

Abstract
Microphthalmia and anophthalmia are at the severe end of the spectrum of abnormalities in ocular development. Mutations in several genes have been involved in syndromic and non-syndromic anophthalmia. Previously, RAX recessive mutations were implicated in a single patient with right anophthalmia and left microphthalmia and sclerocornea. Here, we report the findings of novel compound heterozygous RAX mutations in a child with bilateral anophthalmia. Both mutations are located in exon 3. c.664delT is a frameshifting deletion predicted to introduce a premature stop codon (p.Ser222ArgfsX62), and c.909 C>G is a nonsense mutation with similar consequences (p.Tyr303X). This is the second report of a patient with anophthalmia caused by RAX mutations. These findings confirm that RAX plays a major role in the early stages of eye development and is involved in human anophthalmia.
Keywords: Anophthalmos; genetics; pathology; Child, Preschool; Cornea; abnormalities; Eye Proteins; genetics; Female; Homeodomain Proteins; genetics; Humans; Molecular Sequence Data; Orbit; abnormalities; Sequence Analysis, DNA; Transcription Factors; genetics
Keywords: anophthalmia, microphthalmia, OAR transactivation domain, RAX
INTRODUCTION
Microphthalmia and anophthalmia are at the severe end of the spectrum of abnormalities in ocular development. The combined occurrence rate for these two malformations is 1/10000 births (1, 2). Mutations in several genes have been isolated in syndromic and non-syndromic anophthalmia. Heterozygous mutations in SOX2 account for approximately 10 % of anophthalmia (3, 4). Other genes have been identified as causing anophthalmia or extreme microphthalmia in humans (PAX6, OTX2, CHX10, STRA6, BMP4) (5, 6). These latter are implicated in a very small proportion of affected individuals, implying wide genetic heterogeneity to match the phenotypic variability.
The RAX homeobox gene is essential for vertebrate eye development. RAX transcription begins in the anterior neural plate, then simultaneously in the eye field and in the ventral forebrain (7). Even before PAX6, its expression is critical to defining the eye field during early development in animal models (8). The lack of RAX expression hampers optic vesicle formation and leads to brain size reduction in mouse, while ectopic expression induces the appearance and proliferation of retinal pigment epithelium cells in Xenopus (9). The function of the RAX gene in eye development is yet not fully understood but there is additional evidence from animal studies that it is involved in the proliferation of neural and retinal cells (10). In humans, the role of RAX in eye formation is clearly supported by the association of anophthalmia and sclerocornea in a patient bearing a truncating mutation and a missense mutation, both located in the DNA-binding helix of the homeodomain and reducing the DNA binding ability of the resulting protein (11). We report here the case of a new patient with bilateral anophthalmia associated with two distinct and novel truncating mutations of the RAX gene.
PATIENT, MATERIALS AND METHODS
Patient
The proband, a 2 year old girl, is the third child born to non-consanguineous, healthy Algerian parents. There was no relevant familial history of ocular malformation or remarkable disease. The pregnancy was uneventful and the prenatal ultrasonography was not suggestive of anomaly. Delivery occurred at 41 weeks of amenorrhea without neonatal difficulties. Birth weight was 3200g. At birth, bilateral small palpebral features were noted without other malformation or dysmorphic features. Anophthalmia was subsequently confirmed. Psychomotor development was within the normal range with head held up at three months, sitting at ten months, walking at 1 year. Speech developed normally. A slight growth defect was recorded at 14 months, with weight at −0.5 DS (9020 g), height at −1 DS (72 cm) and head circumference at −2 DS (44 cm). Abdominal and pelvic ultrasonography detected no visceral anomalies. Orbital and cranial MRI scan showed bilateral absence of eyes with only fibrous tissue in the orbits (Fig. 1). Optic nerves and chiasma were hypoplastic. Extraocular muscles appeared to be relatively preserved. The hypothalamus and pituitary gland were normal. No cerebral malformation was observed.
Figure 1.
