

Abstract
Pancreatic beta cell function continuously deteriorates in type 2 diabetes despite optimal treatment regimens, which has been attributed to hyperglycemia itself via formation of excess levels of reactive oxygen species (ROS). Glutathione peroxidase GPx), by virtue of its ability to catabolize both H2O2 and lipid peroxides, is uniquely positioned to protect tissues from ROS. The level of this antioxidant in beta cells is extremely low and overexpression of GPx in islets provides enhanced protection against oxidative stress. This suggests that GPx mimetics may represent a valuable ancillary treatment that could add a novel layer of protection for the beta cell.
Introduction
This review addresses the general area of cellular stress through a consideration of the pancreatic islet β-cell and its interactions with reactive oxygen species (ROS) under physiologic conditions, as well as under the pathophysiologic state of diabetes mellitus and attendant hyperglycemia. It focuses on the antioxidant enzyme glutathione peroxidase (GPx), an enzyme that is uniquely positioned in the ROS degradation pathway to protect cells from excessive levels of hydrogen peroxide (H2O2) and intracellular lipid peroxides. In contrast, catalase, which also metabolizes H2O2, does not metabolize lipid peroxides. Pharmacologic insights and therapeutic suggestions are provided from a consideration of molecular mechanisms involved in ROS-induced defective insulin gene expression and beta cell dysfunction.
1. The pancreatic islet: physiology and pathophysiology
The islets of Langerhans are scattered throughout the exocrine pancreas and comprise only 2–3% of the pancreatic mass. Islets contain four major cell types: alpha, beta, delta, and PP cells. These cells secrete glucagon, insulin, somatostatin, and pancreatic polypeptide, respectively. The primary stimulus for insulin secretion is glucose, but it can also be stimulated by amino acids, other hormones, and certain drugs. Insulin secretion ceases when the blood glucose reaches abnormally low levels, thereby preventing worsening of hypoglycemia. The primary regulators for glucagon secretion are just the opposite, i.e. hypoglycemia stimulates glucagon secretion whereas hyperglycemia inhibits the secretion of this hormone. Somatostatin secretion is stimulated by glucose and amino acids. Pancreatic polypeptide is stimulated by hypoglycemia and another hormone, secretin. The interplay of regulators of hormone secretion from the pancreatic islet provides a sophisticated means by which the products of islet cells regulate overall body metabolism. Insulin is the primary regulator of blood glucose and is essential for normal growth and storage of fuels in body tissues. Absence of normal insulin secretion causes very high levels of glucose and leads to diabetes. If excessive insulin is injected from an exogenous source, blood glucose can be lowered to abnormally low levels, termed hypoglycemia. In this situation, glucagon is normally secreted from alpha cells and recovery from hypoglycemia begins. As soon as the blood glucose returns to normal, glucagon secretion is inhibited. Somatostatin is a local regulator of both insulin and glucagon secretion by virtue of its capacity to inhibit secretion from the beta and alpha cell. The function of pancreatic polypeptide in humans is unknown.
Both insulin and glucagon have important metabolic targets. The major function of these two partners is to finely regulate blood glucose levels so that neither prolonged hypoglycemia or hyperglycemia are allowed to develop. Insulin inhibits and glucagon stimulates glycogenolysis in the liver, a process by which stored glycogen is broken down into glucose for release into the bloodstream. Insulin inhibits and glucagon stimulates gluconeogenesis, a biochemical process by which amino acids are used to synthesize glucose. Insulin inhibits and glucagon stimulates lipolysis, a process by which stored triglyceride within fat cells is released into the blood stream in the form of fatty acids and glycerol. Insulin inhibits and glucagon stimulates ketogenesis, a biochemical process in which free fatty acids form ketones in the liver for release into the bloodstream.
2. Physiology of ROS formation and regulation
The term ROS designates a large group of reactive oxygen species that in physiological concentrations help maintain homeostasis, for example, in regulation of gene transcription and as participants in white blood cell defense against infection. It is also reasonable to postulate that physiologic amounts of ROS regulate islet gene expression and facilitate regulation of beta cell function. However, when ROS accumulate in excess for prolonged periods of time, they cause chronic oxidative stress and potentially adverse effects. Antioxidant enzymes contained within host tissues are the first line of defense against excessive levels of ROS (Fig 1). An important consideration is the level of antioxidant host defenses within the islet. Generally speaking, the highest levels of antioxidant enzyme expression are in the liver, a major detoxification center. In contrast, it has been reported that the islet is among the least well-endowed tissues in terms of intrinsic antioxidant enzyme expression and activity [1, 2], including superoxide dismutases (SOD-1, SOD-2), catalase, and glutathione peroxidase (GPx). In contrast, gene expression of the catalytic subunit of γ-glutamylcysteine ligase (GCLC), the rate-limiting enzyme for glutathione (GSH) synthesis, is well expressed in islets [3]. GSH is the major intrinsic antioxidant in cells, as well as being a cofactor for GPx activity. Levels of GCLC mRNA are comparable to those found in liver and greater than those found in muscle, lung and fat [3]. It has been reported, however, that long-term exposure to high glucose concentrations decreases GCLC expression in mesangial as well as retinal cells, and that this is associated with a decrease in GSH levels [4, 5].
