

Abstract
Alterations in chromatin play an important role in oncogenic transformation although the underlying mechanisms are often poorly understood. The SWI/SNF complex contributes to epigenetic regulation by utilizing the energy of ATP hydrolysis to remodel chromatin and thus regulate transcription of target genes. SNF5, a core subunit of the SWI/SNF complex, is a potent tumor suppressor that is specifically inactivated in several types of human cancer. However, the mechanism by which SNF5 mutation leads to cancer and the role of SNF5 within the SWI/SNF complex remains largely unknown. It has been hypothesized that oncogenesis in the absence of SNF5 occurs due to a loss of function of the SWI/SNF complex. Here we show, however, distinct effects for inactivation of Snf5 and the ATPase subunit Brg1 in primary cells. Further, using both human cell lines and mouse models, we show that cancer formation in the absence of SNF5 does not result from SWI/SNF inactivation but rather that oncogenesis is dependent upon continued presence of BRG1. Collectively, our results demonstrate that cancer formation in the absence of SNF5 is dependent upon the activity of the residual BRG1-containing SWI/SNF complex. These findings suggest that, much like the concept of oncogene addiction, targeted inhibition of SWI/SNF ATPase activity may be an effective therapeutic approach for aggressive SNF5 deficient human tumors.
Keywords: SNF5, BRG1, SWI/SNF, tumorigenesis, epigenetics
Introduction
SNF5 (SMARCB1/INI1/BAF47) has potent tumor suppressor activity. Specific inactivating mutations in SNF5 are present in the large majority of malignant rhabdoid tumors (MRTs), highly aggressive cancers that strike young children (1, 2). These tumors have a median age of onset of 11 months and most of children die from the disease within one year of diagonosis, despite the use of intensive therapies. Familial cases occur and arise due to inheritance of a mutant SNF5 allele, a condition termed the “rhabdoid predisposition syndrome” (3). Biallelic inactivating mutations in SNF5 with resultant loss of protein expression also occur in epithelioid sarcomas small cell hepatoblastomas, extraskeletal myxoid chondrosarcomas, and undifferentiated sarcomas (4-7). Loss of SNF5 protein was also recently found to occur in all tested cases of renal medullary carcinoma, possibly due to epigenetic silencing (8). Inheritance of mutations predicted to have a milder effect upon SNF5 function has recently been identified as the basis of familial schwannomatosis (9). Additionally, moderately reduced expression of SNF5, without mutation, is associated with steroid resistant refractory acute lymphoblastic leukemia (10).
In mice, homozygous deletion of Snf5 results in early embryonic lethality while heterozygotes, similar to humans, are predisposed to develop aggressive sarcomas (11-13). Conditional inactivation of Snf5 results in profound cancer susceptibility with all mice developing cancer at a median of 11 weeks (14). The rapidity with which cancer develops is remarkable for inactivation of a single gene. In comparison, p53 loss leads to cancer at 20 weeks, p19Arf deficiency at 38 weeks and p16Ink4a deficiency at 60 weeks. Thus, the 11-week median cancer onset demonstrates a potent role for Snf5 in tumor suppression.
SNF5 is a core member of the SWI/SNF chromatin remodeling complex, which is a conserved modulator of chromatin structure. In vitro, Swi/Snf complexes are capable of disrupting DNA-histone contacts but the complexes have also been implicated in larger scale regulation of chromatin structure in vivo (15). Mammalian SWI/SNF complexes consist of at least nine subunits, including either of two mutually exclusive core ATPases, BRG1 or BRM, invariant core subunits including SNF5, BAF155, and BAF170, as well as a variety of lineage-restricted subunits (16, 17). While the BRG1 or BRM ATPase subunit of the SWI/SNF complex is itself capable of remodeling nucleosomes in vitro (18), the function of SNF5 within the complex and the mechanism by which SNF5 loss contributes to oncogenic transformation remains unclear. In yeast, Snf5 mutants behave similarly to other Swi/Snf mutants and strains carrying mutations in multiple Swi/Snf genes also have similar phenotypes (19), suggesting that inactivation of Snf5 is akin to inactivating the entire complex. In mammals SNF5 is present in stoichiometric amounts and essentially all SNF5 co-purifies with the SWI/SNF complex (20). Further, both Brg1 and Snf5 inactivation in mice lead to early embryonic lethality (11-13, 21). Therefore, it has been hypothesized that oncogenesis in the absence of SNF5 occurs due to loss of function of the SWI/SNF complex. However, this view has been challenged by several findings. First, Snf5 loss leads to effects more frequently associated with oncogene activation than tumor suppressor loss. Loss of function of most tumor suppressors confers either an immediate proliferative advantage or a later selective advantage in primary cells, while Snf5 loss leads to cell cycle arrest and apoptosis (22, 23). These inhibitory effects are more commonly associated with gain of function activation of oncogenes. For example, overexpression of MYC or mutant RAS triggers cell cycle checkpoints and leads to senescence or apoptosis in primary cells (24, 25). Second, while a comprehensive analysis of human tumors looking for BRG1 and BRM mutations has yet to be published, so far mutations of BRG1/BRM or SNF5 are correlated with different tumor spectrums and BRG1/BRM mutations have not been detected in MRTs (14, 21). Third, BRG1 can associate with at least some other SWI/SNF subunits in the absence of SNF5 and some genes thought to be dependent upon BRG1 for expression are expressed in MRT cell lines (26). We therefore hypothesized that SNF5 loss does not equate to inactivation of the SWI/SNF complex but rather results in residual activity of the complex that promotes oncogenic transformation.
Here we have tested the activities of SNF5 within the SWI/SNF complex. We show, in primary mouse embryonic fibroblasts (MEFs) and T cells, that deletion of Brg1 is not redundant to Snf5 loss but instead exacerbates the effects of Snf5 loss. Such synergy between Brg1 loss and Snf5 loss suggests functional activity of the residual Swi/Snf complex in the absence of Snf5. With respect to oncogenesis, if SNF5 loss impaired the function of the complex, it would be predicted that inactivation of BRG1 should either be redundant or even lead to synergistic tumor formation by removing any residual function. In contrast, we find that BRG1 loss is antagonistic to the oncogenesis caused by SNF5 loss. Lastly, we demonstrate that Brg1 is essential for tumor formation caused by Snf5 loss in vivo.
