a folding transition from random coil to α-helix and, therefore, the binding thermodynamics may be expected to be quite different from those measured using short BH3 peptide analogs. Solution NMR studies with short peptides spanning the BID BH3 domain have shown that the peptides which are unstructured in solution become α-helical upon binding pro-survival BCL-XL or BCL-W (26, 29), however, no structural or thermodynamic information is available for the interaction of full-length tBID with its pro-survival partners.
Here we describe the interactions of full-length tBID with BCL-XL in aqueous solution. tBID forms a stable specific complex with BCL-XL by engaging the BH3-binding hydrophobic groove on the surface of BCL-XL with its BH3 domain. This association stabilizes the 20-residue helix spanning the BH3 domain. Characterization of the binding thermodynamics by isothermal titration calorimetry (ITC) reveals that the tBID/BCL-XL interaction is driven by enthalpy and disfavored by the entropy cost associated with the conformational order induced by the association with BCL-XL.
Materials and Methods
Sample Preparation and Characterization
Full-length human BID and human tBID (residues 61-195) were expressed and purified as described previously (22, 32). The 44-residue BID61-104 peptide, spanning residues 61-104 of human BID, was expressed and purified using the TrpLE fusion protein as described for tBID (22). The amino acid sequences are shown in Figure 1. Human BCL-XL, lacking residues 45-84 in the unstructured loop and residues 213-233 in the C-terminus, was expressed and purified as described previously (33). Uniformly 15N or 15N/13C labeled proteins were prepared by growing E. coli cells in M9 minimal media with (15NH4)2SO4 and 13C-glucose (Cambridge Isotope Laboratories) as the sole nitrogen and carbon sources. The NMR samples contained 0.2-0.5 mM protein in 25 mM sodium phosphate at pH 7 or pH 4.
Analytical Size Exclusion Chromatography
Analytical size exclusion chromatography was performed on a Waters HPLC chromatography system with a flow rate of 0.4 mL/min, using a Superdex 75 column (GE Healthcare). Protein samples were dissolved in 25 mM sodium phosphate buffer at pH 4, supplemented with 150 mM NaCl, 1 mM EDTA and 2 mM DTT, at a concentration of 0.5 mg/mL. To detect protein-protein interactions, BID or tBID were mixed with BCL-XL in equimolar ratios before column injection. The column was calibrated with ovalbumin (43 kDa), chymotryspinogen (25 kDa) and lysozyme (16 kDa) molecular weight standards.
Isothermal Titration Calorimetry Experiments
ITC experiments were performed at 30°C using an iTC200 instrument (MicroCal, Amherst, MA). Samples were prepared in 25 mM sodium phosphate at pH 7 or pH 4. Protein concentration was determined by measuring absorbance at 280 nm. The concentration of BCL-XL in the ITC cell was between 15-30 μM; the concentrations of BID61-104 and tBID in the injection syringe were between 0.3-0.5 mM. The thermodynamic data were processed with the ORIGIN program (Microcal). The values of ΔH are measured experimentally for each titration, and fitting the binding isotherms with a one-site binding model yields the values of the association constant (Ka). The binding free energy (ΔG), entropy (ΔS), and dissociation constant (Kd) were calculated from the experimentally determined values of ΔH and Ka, using Equations 1 and 2,
| (Equation 1) |
| (Equation 2) |
where R is the gas constant (1.987 cal/mol•°K) and T is the working temperature (303°K) (34).
Circular Dichroism Experiments
CD experiments were carried out on a Jasco J-810 CD System purged with nitrogen and equipped with a Pelletier temperature controller. CD spectra were acquired with 0.5 nm increments between 195 nm and 260 nm in a cuvette with a 2 mm path length. Raw data, acquired on the instrument, were baseline subtracted and converted to mean residue ellipticity.
NMR Experiments
NMR experiments were performed on Bruker AVANCE 600 or 800 MHz spectrometers at 40°C. Backbone resonances were assigned for BID61-104 in aqueous solution at pH 4, and for BCL-XL in aqueous solution at pH 4 and pH 7. Partial assignments of the spectra of tBID in water at pH 4 were obtained by comparison with the spectra from BID61-104. Resonance assignments were obtained using HNCA and HNCACB experiments with gradient selection, water flip back and sensitivity enhancement (35-37), and were aided by comparison with the previously determined assignments for BCL-XL at neutral pH (38). The chemical shifts were referenced as described (39). The NMR data were processed and analyzed using NMRPipe (40) and Sparky (41). Chemical shift mapping experiments were performed by monitoring peak frequency changes in the two dimensional 1H/15N HSQC (42) spectrum of each 15N-labeled protein in the presence of its unlabeled partner. The total change in chemical shift (ΔTOT) for each residue was calculated by adding the changes in 1H (ΔH) and 15N (ΔN) chemical shifts using to the equation ΔTOT = [(ΔH)2 + (ΔN/5)2]1/2, where the 15N chemical shift is scaled by 1/5 to account for the 5-fold difference between the chemical shift dispersions of 15N and 1H (43, 44). Secondary structure information was obtained by analyzing the chemical shifts using TALOS (40) and in terms of the combined deviations of CA and CB chemical shifts from their random coil values (ΔCA - ΔCB) (45, 46).