Figure 1.
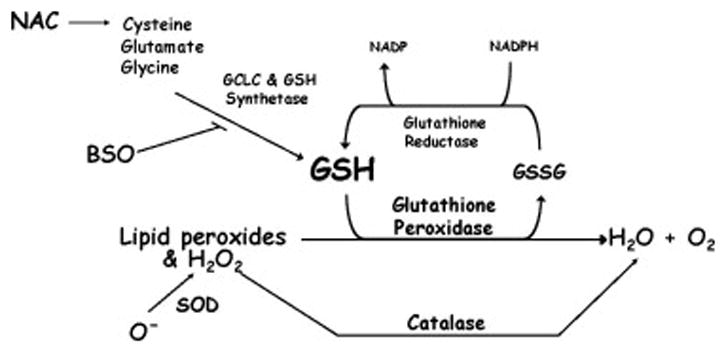
Glutathione (GSH) metabolism. Cysteine, the rate limiting aminoacid for GSH synthesis, along with glutamate and glycine, are used by glutathione synthetase and the catalytic subunit of glutathione cysteine ligase (GCLC) to synthesize GSH. Buthionine sulfoximine (BSO) inhibits GCLC activity. GSH is used by glutathione peroxidase (GPx) in a reduction reaction to metabolize hydrogen peroxide (H2O2) and lipid peroxides into elemental oxygen and water. Catalase can also metabolize H2O2, but not lipid peroxides. Superoxide dismutases 1 and 2 catabolize superoxide ions to form H2O2.
3. Hyperglycemia and oxidative stress
A. Clinical evidence in humans: markers of oxidative stress
Many clinical studies have documented that patients with type 2 diabetes are subjected to chronic oxidative stress [6–11]. Measurements using sophisticated laboratory techniques, including high performance liquid chromatography, gas chromatography/mass spectrometry, and immunostaining of pancreas have been used. Markers for oxidative tissue damage, such as 8-hydroxy-deoxyguanine; 4-hydroxy-2-nonenal; 8-epi-PGF2α; hydroperoxides; and oxidized DNA bases have been reported to be elevated in serum, plasma, white blood cells, and pancreas biopsies of patients with type 2 diabetes. Glutathione, the primary intracellular antioxidant, has been studied xtensively. Murakami et al. reported that red blood cells from diabetic patients contain low levels of the reduced form of glutathione (GSH), high levels of the oxidized form (GSSG), and a 51% reduction in the GSH/GSSG ratio [12]. Sharma et al. also reported that type 2 diabetic patients in poor glycemic control had depressed red blood cell GSH levels [13]. After decreasing red blood cell HbA1c levels by sulfonylurea treatment, the red blood cell GSH levels increased two-fold, almost reaching the values found in a non-diabetic control group. Similar data were reported by Yoshida et al. who, in addition, reported that activity of the rate-limiting enzyme of GSH synthesis, γ-glutamylcysteine synthase, increased with improved glycemic control [14]. This group also reported that red blood cell thiol transport was significantly and inversely correlated with levels of HbA1c.
B. In vivo evidence in animals: effects of antioxidants
The Zucker diabetic fatty (ZDF) rat is homozygous for deficiency of the leptin receptor, spontaneously develops obesity and hyperglycemia, and has decreased insulin gene expression after 6 to 7 weeks of age [15–19]. Seufert et al. [20] have found that the development of hyperglycemia in this animal model is associated with the loss of an important insulin promoter activator, PDX-1. We designed experiments to examine expression of PDX-1 (STF-1) and insulin in the ZDF animal and ascertained whether prevention of hyperglycemia would alter the deleterious changes in insulin gene expression and PDX-1 binding to the insulin promoter. We designed studies in which ZDF animals were either treated or not treated with troglitazone given orally [21]. The drug was begun at 6 weeks of age. By 16 weeks of age the untreated ZDF animals had hyperglycemia and marked decreases in islet PDX-1 mRNA levels, PDX-1 binding to DNA, insulin mRNA levels, and glucose-induced insulin secretion. In contrast, the ZDF animals treated with troglitazone did not develop as severe hyperglycemia and had preserved levels of PDX-1 mRNA and nuclear protein binding, insulin mRNA, and glucose-stimulated insulin secretion [21]. These findings are consistent with the hypothesis that progressive hyperglycemia in this animal model causes secondary effects on insulin gene expression that are associated with a decrease in PDX-1 gene expression. However, since troglitazone also has been reported to decrease circulating triglycerides and free fatty acids, it is also possible that its beneficial effects on insulin gene expression could be partially explained by the avoidance of another pathophysiologic state, lipotoxicity [22–4].