Materials and Methods
Cell culture
Primary MEFs were harvested and cultured as previously described (22). All cell lines have been in the possession of the Roberts laboratory for several years. All MRT cell lines have been validated as SNF5 deficient and the SW13 lines as BRG1/BRM deficient by use of immunoblots (Figure 4).
Figure 4. Brg1, not Brm, is present and binds to other Swi/Snf components in Snf5 deficient tumors.
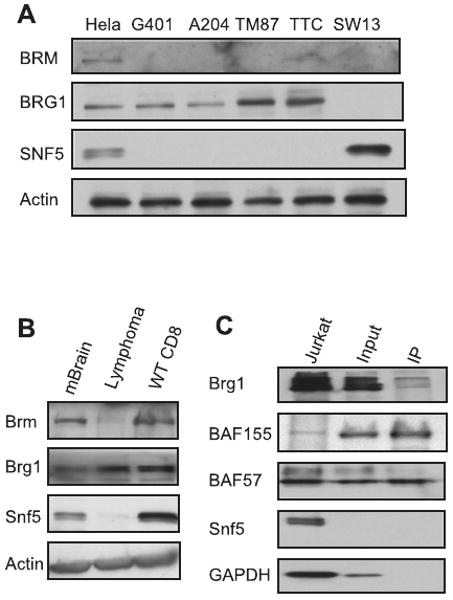
(A) Western analysis of BRG1, BRM and SNF5 in human MRT cell lines. Hela cells were used as a positive control for expression. (B) Western analysis of Brg1, Brm and a Snf5 in Snf5-deficient murine lymphoma. CD8+ T cells and murine brain extracts were used as positive controls. Brg1 is present while Brm is not. (C) Brg1 binds to other Swi/Snf components in Snf5 deficient lymphomas. Whole cell extracts from murine Snf5 deficient lymphomas were subjected to Brg1 IP followed by immunoblotting. Jurkat cells were used as positive controls.
Antibodies used for western blot
Anti-Snf5 antibody was purchased from BD Biosciences, anti-Brg1, BAF155, and BAF170 antibodies were purchased from Santa Cruz, anti-Brm and anti-Actin antibodies were purchased from Abcam.
FACS analysis of T cells
Fluorochrome-conjugated mAbs against B220 (2D1), CD3ε (145-2C11), CD4 (RM4-5), and CD8α (53-6.7) were purchased from BD Pharmingen. Data was collected using a Facscalibur cytometer (Becton-Dickinson) and analyzed with FlowJo software (Tree Star, Ashland, OR).
RNAi and siRNA
The RNAi construct against BRG1/BRM and the control virus were obtained from Stephen Smale (UCLA). The siRNA reagents were ordered from Dharmacon and the sequences are available upon request.
RNA purification and RT-PCR
Total RNA was extracted using Trizol reagent (Invitrogen) and reverse transcribed by the Reverse Transcription System (Promega). Gene expression was normalized to RPS8. Error bars in RNA analysis represent standard deviations of mean expression or fold changes based on at least three independent RNA isolations, or as indicated in figure legends. Primer sequences are available upon request.
Results
Residual Swi/Snf activity in the absence of Snf5
We first asked whether loss of Snf5 equates to inactivation of the Swi/Snf complex in primary cells. All Swi/Snf complexes contain one of two mutually exclusive ATPases: Brg1 or Brm. Brg1 is expressed in proliferating cells and is essential for mouse development while Brm tends to be expressed in differentiated cells and is dispensable for mouse development and survival (21, 27). We evaluated the effects of Snf5 deletion, Brg1 deletion or deletion of both in conditionally targeted primary MEFs (Figure 1A). Loss of either Snf5 or Brg1 was detrimental to proliferation of MEFs (Figure 1B). Both Snf5 deficient and Brg1deficient cells stopped proliferating beginning five days after deletion. Importantly, following several days of arrest, Brg1deficient cells ultimately re-entered the cell cycle and proliferated well (Figure 1C and Supplemental Figure 1). In contrast, cells deficient in Snf5 never re-entered the cell cycle. Although we observed occasional colonies growing on the Snf5fl/fl + Cre plates after several weeks, in all cases these colonies were derived from cells that had escaped deletion and still expressed Snf5 (data not shown). Differences between Snf5 loss and Brg1 loss were also detectable at the gene expression level. We examined expression of a panel of 6 randomly selected genes that had been previously used as a validation cohort for genes up-regulated following Snf5 inactivation in MEFs (22). While four of the six genes displayed similar changes following loss of either Snf5 or Brg1, the other two did not. In particular, Fmod was up-regulated 12-fold following Snf5 loss but down regulated 5-fold following Brg1 loss (Figure 1D).
Figure 1. Inactivation of BRG1 and SNF5 have distinct effects upon cell survival and gene expression.
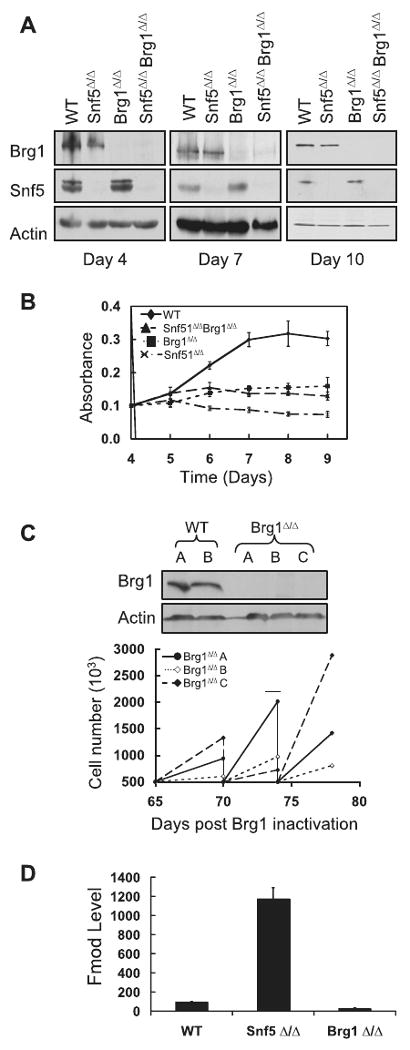
(A) Immunoblots showing that Snf5 and Brg1 protein are efficiently ablated in Snf5fl/fl or Brg1fl/fl cells exposed to Cre recombinase. (B) Following treatment with Cre, proliferation of wild-type (WT), Snf5-deficient (Snf5Δ/Δ), Brg1-deficient (Brg1Δ/Δ), and Brg/Snf5 doubly-deficient cells (Snf5Δ/Δ Brg1Δ/Δ)were monitored. While cells lacking either Brg1 or Snf5 alone underwent growth arrest, combined loss of both Brg1 and Snf5 led to rapid cell death. (C) Primary MEFs can survive and proliferate in the absence of Brg1. MEFs isolated from three Brg1fl/fl embryos (A, B, C) were exposed to a retrovirus expressing Cre-recombinase as in panel A and then maintained in selection. MEFs continue to stably proliferate in the absence of Brg1. To establish that MEFs continue to stably proliferate long-term in the absence of Brg1, MTT assays were performed on Brg1-deficinet and control MEFs. Representative proliferation data, from the day 65 to 78 time frame, is shown including serial re-plating on days 70 and 74. Immunoblots of Brg1 and actin at day 80 post infection is shown to confirm Brg1 absence. (D) Expression of Fmod reveals differences in Snf5 loss compared to Brg1 loss. Following Adeno-Cre treatment of conditional MEFs, real-time PCR reveals that Fmod expression is increased in Snf5 deficient MEFs but decreased in Brg1 deficient MEFs. Data represents level of expression of Fmod normalized to Actin message in three independent experiments ± S.E.