Results and Discussion
Size exclusion characterization of the tBID/BCL-XL complex
To characterize the interactions between tBID and BCL-XL, we analyzed the free and mixed proteins using analytical size exclusion chromatography and solution NMR spectroscopy. All of the binding studies were carried out at pH 4 since human tBID has very limited solubility at pH values greater than 5. This reflects the dramatic changes in the physical properties of the protein that accompany caspase-8 cleavage: removal of residues 1-60 shifts the calculated pI from 5.3 to 9.3 and the net charge from -7 to +2, and exposes hydrophobic residues in helix h3, containing the BH3 domain and in the central core helices h6 and h7 (18, 19, 31). The association of BCL-XL and BID with lipid bilayer membranes is promoted by acidic pH (31, 47) and involves a conformational change of the proteins (22, 33, 48). Nevertheless, although acidic pH is important for membrane insertion, BCL-XL has been reported to retain its globular fold upon acidification from pH 7.4 to 4.9 (49). We find that BID, like BCL-XL, is highly stable at pH 4, as evidenced by the 1H-15N HSQC NMR spectra which exhibit no evidence of unfolding compared to the spectra at pH 7 (Figure 2).
Figure 2.
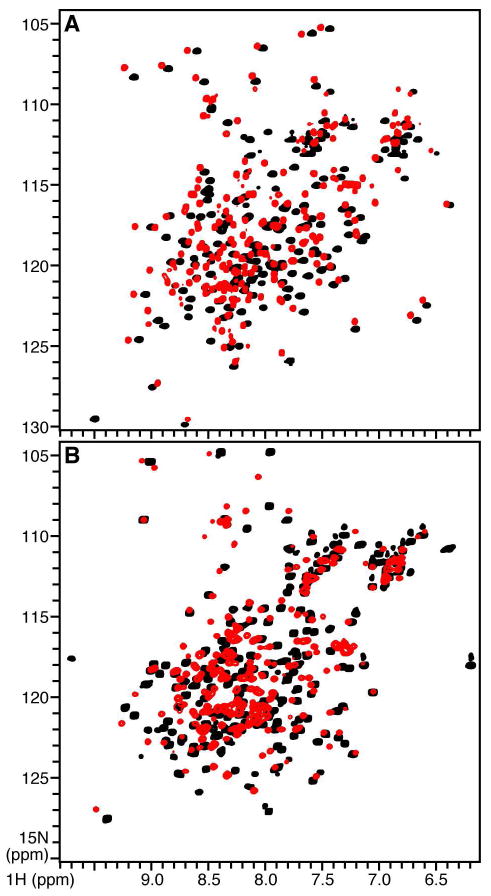
1H/15N HSQC NMR spectra of 15N-labeled (A) BCL-XL and (B) BID at pH 7 (black) or pH 4 (red).
The data from analytical size exclusion chromatography (Figure 3) show that the free proteins elute at apparent molecular weights corresponding to those calculated from their respective amino acid sequences (BID: 23 kDa; BCL-XL: 21 kDa; tBID 15 kDa), reflecting their monomeric states in solution. A mixed sample of equimolar BID and BCL-XL elutes at an apparent molecular weight near 20 kDa, as observed for the individual components and, therefore, shows no evidence of complex formation. In contrast, a sample containing equimolar tBID and BCL-XL elutes at a distinctly higher molecular weight, close to the added molecular weights of the individual proteins (∼36 kDa). This result is in agreement with the finding that tBID has a 10-fold greater binding affinity than BID for BCL-XL (9, 10) and demonstrates that tBID and BCL-XL form a long-lived complex in solution, while no stable interaction is observed between BID and BCL-XL.
Figure 3.
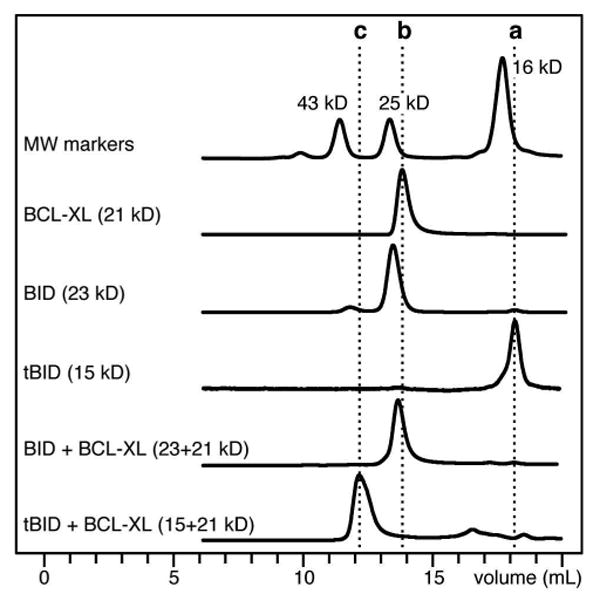
Size exclusion chromatography showing formation of the tBID/BCL-XL complex in aqueous solution. The dotted lines indicate elution of (a) tBID, (b) BCL-XL, and (c) tBID+BCL-XL.
Structural characterization of the tBID/BCL-XL interaction
The 1H and 15N chemical shift frequencies in the HSQC NMR spectra of proteins are highly sensitive to the chemical environment and conformation of their corresponding15N-labeled protein site, and the peak line widths and shapes reflect protein backbone dynamics, conformational order, and aggregation state. In addition, the changes in chemical shifts can be used to monitor changes in protein structure and ligand binding. Although the CD spectrum of tBID in water shows that it adopts α-helical secondary structure like its intact precursor (Figure 4A), its 1H/15N HSQC spectrum exhibits the hallmark features of dynamically disordered proteins (Figure 5, black). Most of the peaks cannot be detected in the spectrum, indicating that the protein adopts multiple conformations and undergoes dynamic conformational exchange in the intermediate range of the μsec-msec NMR time scale. Protein aggregation, which could also result in line broadening and degradation of the NMR spectrum, is ruled out by the size exclusion chromatography profile showing that tBID elutes as a single peak, at an apparent molecular weight corresponding to that of the monomeric protein. The few peaks that are visible in the HSQC spectrum can be assigned to residues in the unstructured N-terminal loop of the protein (Figure 6A), and their chemical shift frequencies reflect the highly dynamic random coil conformation of this segment.