To differentiate glucotoxicity from lipotoxicity in the ZDF animal, we treated animals with either bezafibrate, a lipid-lowering drug that does not affect plasma glucose levels, or phlorizin, a drug that reduces plasma glucose without lowering lipid levels. Despite lowering plasma triglyceride, bezafibrate was not effective in preventing increased islet triglyceride content and did not prevent the associated decrease in insulin mRNA levels [25]. On the other hand, phlorizin treatment prevented hyperglycemia, lowered islet triglyceride content, and preserved insulin mRNA levels without preventing hypertriglyceridemia. We concluded that glucose toxicity rather than lipotoxicity was the more dominant force in the development of progressive loss of insulin mRNA and insulin secretion in ZDF animals.
Several studies suggest that glucose toxicity and lipotoxicity are interrelated. In vitro, prolonged exposure of isolated islets to elevated levels of fatty acids impairs insulin gene expression only in the presence of high glucose concentration [26]. Glucose is rate-limiting for the incorporation of fatty-acids into triglycerides [27] and forcing triglyceride synthesis in islets impairs glucose-induced insulin secretion [28]. High-fat feeding impairs insulin secretion in islets from hyperglycemic Goto-Kakizaki (GK) rats, but not in islets from normoglycemic Wistar rats or GK rats treated with insulin [29]. Thus, we proposed that chronic hyperlipidemia might be deleterious only in the context of concomitant hyperglycemia, whereas chronic hyperglycemia can cause islet dysfunction in the absence of hyperlipidemia [30].
In other experiments, the ZDF rat model was used to evaluate the hypothesis that the glucose toxicity observed in our experiments was at least partially caused by chronic oxidative stress. Hyperglycemia is known to be associated with oxidative stress in clinical diabetes [6–14] and auto-oxidation of glucose is known to generate reactive oxygen species [31]. Thus, we treated ZDF animals with either N-acetylcysteine (NAC), a powerful antioxidant [32], or placebo injection. Treatment was begun at 6 weeks of age and carried through 12 weeks of age. Measures of chronic oxidative stress were higher in the ZDF rats that developed hyperglycemia compared to ZDF rats treated with NAC. Associated with the increase of oxidative stress markers was the development of accelerated hyperglycemia, decreased glucose tolerance, decreased insulin secretion, decreased PDX-1 binding to the insulin promoter, and decreased insulin gene expression. In contrast, the ZDF animals treated with NAC developed much less hyperglycemia (Fig 2) and had preserved PDX-1 DNA binding, insulin gene expression, and nearly normal insulin secretion and glucose tolerance. Similar findings were observed in parallel studies using aminoguanidine, a drug that is both an antioxidant and an inhibitor of glycosylation [33]. Control studies using an inhibitor of nitric oxide synthase did not demonstrate prevention of hyperglycemia and preservation of insulin gene expression. These findings point to chronic oxidative stress as a potential mechanism of action for glucose toxicity. In this context, it is interesting to note that troglitazone, a drug shown to prevent diabetes in the ZDF animal, is also an antioxidant.
Figure 2.
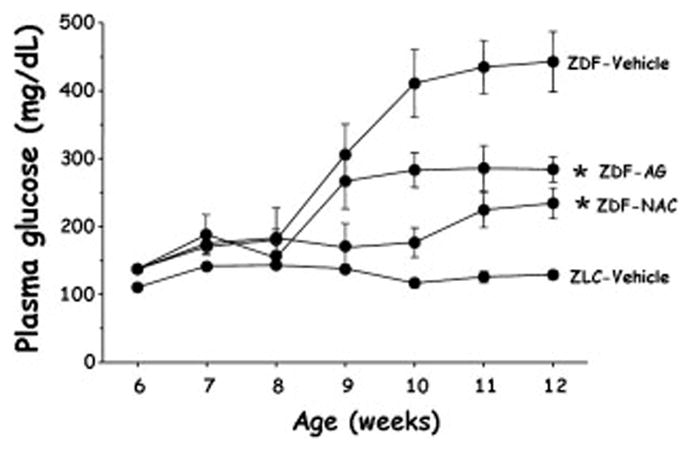
Plasma glucose levels in Zucker Diabetic Fatty (ZDF) rats, a genetic model of type 2 diabetes, who were treated with placebo, n-acetylcysteine, or aminoguanidine beginning at 6 weeks of age. Both drugs are antioxidants and both drugs ameliorated the degree of hyperglycemia developed by the animals. Zucker lean controls (ZLC) that do not develop hyperglycemia are shown for comparison. Taken from Ref. 32.