To investigate the mechanism underlying the differential effect of Snf5 and Brg1 inactivation upon proliferation, we examined Brm, the other Swi/Snf ATPase. The proliferation of Brg1 deficient MEFs was accompanied by up-regulation of Brm, suggesting that Brm can partially compensate for Brg1 loss (Figure 2A). Consistent with this observation, MEFs in which Brg1 and Brm were simultaneously inhibited by RNAi were non-viable (Figure 2B and C).
Figure 2. Brm can partially compensate for Brg1 loss in MEFs.
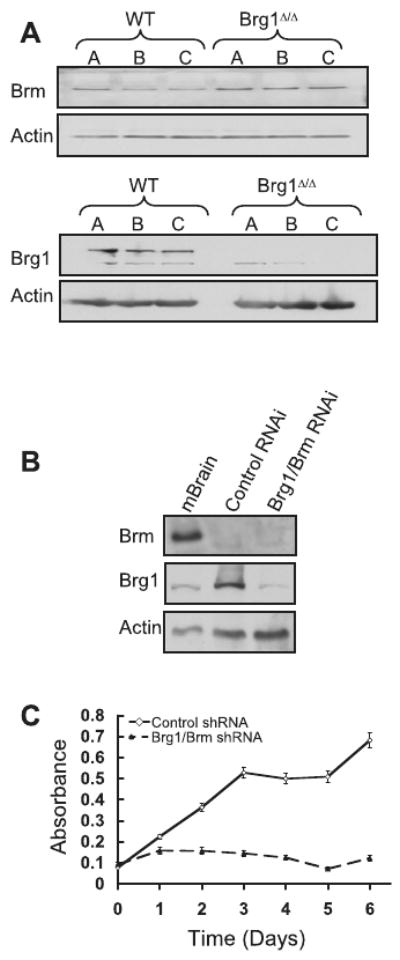
(A) Brm protein levels modestly increase following the knockdown of Brg1 in MEFs. MEFs isolated from three wild-type (WT) and three Brg1fl/fl embryos were exposed to a retro-virus expressing Cre-recombinase and under selection for 40 days. Immunoblots of Brm and Brg1 are shown. (B) MEFs isolated from three WT embryos were infected with either an RNAi-GFP-puror virus against both Brg1/Brm or a control GFP-puror virus. The following day, GFP+ cells were sorted by flow cytometry, and selected in puromycin for 2 days. Immunoblots show that the Brg1/BRM RNAi effectively suppresses Brg1 and Brm expression. Mouse brain was used as a positive staining control. (C) MEFs deficient for both Brg1 and Brm failed to proliferate and died. Graph represents the average of thee independent experiments ± S.E.
Next we further examined whether Brg1 had functional activity in the absence of Snf5, we deleted Brg1 to determine whether this would have any effect upon Snf5-deficient MEFs. Co-inactivation of Brg1 had an immediate negative effect upon Snf5-deficient MEFs and lead to rapid cell death (Figure 1B and 3A). This synergy also occurred in T cells where deletion of Brg1 exacerbated the negative effects of Snf5 loss (Figure 3B).
Figure 3. Co-inactivation of BRG1 and SNF5 affects cell survival and proliferation more severely than loss of either one alone.

(A) Loss of Brg1 exacerbates the negative effects of Snf5 loss upon MEF survival. Graph represents the average survival ± S.E. from six independent experiments. The asterisk indicates statistical significance, p < 0.002. (B) Inactivation of Brg1 in T cells in vivo similarly exacerbates the effects of Snf5 loss. Splenocytes were isolated from CD4-Cre Brg1fl/fl, CD4-Cre Snf5fl/fl, and CD4-Cre Snf5fl/fl, Brg1 fl/fl mice and CD3+ T cells were counted by flow cytometry. The graph represents the average number of cells ± S.E. from four mice of each genotype. The asterisk indicates statistical significance, p < 0.002.
SNF5 deficient tumor cells are BRG1 dependent
We next evaluated whether BRG1 had a role in tumorigenesis. While BRM is absent in both human cancer cell lines and primary murine tumors, BRG1 protein is readily detectable and binds to other SWI/SNF components in the absence of SNF5 (Figure 4). We inactivated BRG1 in human SNF5-deficient MRT cell lines using a retroviral-mediated RNA interference (RNAi) strategy. This approach results in the stable integration and expression of short RNAi hairpins accompanied by expression of a GFP reporter and puromycin resistance. To exclude the possibility that up-regulation of BRM might compensate for the loss of BRG1, we used an RNAi designed to target both BRM and BRG1 simultaneously (28).