Figure 4.
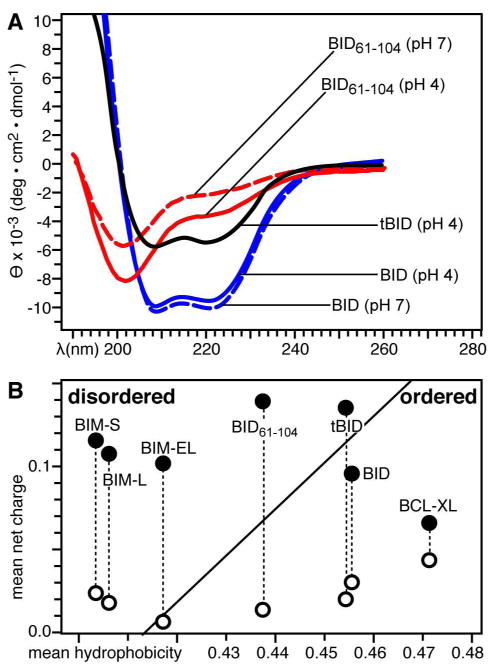
Secondary structure and structural order of tBID and BID61-104. (A) CD spectra obtained at pH 4 or pH 7. (B) Predicted structural order for BID, tBID, BID61-104, BCL-XL, and BIM at pH 4 (●) or pH 7 (○).
Figure 5.
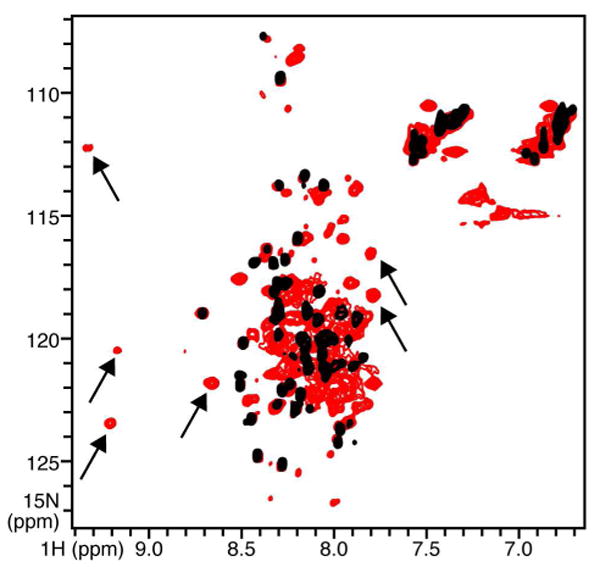
HSQC NMR spectra of 15N-labeled tBID at pH 4, free (black), or bound to equimolar BCL-XL (red). Arrows point to examples of new peaks that appear in the spectrum of the complex.
Figure 6.

HSQC NMR spectra of 15N-labeled tBID and BID61-104, free or bound to BCL-XL. (A) Free tBID (black) and free BID61-104 (red). Selected peaks from the loop and from the BH3 domain (boxes) are labeled. (B) BCL-XL-bound tBID (black) and BCL-XL-bound BID61-104 (red). (C) Free BID61-104 (black) and BCL-XL-bound BID61-104 (red); all peaks are labeled.
To see whether the tertiary structure of tBID could be stabilized by association with BCL-XL, we examined its NMR spectrum within the complex (Figure 5, red). Overall, the association of tBID with BCL-XL produces little improvement in its NMR spectrum, however, binding BCL-XL does cause many peaks to appear anew in the spectrum. These peaks have dispersed 1H and 15N chemical shift frequencies consistent with stable secondary and tertiary structures of proteins, indicating that the structure of at least a region of tBID is indeed stabilized upon binding BCL-XL. To assign these peaks, and map the interaction of tBID with BCL-XL, we compared the spectra of tBID with those of a shorter polypeptide, BID61-104, spanning residues 61 to 104 which include part of the unstructured loop and the entire BH3 domain of the protein. In the NMR spectrum of free BID61-104 (Figure 6A, red) the peaks from residues in the N-terminal loop (e.g. N62, R63, H66, R68, E73, A74) perfectly overlap the peaks from the same residues in the spectrum of free tBID (Figure 6A, black). In contrast, peaks from the BH3 domain (e.g. S78, I82, A87, L90, A91, G94, D95, S96, L97, G104), which are clearly resolved in the spectrum of BID61-104, cannot be seen in the spectrum of tBID. In the spectrum of free BID61-104, peaks from the BH3 domain have chemical shifts that are consistent with random coil conformation, indicating that the peptide is unstructured in water. This is further supported by the CD spectra of BID61-104 showing that the polypeptide in water is a random coil at both pH 4 and pH 7, while intact tBID is α-helical (Figure 4A).