4. Molecular mechanisms of action of glucose toxicity leading to oxidative stress
Much of the early work addressing molecular mechanisms of glucose toxicity in beta cells was performed with the HIT-T15 cell line. We observed that HIT-T15 cells chronically cultured for 6 months in media containing 11.1 mM glucose, a concentration exceeding that necessary to elicit maximal insulin responses, caused loss of insulin mRNA, greatly diminished levels of insulin content, and almost complete disappearance of insulin secretion [33]. In contrast, HIT-T15 cells cultured serially in media containing 0.8 mM glucose (minimally stimulatory for insulin secretion) for 6 months retained insulin mRNA, insulin content, and insulin responsivity to glucose. This suggested that glucose toxicity might be explained by molecular mechanisms involving insulin gene expression and subsequent insulin synthesis. In further studies, HIT-T15 cells were transiently transfected with a chloramphenicol acetyl transferase (CAT) reporter gene controlled by the 5′-regulatory region of the human insulin gene (INSCAT). Early passages of HIT cells cultured in supraphysiologic glucose concentrations readily expressed INSCAT, whereas late passages of cells expressed only 29% of the CAT activity expressed in earlier passages [34]. In contrast, late passages of cells cultured in 0.8 mM glucose retained the ability to express reporter gene activity to a level of 70% to 100% of the activity observed in earlier passages. As part of our studies, we mutated the motif in the insulin promoter to which PDX-1 binds and observed marked decreases in INSCAT activity in early and late passages of HIT-T15 cells, thereby demonstrating its importance as a regulator of transcription. Northern analysis indicated a marked reduction in the steady-state levels of PDX-1 mRNA in HIT-T15 cells chronically cultured in supraphysiologic glucose concentration (Fig 3), which could not be accounted for by changes in the rate of PDX-1 gene transcription assessed by nuclear run-on analysis. We concluded that the molecular mechanism by which chronic exposure of pancreatic islet β-cells to high glucose concentrations can paradoxically decrease insulin gene. transcription (glucose toxicity) involves alteration of the ability of PDX-1 to bind to the insulin gene promoter
Figure 3.
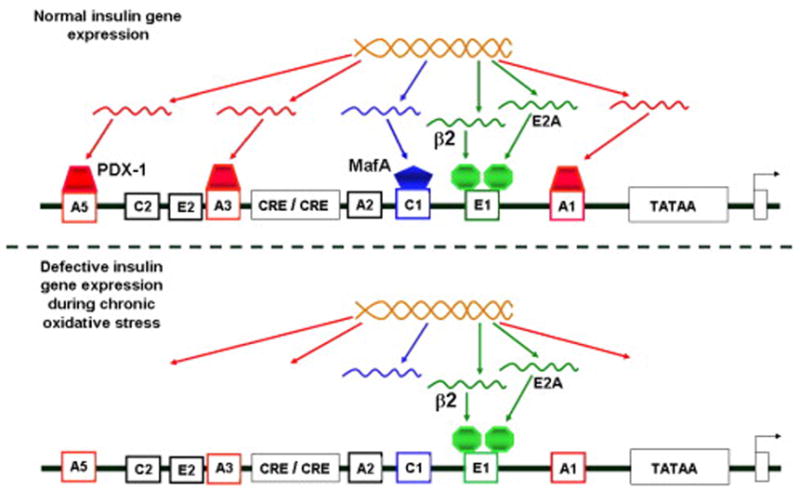
Molecular mechanisms of actions leading to defective insulin gene expression in glucotoxic beta cells. In this model, insulin gene transcription is intrinsically normal, but gene expression of two critical transcription factors, PDX-1 and MafA, is not. Normally, PDX-1 binds to the insulin promoter at three sites, A1, A3, and A5. MafA binds to C1 only. During the development of glucose toxicity, PDX-1 fails to become expressed because of a post-transcriptional defect, and MafA fails to become expressed because of a post-translational defect. Consequently, insulin gene expression at the mRNA level decreases, as does insulin stores and glucose-induced insulin secretion. Taken from Ref. 43.
We also found that decreased levels of RIPE-3b1-activator, recently identified as MafA [35–37], also plays a role in glucose toxicity of the β-cell. We demonstrated that the binding and trans-activation functions of MafA were reduced in HIT-T15 cells chronically cultured in media containing 11.1 mM glucose [39]. In contrast, there was no difference in another important transcription factor, the insulin control element (ICE) activator. Using a luciferase reporter vector, we demonstrated that mutation of MafA binding site caused marked diminution of reporter gene activity in HIT-T15 cells. We concluded that glucotoxic consequences on insulin gene expression involve deficiencies of both MafA and PDX-1 proteins.
Subsequent work by Harmon et al. [39] assessed the temporal sequence of the loss of the PDX-1 and MafA in HIT-T15 cells chronically cultured in supraphysiologic glucose concentrations. MafA binding activity to DNA was decreased below 25% of control by passage 81, whereas PDX-1 binding remained at normal levels through passage 100 and did not decrease to 25% of control levels until passage 106. This cell line begins to lose insulin gene expression by passage 80 to 85, suggesting that loss of MafA may play an early and dominant role in glucotoxic events. Other studies examined whether reconstitution of late-passage glucotoxic cells with PDX-1 cDNA would restore insulin promoter activity. We observed that reconstitution with PDX-1 cDNA of HIT-T15 cells chronically cultured in media containing 11.1 mM glucose partially regained insulin promoter activity [39]. In later work, Northern analysis of glucotoxic HIT-T15 cells revealed normal amounts of MafA mRNA, but Western analysis demonstrated a 97 +/− 1% reduction in MafA protein [40]. Since antioxidants had already been shown to prevent the adverse effects of glucose toxicity on beta cell function both in vivo and in vitro, we chronically cultured HIT-T15 cells with the antioxidant N-acetylcysteine (NAC) and observed prevention of MafA protein loss in late passages cultured in media containing high glucose concentrations. Additionally, transient transfection of PDX-1 and MafA cDNA into glucotoxic cells increased PDX-1 and MafA protein levels. They individually increased insulin promoter activity, and in combination transfection completely restored insulin promoter activity. This recovery of promoter activity following transient transfection had no effect on endogenous insulin mRNA. However, adenoviral infection of MafA and PDX-1 significantly increased endogenous insulin mRNA levels by 93% [40].