The RNAi construct efficiently blocked expression of both BRG1 and BRM proteins (Figure 5A). To evaluate function in the absence of SNF5, we quantified the mRNA levels of two genes known to be dependent upon BRG1 for expression, CSF1 and SPARC (26, 29). Both genes were expressed in the absence of SNF5 and were down-regulated following the inactivation of BRG1 demonstrating that BRG1 retained activity in the absence of SNF5 (Figure 5B). We next evaluated the effects of BRG1 loss upon the proliferation of SNF5 deficient MRT cells and other cell lines. While knockdown of BRG1 had no effect upon cell lines SW13 (BRG1/BRM deficient, SNF5 positive adrenal carcinoma control to rule-out off target effects), HeLa (BRG1/BRM and SNF5 positive cervical cancer), or MDA231 (BRG1/BRM and SNF5 positive breast cancer), knockdown in SNF5-deficient MRT cell lines resulted in reduced proliferation (Figure 5C) accompanied by decreased BrdU incorporation, G1 arrest and cell death (Figure 5D).
Figure 5. Knockdown of BRG1 is lethal to SNF5-deficient human tumor cell lines.
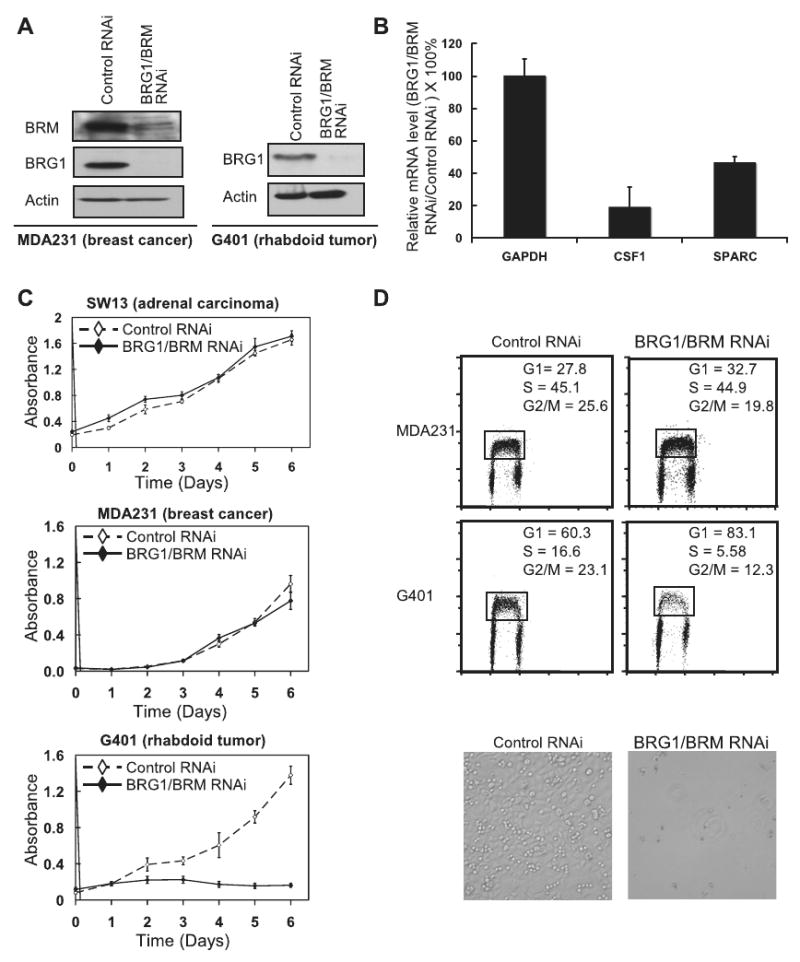
(A) RNAi against BRG1 and BRM leads to stable knockdown of protein expression. Control SNF5 positive MDA321 breast adenocarcinoma cell lines or SNF5-deficient G401 MRT tumor cells lines were infected with a retrovirus containing an RNAi against BRG1/BRM or with a control retrovirus. Knock-down of BRG1 and BRM were confirmed by western following infection. BRM expression is not detectable in these cells (Figure 4). (B) CSF1 and SPARC remain dependent upon BRG1 for expression even in the absence of SNF5. Real-time PCR was used to detect CSF1 and SPARC gene expression following transduction of SNF5-deficient G401 cells with either BRG1/BRM or control RNAi. Data represents level of expression in presence of BRG1/BRM RNAi compared to control RNAi normalized to GADPH message in three independent experiments ± S.E. (C) SNF5 deficient MRT cells are dependent on the presence of BRG1 for cell growth. G401 MRT cells, or SNF5 positive SW13 and MDA231 cells were infected with either BRG1/BRM or control RNAi and monitored for proliferation. Data represent the means of four independent experiments ± SE. (D) Top panel: BrdU cell cycle analysis reveals that loss of Brg1 in SNF5-deficient G401 MRT cells, but not control MDA231 breast cancer cells, leads to decreased S phase (p = 2×10-7) and G2/M (p = 0.001) phases accompanied by a increased G1 phase (p = 2×10-6), indicating a G1 cell cycle arrest. Bottom panel: MRT cells die upon BRG1 knockdown. Photomicrographs following infection with either BRG1/BRM RNAi or control vector and seven days of selection.
To confirm that the growth arrest effects were not due to off-target toxicity, we transduced G401, A204 (another SNF5 deficient MRT cell line), and SW13 with two different synthetic short interfering RNAs (siRNA) against BRG1. Addition of either siRNA led to growth arrest of G401 and A204, but not SW13 cells (Supplemental Figure 2). Thus, inactivation of BRG1 was not redundant with SNF5 loss but instead had specific synergistic effects upon SNF5-deficient cancer cell lines.
Brg1 inactivation prevents the tumor formation following Snf5 loss in mice
To determine whether expression of Brg1 was essential for tumor formation in vivo, we turned to our mouse models. We have previously shown that inactivation of Snf5 in mice using the inducible Mx-Cre transgene leads to the rapid onset of lymphomas or rhabdoid tumors (14). Restricting inactivation of Snf5 to the T cell lineage using the Lck-Cre transgene similarly results in rapid lymphoma onset with complete penetrance4. To investigate whether Brg1 played a role in tumorigenesis caused by Snf5 loss, we bred Brg1fl/fl to Lck-Cre Snf5fl/fl to create three strains: Lck-Cre Snf5fl/fl; Lck-Cre Brg1fl/fl; and Lck-Cre Brg1fl/fl Snf5fl/fl. We aged cohorts of mice to monitor tumor formation. As with our previous report, Lck-Cre Snf5fl/fl mice were cancer prone and 100% developed aggressive lymphoma with an early median onset of only 14 weeks (4 and Figure 6A). In contrast, none of the Lck-Cre Brg1fl/fl mice developed tumors in their two-year lifespan (Figure 6A). This all (Snf5) or none (Brg1) cancer effect was also observed when we used CD4-Cre transgene to induce loss of Snf5 or Brg1 in T cells (Supplemental Figure 3).