The addition of BCL-XL induces identical changes in the spectra of BID61-104 and tBID (Figure 6B). Resonance assignments of the spectra from free and bound BID61-104 (Figure 6C) show that the peaks which experience frequency changes in the tBID/BCL-XL complex correspond to residues in the BH3 domain and in helix h3 identified in the solution NMR structure of full-length human BID (18). In contrast, peaks from residues outside this region do not change in the spectrum of BID61-104, and remain undetected in the spectrum of tBID, suggesting that binding to BCL-XL does not induce a major conformational change of tBID in water.
The distinct effects of BCL-XL on the spectra of tBID and BID61-104 reflect changes in both protein conformation and dynamics. The peaks that experience the largest frequency changes map to residues in helix h3, particularly, R84, A87, R88, L90, A91, and D95, which line up along the length of helix h3 in the structure of BID (Figure 7A). These residues are highly conserved in the BH3 domains of the pro-apoptotic BCL-2 proteins (50), and are crucial for the interactions of the BH3 domain with pro-survival proteins (23, 24). For example, in the structure of a BAK BH3 peptide bound to BCL-XL, they make key contacts with residues in the hydrophobic binding groove of BCL-XL, including key hydrogen bonds between BAK D83 (corresponding to BID D95) and the BCL-XL R139 guanidinium group (23). Our results demonstrate that the same BH3 domain residues are involved in the interaction of tBID with BCL-XL.
Figure 7.
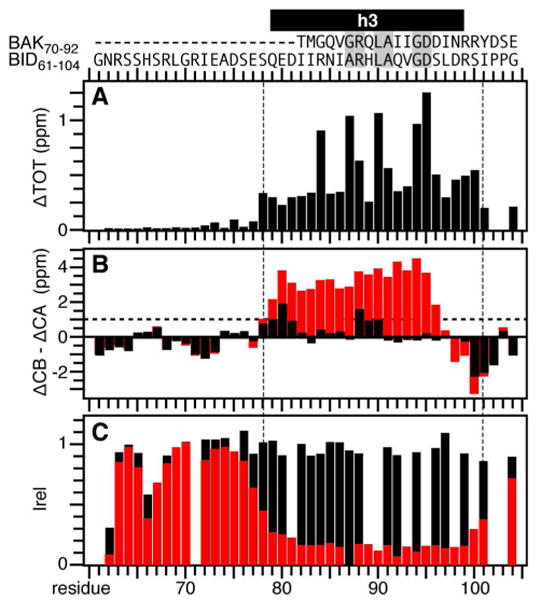
Mapping the effects of BCL-XL on tBID. (A) Total change in BID61-104 amide chemical shifts induced by BCL-XL. (B) Helicity of free (black) and BCL-XL-bound (red) BID61-104, measured as the difference between experimentally measured and random coil values of the CA or CB chemical shifts (ΔCA-ΔCB). The horizontal line at ΔCA-ΔCB = 1 ppm denotes the threshold for helicity. (C) Normalized 1H/15N HSQC peak intensities measured for free (black) and BCL-XL-bound (red) BID61-104. The amino acid sequences of BID61-104 and BAK (residues 70-92) are shown at the top with conserved BH3 residues in gray boxes. Helix h3, identified in the solution NMR structure of human BID, is in black.
The NMR data also show that BID61-104 undergoes a major conformational change upon binding BCL-XL, from random coil and highly dynamic in its free form, to helical and ordered when bound to BCL-XL (Figure 7B, 7C). Carbon chemical shifts are strongly dependent on backbone torsion angles and can be analyzed to obtain useful of indices of polypeptide secondary structure. Analysis of the CA and CB chemical shifts in the spectra of the free and bound peptide show that BCL-XL binding induces the formation of a well-defined α-helix between residues 78 to 97, which correspond to helix h3 in the solution NMR structure of BID.
The frequency changes and helix formation observed upon binding BCL-XL are accompanied by a sharp decrease in HSQC peak intensity, for residues 78 to 101, reflecting the change from a free highly dynamic conformation to a more rigid bound state (Figure 7C). This indicates that the entire 20-residue helical segment, and not just the 14-residue BH3 domain previously identified for the shorter BAK peptide, associates with the binding pocket of the pro-survival partner, consistent with the results reported for a short BID BH3 peptide bound to BCL-XL (29) or to BCL-W (26). Since the spectra of BCL-XL-bound BID61-104 and BCL-XL-bound tBID are fully overlapped in this region, the results in Figure 7 also mirror the interaction of the entire tBID molecule with BCL-XL. Taken together, the data show that the three-dimensional conformation of tBID is highly disordered in water compared to the well-defined globular structure of its full-length precursor, and that binding to BCL-XL induces localized but not global structural ordering of tBID.
To examine the binding site of tBID on BCL-XL we mapped the frequency changes caused by tBID on the 1H/15N HSQC spectrum of 15N-labeled BCL-XL. Addition of tBID causes a subset of peaks in the spectrum of BCL-XL to shift position, while many others exhibited no or only minor changes, indicating that complex formation does not induce a major structural rearrangement of BCL-XL (Figure 8A). The largest changes are observed for peaks from residues in helices h2, h3, h4 and h5 (Figure 8B), and when these changes are mapped onto the structure of BCL-XL determined in complex with the BAK BH3 peptide (23) they clearly highlight the same hydrophobic groove formed by the BH1, BH2, and BH3 regions of BCL-XL, that was previously identified as the BH3-binding site (Figure 8C). Short BH3 peptides from BAD and BAK bind BCL-XL in the same binding region and with similar helicities, and structural studies of 25- or 16-residue BAD BH3 peptides bound to BCL-XL show that the greater affinity of the longer peptide is attributed to the formation of additional interactions with BCL-XL, and to its increased helix propensity (23, 24). Furthermore, a short BIM BH3 peptide forms a C-terminally extended helix upon binding BCL-XL (25).