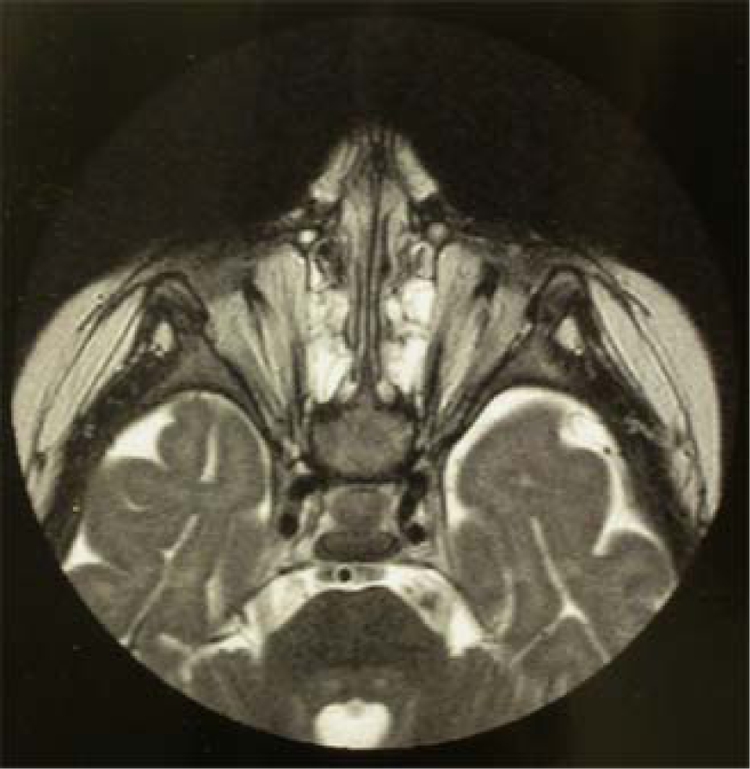
MRI scan of the proband. Note absence of ocular structures replaced by fibrous tissue.
Molecular analysis
Parents gave their informed consent, according to French law, to participate in this study. DNA was isolated by standard procedures from peripheral white blood cells of the proband. Routine examination ruled out rearrangements or point mutations of SOX2 and PAX6 genes. The three RAX exons, with exon-intron borders, were amplified by PCR using previously published primers (11). PCR fragments were subsequently purified with QIAquick Gel Extraction kit (QIAGEN SA France), and both strands sequenced using Big Dye DNA sequencing kit (Applied Biosystems, UK). Reactions were analyzed in an ABI3100 sequencer (Applied Biosystems, UK). Sequence variations were numbered considering adenine of the ATG initiation codon as the first nucleotide (GenBank accession no. NM_013435.2). The changes were verified by performing independent PCR and sequencing reactions on the proband’s DNA.
Exon 3 of the RAX gene was PCR amplified from the patient’s DNA as above (11). The resulting 602 bp fragments were cloned into the pGEM-T vector (Promega, Charbonnières, France). JM109 competent cells were transformed and grown on LB agar plates. DNAs from 10 expanded LacZ-deficient clones were extracted using Promega Wizard miniprep purification system. Further sequencing was performed using the ABI-Big Dye terminator 3.1 on an ABI 3100 sequencer (Applied Biosystems, UK)
RESULTS
Sequence analysis of the proband’s DNA revealed two novel mutations, both located in exon 3 of the RAX gene. c.664delT frameshifting deletion generates a premature stop codon (p.Ser222ArgfsX62). c.909C>G is a nonsense mutation, changing a tyrosine at position 303 to a stop codon (p.Tyr303X). These mutations were not found in a panel of 96 control chromosomes. Both are predicted to lead to a truncated protein so that, if not submitted to nonsense-mediated mRNA decay, the predicted RAX proteins lack the putative OAR (Otp,Aristaless,Rax) transactivation domain and are non functional (7).
As this family left the country, DNA from the proband’s parents was unavailable and thus segregation analysis of these two mutations was impossible. Nevertheless, the c.664delT and the c.909C>G mutations were shown to lie in trans, after sequencing of the cloned products of the patient’s RAX exon 3 (Figure 2).
Figure 2.
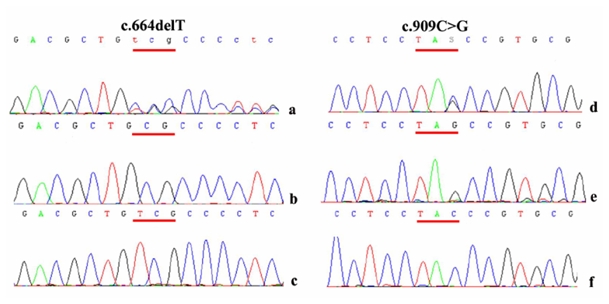
Electropherograms showing the two mutations on RAX exon 3 (a, d) in comparison with wild type sequence (c, f). Sequencing of cloned patient’s exon 3 amplimers in a pGEM-T vector (b, e) demonstrated that mutations were not located on the same alleles.