Figure 6. Inactivation of Brg1 blocks tumor formation in Snf5-conditional mice.
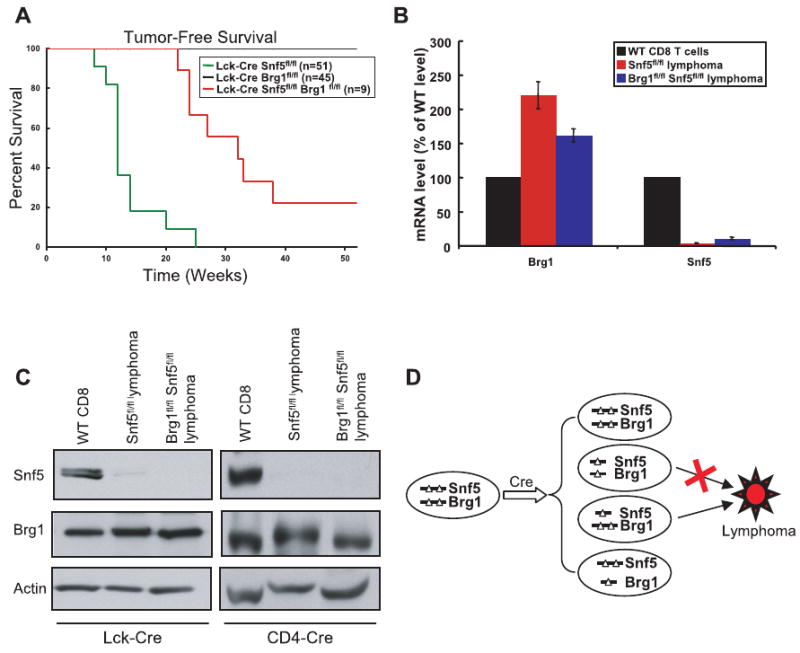
(A) Tumor-free survival curves of Lck-Cre Snf5fl/fl (n=51), Lck-Cre Brg1fl/fl (n=45), and Lck-Cre Snf5fl/fl Brg1fl/fl (n=9) mice. (B) Relative mRNA expression of Brg1 and Snf5 in CD8+ lymphomas obtained from Snf5fl/fl or Snf5fl/fl Brg1fl/fl mice compared to expression in WT CD8+ T cells. While Snf5 expression is gone in the lymphomas, Brg1 is maintained. qPCR data is shown as the mean of two independent experiments ± S.E normalized to RPS8 control message. (C) Immunoblot showing that the lymphomas that ultimately arise in Snf5fl/fl Brg1fl/fl conditional mice are derived from cells in which Snf5 has been deleted but Brg1 retained. (D) A model showing that Cre induction in Lck-Cre or CD4-Cre Snf5fl/fl Brg1fl/fl mice can lead to four possible outcomes: 1) neither Snf5 nor Brg1 get excised, 2) both Snf5 and Brg1 get excised, 3) only Snf5, but not Brg1, gets excised, or 4) only Brg1, but not Snf5, gets excised following Cre induction. Our data reveals that lymphoma can only develop from the rare population of cells in which Snf5, but not Brg1, is excised and inactivated.
To evaluate whether Brg1 loss would synergize, antagonize or have no effect upon oncogenesis in the absence of Snf5, we evaluated tumor formation in the Lck-Cre Brg1fl/fl Snf5fl/fl conditional mice. The presence of the Brg1 conditional alleles markedly impaired tumor formation caused by Snf5 loss. These mice displayed a reduced penetrance (78%) and prolonged onset (27 weeks) compared to the Snf5fl/fl mice (Figure 6A). Strikingly, in the tumors that did develop, Brg1 expression was retained at both the RNA and protein levels (Figure 6B, and left panel of 6C). Thus, these tumors are derived from rare cells that underwent Cre-mediated recombination at the Snf5 locus but not at the Brg1 locus (Figure 6D). This model is further supported by the finding that in addition to retention of Brg1 in the Lck-Cre model, the cancers from Snf5fl/fl Brg1fl/fl mice in both the CD4-Cre and Mx-Cre models have lost Snf5 but retained Brg1. (Figure 6C, right panel and data not shown). Thus, oncogenesis in the absence of SNF5 is dependent upon BRG1 expression.
Discussion
It has become increasingly clear that epigenetic misregulation plays a critical role in oncogenesis. For example, proteins functioning in DNA methylation, histone modifications, and chromatin remodeling have been extensively implicated in tumorigenesis. By utilizing the energy of ATP hydrolysis to remodel chromatin and modulate transcription, the SWI/SNF complex is a key player in the process of epigenetic regulation. SNF5 and the SWI/SNF complex serve particularly potent roles as tumor suppressors as evidenced by the variety of cancers involved, by the extremely rapid cancer onset that occurs following Snf5 inactivation and by the fact that both human and murine Snf5 deficient cancers, despite being highly aggressive, frequently metastatic and rapidly fatal, lack genome instability (30). This latter fact suggests that the epigenetic changes induced by Snf5 loss are extremely potent transforming events and do not require genome instability.
In spite of its potent tumor suppressor activity, the role of SNF5 within the SWI/SNF complex and the mechanism by which SNF5 mutation leads to cancer remain largely unknown. The structure of SNF5 lends little insight into its function. It's most evolutionary conserved feature consists of two imperfect repeat domains, to which protein-protein interactions map, but otherwise lack known function (31). The remainder of the protein also lacks obvious functional domains. Additionally, there are no paralogs of SNF5 in metazoans thus precluding insight from comparison to related genes. Based upon data from yeast in which inactivation of Snf5 results in phenotypes similar to inactivation to the SWI2 ATPase subunit (19) and upon data from mice in which both Brg1 and Snf5 inactivation lead to early embryonic lethality (11-13, 21), it has been hypothesized that the function of SNF5 is integral to SWI/SNF function. However, our findings show that both Snf5-deficient MEFs and T cells are further impaired by Brg1 deletion. In addition, knockdown of BRG1 affects the expression of BRG1-depenent genes even in the absence of SNF5 (Figure 5B). Consequently, our results demonstrate that Brg1 serves important functional roles in the absence of Snf5.