Figure 8.
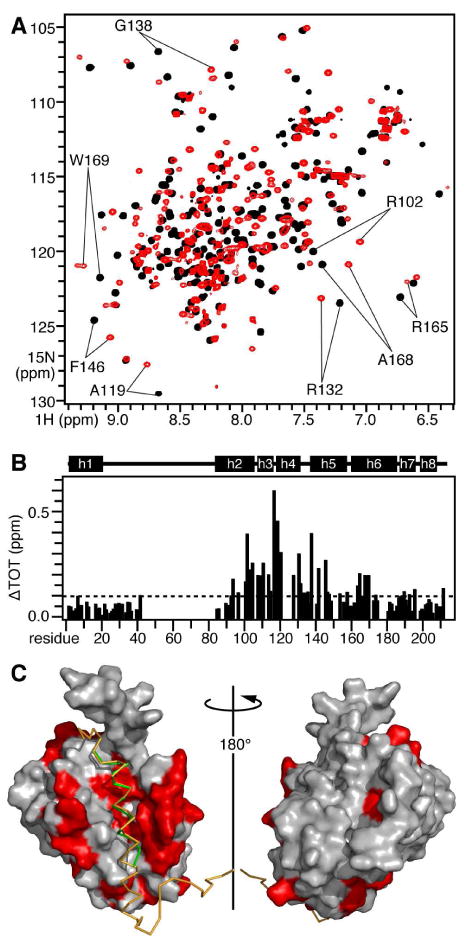
Mapping the effects of tBID on BCL-XL. (A) HSQC NMR spectra of 15N-labeled BCL-XL without (black) or with (red) of unlabeled tBID. (B) Total change in BCL-XL amide chemical shifts induced by equimolar tBID. The helical regions identified in the solution NMR structure of BCL-XL are outlined above the map. (C) Molecular surface representation of the solution NMR structure of BCL-XL bound to a BAK BH3 peptide (green) and to a model BID61-104 peptide (yellow). The atomic coordinates of BCL-XL bound to the BAK BH3 peptide were obtained from the Protein Data Bank (23). The backbone of BID61-104 was modeled in the groove of BCL-XL by alignment with the backbone of the BAK BH3 peptide. Residues in red have HSQC peaks that move by ΔTOT > 0.1 ppm.
The mode of association of tBID with BCL-XL shares similarities as well as subtle differences with those observed for the other BH3-only protein BIM. In both cases, binding to BCL-XL induces selective stabilization of the BH3 domain. BIM-L has an intrinsically unfolded random coil conformation in the absence of pro-survival binding partners, and its BH3 domain becomes α-helical when it binds pro-survival proteins while the rest of the molecule remains unstructured (30). The situation for tBID is somewhat different because the protein maintains the α-helical secondary structure of its precursor, despite its dynamically disordered three-dimensional conformation; in this case, the structure of the BH3 domain is already helical but becomes ordered upon binding BCL-XL while the rest of the molecule remains disordered.
This difference between tBID and BIM is consistent with an analysis of their amino acid sequences based on normalized net charge and mean hydrophobicity (51), indicating a higher degree of structural order for tBID compared to the natively unfolded BIM isoforms (Figure 4B). Like BIM, two other BH3-only proteins BAD and BMF are also predicted to be natively unstructured (30), suggesting that lack of structure, structural disorder, and plasticity may be common features of the BH3-only proteins required for their activity and ability to bind multiple partners within and outside the BCL-2 family, and in both cytosolic and membrane-bound conformations. Indeed, the BIM BH3 domain is capable of binding not only BCL-XL but also BAX (28), albeit at completely different sites of these two structurally similar BCL-2 proteins, indicating that a principal structural requirement for these interactions is the capability of helix formation by the BH3 domain.
Thermodynamics of tBID/BCL-XL association
To further characterize the interaction of tBID with BCL-XL, we performed ITC experiments by titrating both tBID and BID61-104 into BCL-XL. Representative titrations are shown in Figure 9, and the associated thermodynamic parameters for binding are reported in Table 1.
Figure 9.
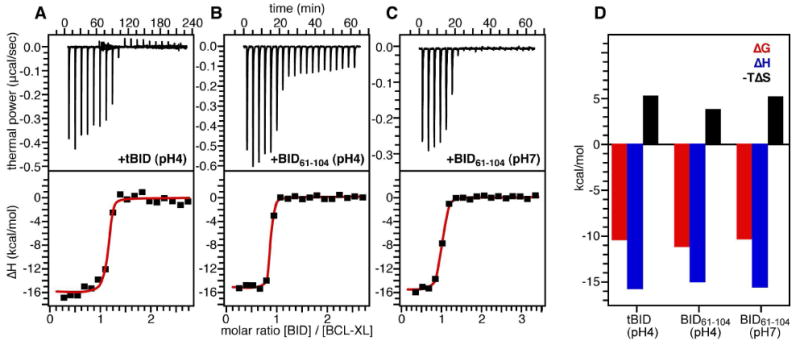
(A-C) ITC titrations of tBID and BID61-104 into BCL-XL. Top panels show calorimetric data. Bottom panels show data obtained by integration of each calorimetric peak, after subtraction of blank titrations and concentration normalization. The red lines trace the fit to a single binding site model used to extract the values of Ka. (D) Thermodynamic signature of BCL-XL binding the BID BH3 domain.