DISCUSSION
This is the second report of human anophthalmia-associated mutations of the RAX homeobox gene (11). While the parents were not carefully examined, they did not complain of any visual impairment at the time their child was evaluated. The proband was demonstrated to bear composite heterozygous mutations on both alleles of the RAX gene. The parents are thus likely to each be healthy carriers of a heterozygous mutation, unless one of these mutations appeared de novo. This would confirm the recessive inheritance of RAX mutations in ocular dysgenesis.
The phenotype, reported here, consisting in bilateral and symmetric anophthalmia is more severe than the one previously described. This first patient had right anophthalmia and left microphthalmia and sclerocornea (11). One of the causative mutations (p.Gln147X) induced, as predicted for the two mutations reported here, a truncation of the protein. The other was a missense p.Arg192Gln, with a milder effect on the protein, which conserved a low activity. This could suggest that the observed phenotypic variability be correlated with the mutation severity. However, definite conclusions cannot be drawn in view of the limited number of observations.
In animal models, all truncating mutations have been reported to have severe effects and lead to the absence of eye development (9, 12, 13). In contrast, antisense or morpholino inhibition in Xenopus act in a dose-dependant manner, leading to graduated phenotypes ranging from eye reduction to anophthalmia (14). In the present report, the location of the mutations in the last exon makes nonsense-mediated mRNA decay unlikely (15). This is in accordance with the observation that, in the cellular model used by Voronina et al (11), the more proximal p.Gln147X mutation allowed translation of a large amount of protein. These facts suggest that the two mutations we report here lead to truncated proteins, both lacking the C-terminal part containing the critical OAR functional domain (Otp,Aristaless,Rax) (7). Absence of RAX C-terminus is known to abolish it’s proliferative effect in Xenopus (14). Furthermore, regulation of transcriptional activity of several other homeobox genes by the OAR domain has been suggested in other studies (7, 16, 17). Thus p.Ser222ArgfsX62 and p.Tyr303X are thought to drastically impair RAX target genes expression. The precise delineation of the mechanistic effects of these mutations must therefore await binding studies and an important goal for future research will be the identification of the putative genes that can modulate RAX activity through direct interaction.
To date, no cerebral malformation has been associated with RAX mutations in man. This is surprising in the light of the observations in insect, batracian, fish and rodent models, where RAX consistently participates in brain development and homozygous null alleles cause severe cerebral malformations (9, 14, 18, 19). A similar situation is seen, however, with respect to the Hesx1 homeobox-containing transcription factor, which in mice has a similar early role and an overlapping domain to that of Rax but is downstream of Pax6 and Otx2 (20) and Rax itself (21). While Hesx1 mouse mutants can demonstrate anophthalmia in addition to cerebral anomalies, human patients have either isolated pituitary malformations or septo-ocular dysplasia, with no further retinal involvement (22). In a complementary fashion and unlike SOX2 or OTX2 mutations, no extraocular malformations have been observed in RAX ocular dysgenesis patients. The patient reported previously by Voronina et al. (11) was diagnosed as autistic. The patient reported here seems to have normal psychomotor development, although she is too young to exclude the possibility of developmental delay and/or autistic features. Thus, RAX phenotypic spectrum is still unclear, and due to the limited number of cases reported so far, the existence of RAX involvement in syndromic forms of anophthalmia cannot be excluded.
Acknowledgments
Authors are grateful to Dr. IBA ZIZEN for providing MRI scan pictures.