As attempts are made to develop targeted therapies for cancers in which SNF5 is mutated, it has not been clear whether a therapeutic goal should be to replace lost function of the SWI/SNF complex or whether to inhibit aberrant function of the residual complex. Our findings address this question by defining relative roles for SNF5 and BRG1 in oncogenesis. Here we show that knock-down of BRG1 in SNF5 deficient MRT cells leads to cell cycle arrest at G0/G1 followed by cell death. Notably, re-introduction of SNF5 into MRT cells similarly leads to G0/G1 arrest followed by cell death (32-35). Further, our in vivo mouse model revealed that inactivation of Brg1 prevents tumor formation that otherwise occurs in the absence of Snf5. Collectively, these data show that SNF5 deficient cancers are dependent upon BRG1. As BRG1 consistently co-purifies in a large complex with other SWI/SNF subunits and still interacts with most of these subunits in the absence of SNF5 (26), our data indicate that oncongenesis is driven not by loss of SWI/SNF complex function but rather by aberrant residual activity of the BRG1-containing complex.
In considering a model for cancer formation driven by perturbation of the SWI/SNF complex, it is noteworthy that other SWI/SNF subunits have been linked to tumor suppression. While data for SNF5 is most definitive, mutations of BAF180 and BAF250 have been identified in subsets of breast cancers and BRM mutations have been found in non-melanomatous skin cancers (36-38). Specific inactivating mutations in BRG1 itself have been identified in some non-small cell lung, breast and prostate cancer cell lines and from some primary tumors (39-42). Brg1 heterozygous mice are predisposed to a low rate of mammary tumors, entirely distinct from the tumors caused by Snf5 loss, in which the remaining allele of Brg1 is always retained. This latter finding suggests a haploinsufficient mechanism whereby effects on the complex caused by low, but not absent, levels of Brg1 can contribute to tumor formation in a mammary lineage (43). Lastly, altered expression of several SWI/SNF subunits has been shown to convey prognostic significance in a variety of cancers(10, 39, 44, 45). Consequently, disruption of individual subunits may predispose to cancers within specific lineages. A recurrent mechanistic theme involving genes mutated during oncogenic transformation is that many are master regulators of development. It is therefore tempting to speculate that lineage specificity derived from mutation of individual SWI/SNF subunits may be related to the role of the complex in control of lineage specific fate decisions. Via combinatorial incorporation of lineage restricted subunits, several hundred variants of the Swi/Snf complex may exist in mammals (46). Mechanistically, incorporation of these variant subunits enables the complex to control differentiation and to determine cellular fate in numerous lineages (46-50). Therefore, based upon our data and supported by the distinct tumor spectra associated with individual subunits, we propose a model whereby perturbation, rather than loss, of SWI/SNF function is a highly oncogenic event driven by aberrant residual function.
Consistent with this, SNF5 possesses some uncommon features for a tumor suppressor. In general, inactivation of tumor suppressor genes typically leads to either increased proliferation or a latent selective advantage that can be uncovered by subsequent mutations. In contrast, deletion of Snf5 results in cell cycle arrest and apoptosis in most primary cells (22, 23). This effect is similar to gain of function activation of oncogenes such as Myc, Ras and others that, when activated alone in primary cells, typically trigger checkpoints and block proliferation (24, 25). Intriguingly, despite the fact that mutation of Snf5 leads to extremely rapid cancer formation in vivo, the negative effects of Snf5 loss upon MEF growth are profound. Consequently, the marked negative effects of Snf5 loss upon cell proliferation and viability, even within the T cell lineage where Snf5 has potent anti-cancer activity4, are highly unusual for a tumor suppressor but are consistent with aberrant activity of the residual Swi/Snf complex predisposing to oncogenic transformation and triggering cell cycle arrest.
Our results establish that SNF5 loss has marked effects on the role of the SWI/SNF complex in gene regulation (22). SWI/SNF dependent genes can be divided into two groups: SNF5 dependent genes such as P16INK4A and Cyclin D1 (33) and SNF5 independent genes such as CD44 and CSF1 (Figure 3B and (26). Loss of SNF5 therefore causes imbalance between these two groups. BRG1 inactivation in SNF5 deficient cancers additionally disrupts SNF5 independent targets and this may be the mechanism by which it blocks tumor formation.
Accordingly, deregulation of epigenetic transcriptional regulatory activities of the SWI/SNF complex due to SNF5 loss contributes to cancer initiation. This epigenetic effect may be equivalent to multiple genetic mutations and undergo similar selective processes during tumorigenesis. In this model, SNF5 loss may result in defective regulation of nucleosome positioning and thus give rise to “epigenetic instability” in contrast to genetic instability. Oncogenic clonal selection, driven by the epigenetic state rather than genetic state of a cell, may then explain the emergence of malignant cells from the otherwise widespread death caused by SNF5 loss in normal cells. This situation, dependence of SNF5-deficient tumors upon the aberrant activity of the residual SWI/SNF complex, an effect not seen in primary MEFs, is in many ways analogous to the phenomenon of oncogene addiction. Our data suggest that inhibition of residual SWI/SNF activity, perhaps via small molecule inhibition of the BRG1 ATPase, may have therapeutic benefit for SNF5 mutant cancers.
Supplementary Material
Acknowledgments
We thank Dr. Stephen Smale for supplying the BRG1/BRM RNAi constructs; Ms. Beth Beighlie for helps on figures, and Drs. Jerry Workman, Boris Wilson and Jian Xu for for critical discussion. C.W. M. R. was supported by the Garrett B. Smith Foundation, the Claudia Adams Barr Foundation, and PHS award R01CA113794. C.G.S. was supported by PHS awards F32CA123776 and the Hope Street Kids foundation. The authors declare no competing financial interests.