Table 1.
Dissociation constants and thermodynamic parameters, determined from ITC, for the polypeptides tBID and BID61-104 binding to BCL-XL at 303°K.
| ligand | Kd nM |
ΔG kcal/mol |
ΔH kcal/mol |
TΔS kcal/mol |
|---|---|---|---|---|
| tBID (pH4) | 27.2±0.5 | -10.49 | -15.85±0.36 | -5.36 |
| BID61-104 (pH4) | 7.3±0.2 | -11.28 | -15.15±0.13 | -3.85 |
| BID61-104 (pH7) | 30.6±0.1 | -10.42 | -15.68±0.19 | -5.24 |
The binding affinity of tBID for BCL-XL at pH 4 (Kd=27 nM) is only about 3.5 times weaker than that of BID61-104 at pH 4 (Kd=7.25 nM), and very similar to that of BID61-104 at pH 7 (Kd=30.58 nM). These values are also within the range of affinities reported in literature for short BH3 peptides binding to BCL-XL and other anti-apoptotic BCL-2 proteins (23, 27, 29, 52), indicating that the binding interaction of tBID with BCL-XL does not involve additional regions of tBID other than residues in and flanking the BH3 domain.
For both tBID and its short BH3 analog, BID61-104, the association with BCL-XL is enthalpy-driven. The binding thermodynamics are characterized by very similar and opposing terms of ΔH and ΔS (Figure 9E), with favorable enthalpy (ΔH<0) and unfavorable entropy (ΔS<0), as previously observed for a series of short BH3 peptides binding the anti-apoptotic BCL-2 family protein MCL-1 (27). The overall binding enthalpy (ΔH) includes a term for intrinsic enthalpy (ΔHi) that reflects the selectivity of the specific interactions between ligand and target, terms associated with conformational change of the target (ΔHct) or of the ligand (ΔHcl) upon binding, and a term (ΔHH) associated with protonation/deprotonation events (53). The overall binding entropy (ΔS) includes a term for the hydrophobic effect (ΔSh) that contributes an entropy increase through the generation of free water molecules, and terms associated with the loss of conformational flexibility of the target (ΔSct) and of the ligand (ΔScl) both of which contribute an entropy decrease.
Since both tBID and BID61-104 bind the same groove of BCL-XL through the same BH3 sequence, and induce a similar conformational change in BCL-XL, similar values of ΔHi, ΔHct, ΔSh and ΔSct are expected for their association with BCL-XL. Thus, any small differences between the affinities of tBID and BID61-104 would be due to differences in the thermodynamic terms associated with ligand conformation (ΔHcl, ΔScl). Protonation/deprotonation effects do not contribute for measurements made at the same pH, however, the slightly greater affinity observed for BID61-104 at pH 4 compared to pH 7 may be related to protonation strengthening the hydrogen bonds between the ligand and BCL-XL and/or promoting helix formation of the BID ligand. In the case of tBID, this effect may be countered by the overall greater unfavorable entropy associated with inducing conformational order in the bigger full-length molecule, resulting in similar thermodynamic parameters for tBID at pH 4 and BID61-104 at pH 7. It is possible that the BH3 domain of tBID retains some interactions with other regions of the protein, as it does in the precursor BID. In this case, binding the hydrophobic groove of BCL-XL would first require release of the tBID BH3 domain. The overall favorable free energy ΔG associated with tBID binding to BCL-XL indicates that BCL-XL-bound tBID is thermodynamically more stable than free tBID. Nevertheless, we note that the close similarity in the affinities of tBID and BID61-104 for BCL-XL overshadows any minor differences, and demonstrates that the interaction is limited to the tBID BH3 domain and the BCL-XL hydrophobic cleft.
Unlike full-length tBID, where the NMR and CD spectra reflect the dynamic exchange of conformations with α-helical secondary structure, BID61-104 and all of the small BH3 ligands are random coils in their free state, and adopt α-helical conformation only upon binding their pro-survival target. This is consistent with the finding that binding of BH3 domains to their pro-survival targets is favored by propensity toward helical structure (24) and that short BH3 peptides chemically modified to promote α-helicity bind with greater affinity (29, 54). Although full-length tBID already has a significant amount of α-helical secondary structure in its free state, the NMR and ITC data show that its three-dimensional conformation is stabilized by binding BCL-XL.
Acknowledgments
We thank Stephen Fesik and Edward Olejniczak (Abbott Laboratories) for sharing their NMR assignments for BCL-XL at pH 7.
Abbreviations
- BH domain
BCL-2 homology domain
- CD
circular dichroism
- HSQC
heteronuclear single quantum correlation
- ITC
isothermal titration calorimetry
Footnotes
This research was supported by a grant from the National Institutes of Health. The NMR studies utilized the Burnham Institute NMR Facility, supported by a grant from the National Institutes of Health (CA030199).