Studies assessing molecular mechanisms for glucose toxicity using isolated islets were performed by Briaud et al. (41) who cultured isolated rat islets for up to 6 weeks in either normal (5.6 mM) or supraphysiologic (16.7 mM) glucose concentrations. Insulin mRNA levels were approximately two-fold lower in islets cultured in high glucose than in islets cultured in normal glucose after 6 weeks of culture.
5. GPx in pancreatic islets
A. In vitro levels: rat and human islets
As indicated above, GPx expression at the mRNA and protein levels has been reported to be very low in rodent islets and absent in a beta cell line [1, 2]. No published GPx data from human islets are available, but recently we have recently detected only minimal amounts of GPx protein and activity in islets isolated from 8 human donors (unpublished data). In contrast, SOD-1 and SOD-2, are detectable in rodent and human islets (1, 2 and our unpublished human islet data), although these levels are lower than levels found in the liver. Catalase expression has been reported to be virtually undetectable in rodent islets [1, 2] and our unpublished work detects only minimal amounts of catalase protein and activity in human islets. These assessments in rodent and human islets lead to the conclusion that a rational strategy for enhancing protection of beta cells against oxidative stress would involve beta cell-specific overexpression of GPx. Overexpression of catalase might also be helpful. However, because it catabolizes only H2O2 and not intracellular lipid peroxides, GPx overexpression would seem to be the better strategy.
B. Beneficial effects of GPx overexpression on insulin gene expression, insulin secretion, and insulin content
To evaluate the hypothesis that GPx overexpression might protect the beta cell from oxidative stress, we first determined whether high glucose concentrations increase intraislet ROS levels [42]. Using flow cytometry and a fluorescent dye detection system, we observed that high glucose concentrations increased intracellular peroxide levels in human islets and HIT-T15 cells. Inhibition of γ-glutamylcysteine ligase by buthionine sulfoximine augmented the increase in islet peroxide and ribose-induced decreases in insulin mRNA levels, content, and secretion in islets and HIT-T15 cells. Adenoviral overexpression of GPx increased GPx activity and protected islets against these adverse effects of ribose (43; Fig 4).
Figure 4.
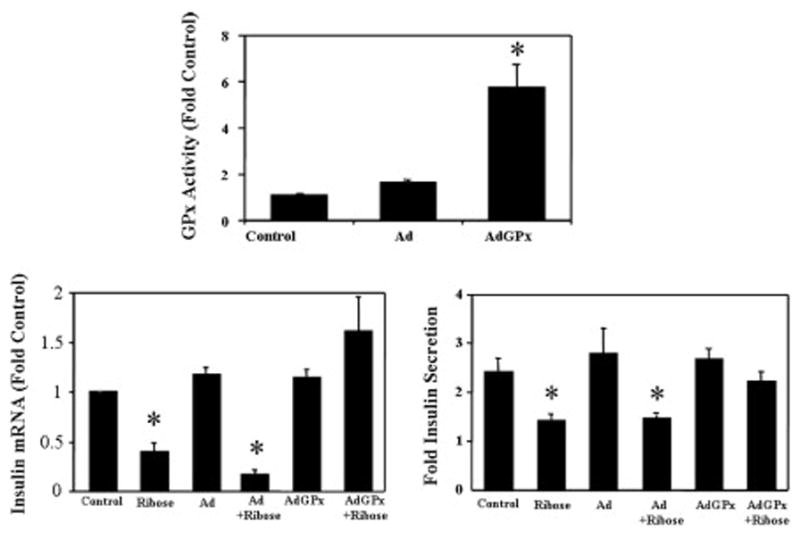
Preventive effects of adenoviral infection of GPx cDNA in isolated islets against the oxidative effects of ribose. Ribose in non-infected cells decreases insulin gene expression and glucose-induced insulin secretion from rat islets in vitro (two lower panels). Adenoviral infection of islets with GPx cDNA increase intrinsic GPx activity in islets 6-fold (top panel) and prevents the adverse effects of ribose on islets (bottom panels). Infection with virus not containing GPx (Ad) has no effects on islets under control conditions or on the adverse effects of ribose on islet function (bottom panels). Taken from Ref. 42
Conclusions
The normal pancreatic islet has intrinsincally low levels of antioxidant enzyme gene expression, protein expression and activities. No explanation is available for this finding, but a possibility is that low levels of ROS facilitate regulation of insulin gene expression and beta cell function. In contrast, excessive concentrations of ROS decrease insulin gene expression and insulin secretion and even damage the islet. High glucose concentrations have been shown to increase intraislet ROS concentrations and clinical measures of oxidative stress are elevated in people with type 2 diabetes mellitus. This raises the question whether prolonged hyperglycemia might create a situation of double jeopardy for the beta cell. In this scenario, the initial genetic cause of type 2 diabetes causes beta cell failure leading to chronic hyperglycemia, which in turn worsens the degree of beta cell failure via further beta cell damage by ROS formed by high glucose concentrations. This hypothesis has been supported by in vitro and in vivo demonstrations that antioxidants protect beta cell lines and rodent models of type 2 diabetes against oxidative stress, thereby preserving insulin gene expression and beta cell function. That GPx overexpression in islets protects beta cells from oxidative stress sets the stage for an assessment of whether transgenic GPx overexpression in beta cells might protect animal models of type 2 diabetes and thereby ameliorate their natural development of hyperglycemia. If so, this will point the way towards the development of GPx mimetics that might be used clinically to provide ancillary protection against the adverse effects of chronic oxidative stress from chronic hyperglycemia in the diabetic patient, who only rarely achieves perfectly controlled glucose levels by conventional anti-diabetic therapy.