Footnotes
X. Wang, M. Werneck, et al. Manuscript in preparation.
References
- 1.Biegel JA, Zhou JY, Rorke LB, Stenstrom C, Wainwright LM, Fogelgren B. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 1999;59:74–9. [PubMed] [Google Scholar]
- 2.Versteege I, Sevenet N, Lange J, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203–6. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- 3.Taylor MD, Gokgoz N, Andrulis IL, Mainprize TG, Drake JM, Rutka JT. Familial posterior fossa brain tumors of infancy secondary to germline mutation of the hSNF5 gene. Am J Hum Genet. 2000;66:1403–6. doi: 10.1086/302833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kohashi K, Oda Y, Yamamoto H, et al. SMARCB1/INI1 protein expression in round cell soft tissue sarcomas associated with chromosomal translocations involving EWS: a special reference to SMARCB1/INI1 negative variant extraskeletal myxoid chondrosarcoma. Am J Surg Pathol. 2008;32:1168–74. doi: 10.1097/PAS.0b013e318161781a. [DOI] [PubMed] [Google Scholar]
- 5.Modena P, Lualdi E, Facchinetti F, et al. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res. 2005;65:4012–9. doi: 10.1158/0008-5472.CAN-04-3050. [DOI] [PubMed] [Google Scholar]
- 6.Kreiger PA, Judkins AR, Russo PA, et al. Loss of INI1 expression defines a unique subset of pediatric undifferentiated soft tissue sarcomas. Mod Pathol. 2009;22:142–50. doi: 10.1038/modpathol.2008.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trobaugh-Lotrario AD, Tomlinson GE, Finegold MJ, Gore L, Feusner JH. Small cell undifferentiated variant of hepatoblastoma: adverse clinical and molecular features similar to rhabdoid tumors. Pediatr Blood Cancer. 2009;52:328–34. doi: 10.1002/pbc.21834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng JX, Tretiakova M, Gong C, Mandal S, Krausz T, Taxy JB. Renal medullary carcinoma: rhabdoid features and the absence of INI1 expression as markers of aggressive behavior. Mod Pathol. 2008;21:647–52. doi: 10.1038/modpathol.2008.44. [DOI] [PubMed] [Google Scholar]
- 9.Hulsebos TJ, Plomp AS, Wolterman RA, Robanus-Maandag EC, Baas F, Wesseling P. Germline mutation of INI1/SMARCB1 in familial schwannomatosis. Am J Hum Genet. 2007;80:805–10. doi: 10.1086/513207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holleman A, Cheok MH, den Boer ML, et al. Gene-expression patterns in drug-resistant acute lymphoblastic leukemia cells and response to treatment. N Engl J Med. 2004;351:533–42. doi: 10.1056/NEJMoa033513. [DOI] [PubMed] [Google Scholar]
- 11.Guidi CJ, Sands AT, Zambrowicz BP, et al. Disruption of Ini1 leads to peri-implantation lethality and tumorigenesis in mice. Mol Cell Biol. 2001;21:3598–603. doi: 10.1128/MCB.21.10.3598-3603.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klochendler-Yeivin A, Fiette L, Barra J, Muchardt C, Babinet C, Yaniv M. The murine SNF5/INI1 chromatin remodeling factor is essential for embryonic development and tumor suppression. EMBO Rep. 2000;1:500–6. doi: 10.1093/embo-reports/kvd129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roberts CW, Galusha SA, McMenamin ME, Fletcher CD, Orkin SH. Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc Natl Acad Sci USA. 2000;97:13796–800. doi: 10.1073/pnas.250492697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roberts CW, Leroux MM, Fleming MD, Orkin SH. Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer Cell. 2002;2:415–25. doi: 10.1016/s1535-6108(02)00185-x. [DOI] [PubMed] [Google Scholar]
- 15.Bazett-Jones DP, Cote J, Landel CC, Peterson CL, Workman JL. The SWI/SNF complex creates loop domains in DNA and polynucleosome arrays and can disrupt DNA-histone contacts within these domains. Mol Cell Biol. 1999;19:1470–8. doi: 10.1128/mcb.19.2.1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roberts CW, Orkin SH. The SWI/SNF complex--chromatin and cancer. Nature Rev Cancer. 2004;4:133–42. doi: 10.1038/nrc1273. [DOI] [PubMed] [Google Scholar]
- 17.Workman JL, Kingston RE. Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annu Rev Biochem. 1998;67:545–79. doi: 10.1146/annurev.biochem.67.1.545. [DOI] [PubMed] [Google Scholar]
- 18.Phelan ML, Sif S, Narlikar GJ, Kingston RE. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol Cell. 1999;3:247–53. doi: 10.1016/s1097-2765(00)80315-9. [DOI] [PubMed] [Google Scholar]
- 19.Winston F, Carlson M. Yeast SNF/SWI transcriptional activators and the SPT/SIN chromatin connection. Trends Genet. 1992;8:387–91. doi: 10.1016/0168-9525(92)90300-s. [DOI] [PubMed] [Google Scholar]
- 20.Wang W, Xue Y, Zhou S, Kuo A, Cairns BR, Crabtree GR. Diversity and specialization of mammalian SWI/SNF complexes. Genes Dev. 1996;10:2117–30. doi: 10.1101/gad.10.17.2117. [DOI] [PubMed] [Google Scholar]
- 21.Bultman S, Gebuhr T, Yee D, et al. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol Cell. 2000;6:1287–95. doi: 10.1016/s1097-2765(00)00127-1. [DOI] [PubMed] [Google Scholar]
- 22.Isakoff MS, Sansam CG, Tamayo P, et al. Inactivation of the Snf5 tumor suppressor stimulates cell cycle progression and cooperates with p53 loss in oncogenic transformation. Proc Natl Acad Sci USA. 2005;102:17745–50. doi: 10.1073/pnas.0509014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klochendler-Yeivin A, Picarsky E, Yaniv M. Increased DNA damage sensitivity and apoptosis in cells lacking the Snf5/Ini1 subunit of the SWI/SNF chromatin remodeling complex. Mol Cell Biol. 2006;26:2661–74. doi: 10.1128/MCB.26.7.2661-2674.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 25.Zindy F, Eischen CM, Randle DH, et al. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998;12:2424–33. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doan DN, Veal TM, Yan Z, Wang W, Jones SN, Imbalzano AN. Loss of the INI1 tumor suppressor does not impair the expression of multiple BRG1-dependent genes or the assembly of SWI/SNF enzymes. Oncogene. 2004;23:3462–73. doi: 10.1038/sj.onc.1207472. [DOI] [PubMed] [Google Scholar]