References
- 1.Reed JC. Bcl-2-family proteins and hematologic malignancies: history and future prospects. Blood. 2008;111:3322–3330. doi: 10.1182/blood-2007-09-078162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 3.Adams JM, Cory S. Bcl-2-regulated apoptosis: mechanism and therapeutic potential. Curr Opin Immunol. 2007;19:488–496. doi: 10.1016/j.coi.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuwana T, Newmeyer DD. Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr Opin Cell Biol. 2003;15:691–699. doi: 10.1016/j.ceb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 5.Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18:157–164. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leber B, Lin J, Andrews DW. Embedded together: the life and death consequences of interaction of the Bcl-2 family with membranes. Apoptosis. 2007;12:897–911. doi: 10.1007/s10495-007-0746-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 8.Wang K, Yin XM, Chao DT, Milliman CL, Korsmeyer SJ. BID: a novel BH3 domain-only death agonist. Genes Dev. 1996;10:2859–2869. doi: 10.1101/gad.10.22.2859. [DOI] [PubMed] [Google Scholar]
- 9.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 10.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 11.Zha J, Weiler S, Oh KJ, Wei MC, Korsmeyer SJ. Posttranslational N-myristoylation of BID as a molecular switch for targeting mitochondria and apoptosis. Science. 2000;290:1761–1765. doi: 10.1126/science.290.5497.1761. [DOI] [PubMed] [Google Scholar]
- 12.Gross A, Yin XM, Wang K, Wei MC, Jockel J, Milliman C, Erdjument-Bromage H, Tempst P, Korsmeyer SJ. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J Biol Chem. 1999;274:1156–1163. doi: 10.1074/jbc.274.2.1156. [DOI] [PubMed] [Google Scholar]
- 13.Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8:705–711. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- 14.Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, Korsmeyer SJ. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell. 2002;2:55–67. doi: 10.1016/s1534-5807(01)00116-2. [DOI] [PubMed] [Google Scholar]
- 15.Eskes R, Desagher S, Antonsson B, Martinou JC. Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol Cell Biol. 2000;20:929–935. doi: 10.1128/mcb.20.3.929-935.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korsmeyer SJ, Wei MC, Saito M, Weiler S, Oh KJ, Schlesinger PH. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000;7:1166–1173. doi: 10.1038/sj.cdd.4400783. [DOI] [PubMed] [Google Scholar]
- 17.Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 18.Chou JJ, Li H, Salvesen GS, Yuan J, Wagner G. Solution structure of BID, an intracellular amplifier of apoptotic signaling. Cell. 1999;96:615–624. doi: 10.1016/s0092-8674(00)80572-3. [DOI] [PubMed] [Google Scholar]
- 19.McDonnell JM, Fushman D, Milliman CL, Korsmeyer SJ, Cowburn D. Solution structure of the proapoptotic molecule BID: a structural basis for apoptotic agonists and antagonists. Cell. 1999;96:625–634. doi: 10.1016/s0092-8674(00)80573-5. [DOI] [PubMed] [Google Scholar]
- 20.Fesik SW. Insights into programmed cell death through structural biology. Cell. 2000;103:273–282. doi: 10.1016/s0092-8674(00)00119-7. [DOI] [PubMed] [Google Scholar]
- 21.Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta. 2004;1644:83–94. doi: 10.1016/j.bbamcr.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 22.Gong XM, Choi J, Franzin CM, Zhai D, Reed JC, Marassi FM. Conformation of Membrane-associated Proapoptotic tBid. J Biol Chem. 2004;279:28954–28960. doi: 10.1074/jbc.M403490200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, Thompson CB, Fesik SW. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science. 1997;275:983–986. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- 24.Petros AM, Nettesheim DG, Wang Y, Olejniczak ET, Meadows RP, Mack J, Swift K, Matayoshi ED, Zhang H, Thompson CB, Fesik SW. Rationale for Bcl-xL/Bad peptide complex formation from structure, mutagenesis, and biophysical studies. Protein Sci. 2000;9:2528–2534. doi: 10.1110/ps.9.12.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu X, Dai S, Zhu Y, Marrack P, Kappler JW. The structure of a Bcl-xL/Bim fragment complex: implications for Bim function. Immunity. 2003;19:341–352. doi: 10.1016/s1074-7613(03)00234-6. [DOI] [PubMed] [Google Scholar]
- 26.Denisov AY, Chen G, Sprules T, Moldoveanu T, Beauparlant P, Gehring K. Structural model of the BCL-w-BID peptide complex and its interactions with phospholipid micelles. Biochemistry. 2006;45:2250–2256. doi: 10.1021/bi052332s. [DOI] [PubMed] [Google Scholar]
- 27.Day CL, Smits C, Fan FC, Lee EF, Fairlie WD, Hinds MG. Structure of the BH3 domains from the p53-inducible BH3-only proteins Noxa and Puma in complex with Mcl-1. J Mol Biol. 2008;380:958–971. doi: 10.1016/j.jmb.2008.05.071. [DOI] [PubMed] [Google Scholar]
- 28.Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, Tu HC, Kim H, Cheng EH, Tjandra N, Walensky LD. BAX activation is initiated at a novel interaction site. Nature. 2008;455:1076–1081. doi: 10.1038/nature07396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ji H, Shekhtman A, Ghose R, McDonnell JM, Cowburn D. NMR determination that an extended BH3 motif of pro-apoptotic BID is specifically bound to BCL-XL. Magn Reson Chem. 2006;44(Spec No):S101–107. doi: 10.1002/mrc.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hinds MG, Smits C, Fredericks-Short R, Risk JM, Bailey M, Huang DC, Day CL. Bim, Bad and Bmf: intrinsically unstructured BH3-only proteins that undergo a localized conformational change upon binding to prosurvival Bcl-2 targets. Cell Death Differ. 2007;14:128–136. doi: 10.1038/sj.cdd.4401934. [DOI] [PubMed] [Google Scholar]