Acknowledgments
Supported by NIH grant R01-3832
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Grankvist K, Marklund SL, Taljedal IB. CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochem J. 1981;199:393–398. doi: 10.1042/bj1990393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tiedge M, Lortz S, Drinkgern J, Lenzen S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes. 1997;46:1733–1742. doi: 10.2337/diab.46.11.1733. [DOI] [PubMed] [Google Scholar]
- 3.Tran PO, Parker SM, LeRoy E, Franklin CC, Kavanagh TJ, Zhang T, Zhou H, Vliet P, Oseid E, Harmon JS, et al. Adenoviral overexpression of the glutamylcysteine ligase catalytic subunit protects pancreatic islets against oxidative stress. J Biol Chem. 2004;279:53988–53993. doi: 10.1074/jbc.M404809200. [DOI] [PubMed] [Google Scholar]
- 4.Catherwood MA, Powell LA, Anderson P, McMaster D, Sharpe PC, Trimble ER. Glucose-induced oxidative stress in mesangial cells. Kidney Int. 2002;61:599–608. doi: 10.1046/j.1523-1755.2002.00168.x. [DOI] [PubMed] [Google Scholar]
- 5.Lu SC, Bao Y, Huang ZZ, Sarthy VP, Kannan R. Regulation of gamma-glutamylcysteine synthetase subunit gene expression in retinal Muller cells by oxidative stress. Invest Ophthalmol Vis Sci. 1999;40:1776–1782. [PubMed] [Google Scholar]
- 6.Ghiselli A, Laurenti O, De Mattia G, Maiani G, Ferro-Luzzi A. Salicylate hydroxylation as an early marker of in vivo oxidative stress in diabetic patients. Free Radic Biol Med. 1992;13:621–626. doi: 10.1016/0891-5849(92)90036-g. [DOI] [PubMed] [Google Scholar]
- 7.Gopaul NK, Anggard EE, Mallet AI, Betteridge DJ, Wolff SP, Nourooz-Zadeh J. Plasma 8-epi-PGF2 alpha levels are elevated in individuals with non-insulin dependent diabetes mellitus. FEBS Lett. 1995;368:225–229. doi: 10.1016/0014-5793(95)00649-t. [DOI] [PubMed] [Google Scholar]
- 8.Nourooz-Zadeh J, Tajaddini-Sarmadi J, McCarthy S, Betteridge DJ, Wolff SP. Elevated levels of authentic plasma hydroperoxides in NIDDM. Diabetes. 1995;44:1054–1058. doi: 10.2337/diab.44.9.1054. [DOI] [PubMed] [Google Scholar]
- 9.Rehman A, Nourooz-Zadeh J, Moller W, Tritschler H, Pereira P, Halliwell B. Increased oxidative damage to all DNA bases in patients with type II diabetes mellitus. FEBS Lett. 1999;448:120–122. doi: 10.1016/s0014-5793(99)00339-7. [DOI] [PubMed] [Google Scholar]
- 10.Shin CS, Moon BS, Park KS, Kim SY, Park SJ, Chung MH, Lee HK. Serum 8-hydroxy-guanine levels are increased in diabetic patients. Diabetes Care. 2001;24:733–737. doi: 10.2337/diacare.24.4.733. [DOI] [PubMed] [Google Scholar]
- 11.Sakuaba H, Mizukami H, Yagihashi N, Wada R, Hanyu C, Yagihashi S. Reduced beta-cell mass and expression of oxidative stress related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia. 2002;45:85–96. doi: 10.1007/s125-002-8248-z. [DOI] [PubMed] [Google Scholar]
- 12.Murakami K, Kondo T, Ohtsuka Y, Fujiwara Y, Shimada M, Kawakami Y. Impairment of glutathione metabolism in erythrocytes from patients with diabetes mellitus. Metabolism. 1989;38:753–758. doi: 10.1016/0026-0495(89)90061-9. [DOI] [PubMed] [Google Scholar]
- 13.Sharma A, Kharb S, Chugh SN, Kakkar R, Singh GP. Evaluation of oxidative stress before and after control of glycemia and after vitamin E supplementation in diabetic patients. Metabolism. 2000;49:160–162. doi: 10.1016/s0026-0495(00)91117-x. [DOI] [PubMed] [Google Scholar]
- 14.Yoshida K, Hirokawa J, Tagami S, Kawakami Y, Urata Y, Kondo T. Weakened cellular scavenging activity against oxidative stress in diabetes mellitus: regulation of glutathione synthesis and efflux. Diabetologia. 1995;38:201–210. doi: 10.1007/BF00400095. [DOI] [PubMed] [Google Scholar]
- 15.Tokuyama Y, Sturis J, DePaoli AM, Takeda J, Stoffel M, Tang J, Sun X, Polonsky KS, Bell GI. Evolution of beta-cell dysfunction in the male Zucker diabetic fatty rat. Diabetes. 1995;44:1447–1457. doi: 10.2337/diab.44.12.1447. [DOI] [PubMed] [Google Scholar]