- 27.Reyes JC, Barra J, Muchardt C, Camus A, Babinet C, Yaniv M. Altered control of cellular proliferation in the absence of mammalian brahma (SNF2alpha) Embo J. 1998;17:6979–91. doi: 10.1093/emboj/17.23.6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramirez-Carrozzi VR, Nazarian AA, Li CC, et al. Selective and antagonistic functions of SWI/SNF and Mi-2beta nucleosome remodeling complexes during an inflammatory response. Genes Dev. 2006;20:282–96. doi: 10.1101/gad.1383206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu R, Liu H, Chen X, Kirby M, Brown PO, Zhao K. Regulation of CSF1 promoter by the SWI/SNF-like BAF complex. Cell. 2001;106:309–18. doi: 10.1016/s0092-8674(01)00446-9. [DOI] [PubMed] [Google Scholar]
- 30.McKenna ES, Sansam CG, Cho YJ, et al. Loss of the epigenetic tumor suppressor SNF5 leads to cancer without genomic instability. Mol Cell Biol. 2008;28:6223–33. doi: 10.1128/MCB.00658-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morozov A, Yung E, Kalpana GV. Structure-function analysis of integrase interactor 1/hSNF5L1 reveals differential properties of two repeat motifs present in the highly conserved region. Proc Natl Acad Sci USA. 1998;95:1120–5. doi: 10.1073/pnas.95.3.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Betz BL, Strobeck MW, Reisman DN, Knudsen ES, Weissman BE. Re-expression of hSNF5/INI1/BAF47 in pediatric tumor cells leads to G1 arrest associated with induction of p16ink4a and activation of RB. Oncogene. 2002;21:5193–203. doi: 10.1038/sj.onc.1205706. [DOI] [PubMed] [Google Scholar]
- 33.Oruetxebarria I, Venturini F, Kekarainen T, et al. P16INK4a is required for hSNF5 chromatin remodeler-induced cellular senescence in malignant rhabdoid tumor cells. J Biol Chem. 2004;279:3807–16. doi: 10.1074/jbc.M309333200. [DOI] [PubMed] [Google Scholar]
- 34.Versteege I, Medjkane S, Rouillard D, Delattre O. A key role of the hSNF5/INI1 tumour suppressor in the control of the G1-S transition of the cell cycle. Oncogene. 2002;21:6403–12. doi: 10.1038/sj.onc.1205841. [DOI] [PubMed] [Google Scholar]
- 35.Zhang ZK, Davies KP, Allen J, et al. Cell cycle arrest and repression of cyclin D1 transcription by INI1/hSNF5. Mol Cell Biol. 2002;22:5975–88. doi: 10.1128/MCB.22.16.5975-5988.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moloney FJ, Lyons JG, Bock VL, Huang XX, Bugeja MJ, Halliday GM. Hotspot mutation of Brahma in non-melanoma skin cancer. J Invest Dermatol. 2009;129:1012–5. doi: 10.1038/jid.2008.319. [DOI] [PubMed] [Google Scholar]
- 37.Wang X, Nagl NG, Jr, Flowers S, Zweitzig D, Dallas PB, Moran E. Expression of p270 (ARID1A), a component of human SWI/SNF complexes, in human tumors. Int J Cancer. 2004;112:636. doi: 10.1002/ijc.20450. [DOI] [PubMed] [Google Scholar]
- 38.Xia W, Nagase S, Montia AG, et al. BAF180 is a critical regulator of p21 induction and a tumor suppressor mutated in breast cancer. Cancer Res. 2008;68:1667–74. doi: 10.1158/0008-5472.CAN-07-5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reisman DN, Sciarrotta J, Wang W, Funkhouser WK, Weissman BE. Loss of BRG1/BRM in human lung cancer cell lines and primary lung cancers: correlation with poor prognosis. Cancer Res. 2003;63:560–6. [PubMed] [Google Scholar]
- 40.Medina PP, Romero OA, Kohno T, et al. Frequent BRG1/SMARCA4-inactivating mutations in human lung cancer cell lines. Hum Mutat. 2008;29:617–22. doi: 10.1002/humu.20730. [DOI] [PubMed] [Google Scholar]
- 41.Wong AK, Shanahan F, Chen Y, et al. BRG1, a component of the SWI-SNF complex, is mutated in multiple human tumor cell lines. Cancer Res. 2000;60:6171–7. [PubMed] [Google Scholar]
- 42.Decristofaro MF, Betz BL, Rorie CJ, Reisman DN, Wang W, Weissman BE. Characterization of SWI/SNF protein expression in human breast cancer cell lines and other malignancies. J Cell Physiol. 2001;186:136–45. doi: 10.1002/1097-4652(200101)186:1<136::AID-JCP1010>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 43.Bultman SJ, Herschkowitz JI, Godfrey V, et al. Characterization of mammary tumors from Brg1 heterozygous mice. Oncogene. 2008;27:460–8. doi: 10.1038/sj.onc.1210664. [DOI] [PubMed] [Google Scholar]
- 44.Pottier N, Cheok MH, Yang W, et al. Expression of SMARCB1 modulates steroid sensitivity in human lymphoblastoid cells: identification of a promoter SNP that alters PARP1 binding and SMARCB1 expression. Hum Mol Genet. 2007;16:2261–71. doi: 10.1093/hmg/ddm178. [DOI] [PubMed] [Google Scholar]
- 45.Pottier N, Yang W, Assem M, et al. The SWI/SNF chromatin-remodeling complex and glucocorticoid resistance in acute lymphoblastic leukemia. J Natl Cancer Inst. 2008;100:1792–803. doi: 10.1093/jnci/djn416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu JI, Lessard J, Crabtree GR. Understanding the words of chromatin regulation. Cell. 2009;136:200–6. doi: 10.1016/j.cell.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaeser MD, Emerson BM. Remodeling plans for cellular specialization: unique styles for every room. Curr Opin Genet Dev. 2006;16:508–12. doi: 10.1016/j.gde.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 48.Ho L, Ronan JL, Wu J, et al. An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proc Natl Acad Sci USA. 2009;106:5181–6. doi: 10.1073/pnas.0812889106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lessard J, Wu JI, Ranish JA, et al. An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron. 2007;55:201–15. doi: 10.1016/j.neuron.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lickert H, Takeuchi JK, Von Both I, et al. Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature. 2004;432:107–12. doi: 10.1038/nature03071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.