- 31.Schendel SL, Azimov R, Pawlowski K, Godzik A, Kagan BL, Reed JC. Ion channel activity of the BH3 only Bcl-2 family member, BID. J Biol Chem. 1999;274:21932–21936. doi: 10.1074/jbc.274.31.21932. [DOI] [PubMed] [Google Scholar]
- 32.Zhou Q, Snipas S, Orth K, Muzio M, Dixit VM, Salvesen GS. Target protease specificity of the viral serpin CrmA. Analysis of five caspases. J Biol Chem. 1997;272:7797–7800. doi: 10.1074/jbc.272.12.7797. [DOI] [PubMed] [Google Scholar]
- 33.Franzin CM, Choi J, Zhai D, Reed JC, Marassi FM. Structural studies of apoptosis and ion transport regulatory proteins in membranes. Magn Reson Chem. 2004;42:172–179. doi: 10.1002/mrc.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Freire E. Do enthalpy and entropy distinguish first in class from best in class? Drug Discov Today. 2008;13:869–874. doi: 10.1016/j.drudis2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ikura M, Kay LE, Bax A. A novel approach for sequential assignment of 1H, 13C, and 15N spectra of proteins: heteronuclear triple-resonance three-dimensional NMR spectroscopy. Application to calmodulin. Biochemistry. 1990;29:4659–4667. doi: 10.1021/bi00471a022. [DOI] [PubMed] [Google Scholar]
- 36.Sattler M, Schleucher J, Griesinger C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog Nucl Magn Reson Spectrosc. 1999;34:93–158. [Google Scholar]
- 37.Grzesiek S, Bax A. Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. J Am Chem Soc. 1992;114:6291–6293. [Google Scholar]
- 38.Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS, Nettesheim D, Chang BS, Thompson CB, Wong SL, Ng SL, Fesik SW. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature. 1996;381:335–341. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- 39.Cavanagh J, Fairbrother WJ, Palmer AG, Skelton NJ. Protein NMR spectroscopy : principles and practice. Academic Press; San Diego: 1996. [Google Scholar]
- 40.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 41.Goddard TD, Kneller DG. SPARKY 3. University of California; San Francisco: 2004. [Google Scholar]
- 42.Mori S, Abeygunawardana C, Johnson MO, Vanzijl PCM. Improved Sensitivity of HSQC Spectra of Exchanging Protons at Short Interscan Delays Using a New Fast HSQC (FHSQC) Detection Scheme That Avoids Water Saturation. J Magn Reson B. 1995;108:94–98. doi: 10.1006/jmrb.1995.1109. [DOI] [PubMed] [Google Scholar]
- 43.Grzesiek S, Bax A, Clore GM, Gronenborn AM, Hu JS, Kaufman J, Palmer I, Stahl SJ, Wingfield PT. The solution structure of HIV-1 Nef reveals an unexpected fold and permits delineation of the binding surface for the SH3 domain of Hck tyrosine protein kinase. Nat Struct Biol. 1996;3:340–345. doi: 10.1038/nsb0496-340. [DOI] [PubMed] [Google Scholar]
- 44.Hoffman RM, Li MX, Sykes BD. The binding of w7, an inhibitor of striated muscle contraction, to cardiac troponin C. Biochemistry. 2005;44:15750–15759. doi: 10.1021/bi051583y. [DOI] [PubMed] [Google Scholar]
- 45.Wang L, Eghbalnia HR, Markley JL. Probabilistic approach to determining unbiased random-coil carbon-13 chemical shift values from the protein chemical shift database. J Biomol NMR. 2006;35:155–165. doi: 10.1007/s10858-006-9022-0. [DOI] [PubMed] [Google Scholar]
- 46.Wishart DS, Sykes BD. Chemical shifts as a tool for structure determination. Methods Enzymol. 1994;239:363–392. doi: 10.1016/s0076-6879(94)39014-2. [DOI] [PubMed] [Google Scholar]
- 47.Schendel SL, Montal M, Reed JC. Bcl-2 family proteins as ion-channels. Cell Death Differ. 1998;5:372–380. doi: 10.1038/sj.cdd.4400365. [DOI] [PubMed] [Google Scholar]
- 48.Losonczi JA, Olejniczak ET, Betz SF, Harlan JE, Mack J, Fesik SW. NMR studies of the anti-apoptotic protein Bcl-xL in micelles. Biochemistry. 2000;39:11024–11033. doi: 10.1021/bi000919v. [DOI] [PubMed] [Google Scholar]
- 49.Thuduppathy GR, Hill RB. Acid destabilization of the solution conformation of Bcl-xL does not drive its pH-dependent insertion into membranes. Protein Sci. 2006;15:248–257. doi: 10.1110/ps.051807706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lanave C, Santamaria M, Saccone C. Comparative genomics: the evolutionary history of the Bcl-2 family. Gene. 2004;333:71–79. doi: 10.1016/j.gene.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 51.Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 52.O'Neill JW, Manion MK, Maguire B, Hockenbery DM. BCL-XL dimerization by three-dimensional domain swapping. J Mol Biol. 2006;356:367–381. doi: 10.1016/j.jmb.2005.11.032. [DOI] [PubMed] [Google Scholar]
- 53.Luque I, Freire E. Structural parameterization of the binding enthalpy of small ligands. Proteins. 2002;49:181–190. doi: 10.1002/prot.10208. [DOI] [PubMed] [Google Scholar]
- 54.Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J, Smith E, Verdine GL, Korsmeyer SJ. A stapled BID BH3 helix directly binds and activates BAX. Mol Cell. 2006;24:199–210. doi: 10.1016/j.molcel.2006.08.020. [DOI] [PubMed] [Google Scholar]