References
- 1.Morrison D, FitzPatrick D, Hanson I, et al. National study of microphthalmia, anophthalmia, and coloboma (MAC) in Scotland: investigation of genetic aetiology. J Med Genet. 2002;39:16–22. doi: 10.1136/jmg.39.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lowry RB, Kohut R, Sibbald B, et al. Anophthalmia and microphthalmia in the Alberta Congenital Anomalies Surveillance System. Can J Ophthalmol. 2005;40:38–44. doi: 10.1016/S0008-4182(05)80115-2. [DOI] [PubMed] [Google Scholar]
- 3.Fantes J, Ragge NK, Lynch SA, et al. Mutations in SOX2 cause anophthalmia. Nat Genet. 2003;33:461–463. doi: 10.1038/ng1120. [DOI] [PubMed] [Google Scholar]
- 4.Ragge NK, Lorenz B, Schneider A, et al. SOX2 anophthalmia syndrome. Am J Med Genet A. 2005;135:1–7. doi: 10.1002/ajmg.a.30642. discussion 8. [DOI] [PubMed] [Google Scholar]
- 5.Bakrania P, Efthymiou M, Klein JC, et al. Mutations in BMP4 cause eye, brain, and digit developmental anomalies: overlap between the BMP4 and hedgehog signaling pathways. Am J Hum Genet. 2008;82:304–319. doi: 10.1016/j.ajhg.2007.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verma AS, Fitzpatrick DR. Anophthalmia and microphthalmia. Orphanet J Rare Dis. 2007;2:47. doi: 10.1186/1750-1172-2-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Furukawa T, Kozak CA, Cepko CL. rax, a novel paired-type homeobox gene, shows expression in the anterior neural fold and developing retina. Proc Natl Acad Sci U S A. 1997;94:3088–3093. doi: 10.1073/pnas.94.7.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang L, Mathers PH, Jamrich M. Function of Rx, but not Pax6, is essential for the formation of retinal progenitor cells in mice. Genesis. 2000;28:135–142. [PubMed] [Google Scholar]
- 9.Mathers PH, Grinberg A, Mahon KA, et al. The Rx homeobox gene is essential for vertebrate eye development. Nature. 1997;387:603–607. doi: 10.1038/42475. [DOI] [PubMed] [Google Scholar]
- 10.Bailey TJ, El-Hodiri H, Zhang L, et al. Regulation of vertebrate eye development by Rx genes. Int J Dev Biol. 2004;48:761–770. doi: 10.1387/ijdb.041878tb. [DOI] [PubMed] [Google Scholar]
- 11.Voronina VA, Kozhemyakina EA, O’Kernick CM, et al. Mutations in the human RAX homeobox gene in a patient with anophthalmia and sclerocornea. Hum Mol Genet. 2004;13:315–322. doi: 10.1093/hmg/ddh025. [DOI] [PubMed] [Google Scholar]
- 12.Tucker P, Laemle L, Munson A, et al. The eyeless mouse mutation (ey1) removes an alternative start codon from the Rx/rax homeobox gene. Genesis. 2001;31:43–53. doi: 10.1002/gene.10003. [DOI] [PubMed] [Google Scholar]
- 13.Loosli F, Staub W, Finger-Baier KC, et al. Loss of eyes in zebrafish caused by mutation of chokh/rx3. EMBO Rep. 2003;4:894–899. doi: 10.1038/sj.embor.embor919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andreazzoli M, Gestri G, Angeloni D, et al. Role of Xrx1 in Xenopus eye and anterior brain development. Development. 1999;126:2451–2460. doi: 10.1242/dev.126.11.2451. [DOI] [PubMed] [Google Scholar]
- 15.Harries LW, Bingham C, Bellanne-Chantelot C, et al. The position of premature termination codons in the hepatocyte nuclear factor -1 beta gene determines susceptibility to nonsense-mediated decay. Hum Genet. 2005;118:214–224. doi: 10.1007/s00439-005-0023-y. [DOI] [PubMed] [Google Scholar]
- 16.Amendt BA, Sutherland LB, Russo AF. Multifunctional role of the Pitx2 homeodomain protein C-terminal tail. Mol Cell Biol. 1999;19:7001–7010. doi: 10.1128/mcb.19.10.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Norris RA, Kern MJ. Identification of domains mediating transcription activation, repression, and inhibition in the paired-related homeobox protein, Prx2 (S8) DNA Cell Biol. 2001;20:89–99. doi: 10.1089/104454901750070292. [DOI] [PubMed] [Google Scholar]
- 18.Eggert T, Hauck B, Hildebrandt N, et al. Isolation of a Drosophila homolog of the vertebrate homeobox gene Rx and its possible role in brain and eye development. Proc Natl Acad Sci U S A. 1998;95:2343–2348. doi: 10.1073/pnas.95.5.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andreazzoli M, Gestri G, Cremisi F, et al. Xrx1 controls proliferation and neurogenesis in Xenopus anterior neural plate. Development. 2003;130:5143–5154. doi: 10.1242/dev.00665. [DOI] [PubMed] [Google Scholar]
- 20.Spieler D, Baumer N, Stebler J, et al. Involvement of Pax6 and Otx2 in the forebrain-specific regulation of the vertebrate homeobox gene ANF/Hesx1. Dev Biol. 2004;269:567–579. doi: 10.1016/j.ydbio.2004.01.044. [DOI] [PubMed] [Google Scholar]
- 21.Martinez-Barbera JP, Rodriguez TA, Beddington RS. The homeobox gene Hesx1 is required in the anterior neural ectoderm for normal forebrain formation. Dev Biol. 2000;223:422–430. doi: 10.1006/dbio.2000.9757. [DOI] [PubMed] [Google Scholar]
- 22.Dattani MT, Martinez-Barbera JP, Thomas PQ, et al. Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nat Genet. 1998;19:125–133. doi: 10.1038/477. [DOI] [PubMed] [Google Scholar]