- 16.Sreenan S, Sturis J, Pugh W, Burant CF, Polonsky KS. Prevention of hyperglycemia in the Zucker diabetic fatty rat by treatment with metformin or troglitazone. Am J Physiol. 1996;271:E742–747. doi: 10.1152/ajpendo.1996.271.4.E742. [DOI] [PubMed] [Google Scholar]
- 17.Ohtani KI, Shimizu H, Sato N, Mori M. Troglitazone (CS-045) inhibits beta-cell proliferation rate following stimulation of insulin secretion in HIT-T 15 cells. Endocrinology. 1998;139:172–178. doi: 10.1210/endo.139.1.5670. [DOI] [PubMed] [Google Scholar]
- 18.Pick A, Clark J, Kubstrup C, Levisetti M, Pugh W, Bonner-Weir S, Polonsky KS. Role of apoptosis in failure of beta-cell mass compensation for insulin resistance and beta-cell defects in the male Zucker diabetic fatty rat. Diabetes. 1998;47:358–364. doi: 10.2337/diabetes.47.3.358. [DOI] [PubMed] [Google Scholar]
- 19.Wang MY, Koyama K, Shimabukuro M, Mangelsdorf D, Newgard CB, Unger RH. Overexpression of leptin receptors in pancreatic islets of Zucker diabetic fatty rats restores GLUT-2, glucokinase, and glucose-stimulated insulin secretion. Proc Natl Acad Sci U S A. 1998;95:11921–11926. doi: 10.1073/pnas.95.20.11921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seufert J, Weir GC, Habener JF. Differential expression of the insulin gene transcriptional repressor CCAAT/enhancer-binding protein beta and transactivator islet duodenum homeobox-1 in rat pancreatic beta cells during the development of diabetes mellitus. J Clin Invest. 1998;101:2528–2539. doi: 10.1172/JCI2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harmon JS, Gleason CE, Tanaka Y, Oseid EA, Hunter-Berger KK, Robertson RP. In vivo prevention of hyperglycemia also prevents glucotoxic effects on PDX-1 and insulin gene expression. Diabetes. 1999;48:1995–2000. doi: 10.2337/diabetes.48.10.1995. [DOI] [PubMed] [Google Scholar]
- 22.Zhou YT, Shimabukuro M, Lee Y, Koyama K, Higa M, Ferguson T, Unger RH. Enhanced de novo lipogenesis in the leptin-unresponsive pancreatic islets of prediabetic Zucker diabetic fatty rats: role in the pathogenesis of lipotoxic diabetes. Diabetes. 1998;47:1904–1908. doi: 10.2337/diabetes.47.12.1904. [DOI] [PubMed] [Google Scholar]
- 23.Gremlich SBC, Waeber G, et al. Fatty acids decrease IDX-1 expression in rat pancreatic islets and reduce GLUT2, glucokinase, insulin, and somatostatin levels. J Biol Chem. 1997;272:30261–30269. doi: 10.1074/jbc.272.48.30261. [DOI] [PubMed] [Google Scholar]
- 24.Shimabukuro M, Zhou YT, Lee Y, Unger RH. Troglitazone lowers islet fat and restores beta cell function of Zucker diabetic fatty rats. J Biol Chem. 1998;273:3547–3550. doi: 10.1074/jbc.273.6.3547. [DOI] [PubMed] [Google Scholar]
- 25.Harmon JS, Gleason CE, Tanaka Y, Poitout V, Robertson RP. Antecedent hyperglycemia, not hyperlipidemia, is associated with increased islet triacylglycerol content and decreased insulin gene mRNA level in Zucker diabetic fatty rats. Diabetes. 2001;50:2481–2486. doi: 10.2337/diabetes.50.11.2481. [DOI] [PubMed] [Google Scholar]
- 26.Jacqueminet S, Briaud I, Rouault C, Reach G, Poitout V. Inhibition of insulin gene expression by long-term exposure of pancreatic beta cells to palmitate is dependent on the presence of a stimulatory glucose concentration. Metabolism. 2000;49:532–536. doi: 10.1016/s0026-0495(00)80021-9. [DOI] [PubMed] [Google Scholar]
- 27.Briaud I, Harmon JS, Kelpe CL, Segu VB, Poitout V. Lipotoxicity of the pancreatic beta-cell is associated with glucose-dependent esterification of fatty acids into neutral lipids. Diabetes. 2001;50:315–321. doi: 10.2337/diabetes.50.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kelpe CL, Johnson LM, Poitout V. Increasing triglyceride synthesis inhibits glucose-induced insulin secretion in isolated rat islets of langerhans: a study using adenoviral expression of diacylglycerol acyltransferase. Endocrinology. 2002;143:3326–3332. doi: 10.1210/en.2002-220402. [DOI] [PubMed] [Google Scholar]
- 29.Briaud I, Kelpe CL, Johnson LM, Tran PO, Poitout V. Differential effects of hyperlipidemia on insulin secretion in islets of langerhans from hyperglycemic versus normoglycemic rats. Diabetes. 2002;51:662–668. doi: 10.2337/diabetes.51.3.662. [DOI] [PubMed] [Google Scholar]
- 30.Poitout V, Robertson RP. Minireview: Secondary beta-cell failure in type 2 diabetes--a convergence of glucotoxicity and lipotoxicity. Endocrinology. 2002;143:339–342. doi: 10.1210/endo.143.2.8623. [DOI] [PubMed] [Google Scholar]
- 31.Wolff SP, Dean RT. Glucose autoxidation and protein modification. The potential role of ‘autoxidative glycosylation’ in diabetes. Biochem J. 1987;245:243–250. doi: 10.1042/bj2450243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanaka Y, Gleason CE, Tran PO, Harmon JS, Robertson RP. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc Natl Acad Sci U S A. 1999;96:10857–10862. doi: 10.1073/pnas.96.19.10857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robertson RP, Zhang HJ, Pyzdrowski KL, Walseth TF. Preservation of insulin mRNA levels and insulin secretion in HIT cells by avoidance of chronic exposure to high glucose concentrations. J Clin Invest. 1992;90:320–325. doi: 10.1172/JCI115865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olson LK, Redmon JB, Towle HC, Robertson RP. Chronic exposure of HIT cells to high glucose concentrations paradoxically decreases insulin gene transcription and alters binding of insulin gene regulatory protein. J Clin Invest. 1993;92:514–519. doi: 10.1172/JCI116596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olbrot M, Rud J, Moss LG, Sharma A. Identification of beta-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc Natl Acad Sci U S A. 2002;99:6737–6742. doi: 10.1073/pnas.102168499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kataoka K, Han SI, Shioda S, Hirai M, Nishizawa M, Handa H. MafA is a glucose-regulated and pancreatic beta-cell-specific transcriptional activator for the insulin gene. J Biol Chem. 2002;277:49903–49910. doi: 10.1074/jbc.M206796200. [DOI] [PubMed] [Google Scholar]
- 37.Matsuoka TA, Zhao L, Artner I, Jarrett HW, Friedman D, Means A, Stein R. Members of the large Maf transcription family regulate insulin gene transcription in islet beta cells. Mol Cell Biol. 2003;23:6049–6062. doi: 10.1128/MCB.23.17.6049-6062.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharma A, Olson LK, Robertson RP, Stein R. The reduction of insulin gene transcription in HIT-T15 beta cells chronically exposed to high glucose concentration is associated with the loss of RIPE3b1 and STF-1 transcription factor expression. Mol Endocrinol. 1995;9:1127–1134. doi: 10.1210/mend.9.9.7491105. [DOI] [PubMed] [Google Scholar]
- 39.Harmon JS, Tanaka Y, Olson LK, Robertson RP. Reconstitution of glucotoxic HIT-T15 cells with somatostatin transcription factor-1 partially restores insulin promoter activity. Diabetes. 1998;47:900–904. doi: 10.2337/diabetes.47.6.900. [DOI] [PubMed] [Google Scholar]
- 40.Harmon JS, Stein R, Robertson RP. Oxidative stress-mediated, post-translational loss of MafA protein as a contributing mechanism to loss of insulin gene expression in glucotoxic beta cells. J Biol Chem. 2005;280:11107–11113. doi: 10.1074/jbc.M410345200. [DOI] [PubMed] [Google Scholar]
- 41.Briaud I, Rouault C, Reach G, Poitout V. Long-term exposure of isolated rat islets of Langerhans to supraphysiologic glucose concentrations decreases insulin mRNA levels. Metabolism. 1999;48:319–323. doi: 10.1016/s0026-0495(99)90079-3. [DOI] [PubMed] [Google Scholar]
- 42.Tanaka Y, Tran PO, Harmon J, Robertson RP. A role for glutathione peroxidase in protecting pancreatic beta cells against oxidative stress in a model of glucose toxicity. Proc Natl Acad Sci U S A. 2002;99:12363–12368. doi: 10.1073/pnas.192445199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robertson RP. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J Biol Chem. 2004;279:42351–42354. doi: 10.1074/jbc.R400019200. [DOI] [PubMed] [Google Scholar]