

Abstract
Many pathogenic bacteria have evolved mechanisms for evading host immune systems. One evasion mechanism is manifest by the surface layer (S-layer), a paracrystalline protein structure composed of S-layer proteins (SLPs). The S-layer, possessed by 2 Campylobacter species (C. fetus and C. rectus), is external to the bacterial outer membrane and can have multiple functions in immune avoidance. C. fetus is a pathogen of ungulates and immunocompromised humans, in whom it causes disseminated bloodstream disease. In C. fetus, the S-layer is required for dissemination and is involved in 2 mechanisms of evasion. First, the S-layer confers resistance to complement-mediated killing in non-immune serum by preventing the binding of complement factor C3b to the C. fetus cell surface. S-layer expressing C. fetus strains remain susceptible to complement-independent killing, utilizing opsonic antibodies directed against the S-layer. However, C. fetus has also evolved a mechanism for avoiding antibody-mediated killing by high-frequency antigenic variation of SLPs. Antigenic variation is accomplished by complex DNA inversion events involving a family of multiple SLP-encoding genes and a single SLP promoter. Inversion events result in the expression of antigenically variant S-layers, which require distinct antibody responses for killing. C. rectus is implicated in the pathogenesis of periodontal disease and also possesses an S-layer that appears to be involved in evading the human system. Although studied less extensively than its C. fetus counterpart, the C. rectus S-layer appears to confer resistance to complement-mediated killing and to cause the down-regulation of proinflammatory cytokines.
Keywords: Campylobacter fetus, Campylobacter rectus, proteins, S-layer, immune systems, antibody formation, inversion events
S-Layers and Immune Evasion
Successful bacterial pathogens must be able to avoid or resist the immune systems of the hosts that they infect. A number of strategies have evolved for immune evasion, including the destruction of immune molecules, expression of antiphagocytic polysaccharide capsules or proteins, regulation of inflammatory mediators such as cytokines, and antigenic variation of immunodominant surface components.1 A specific structure that can result in immune avoidance is the surface layer (S-layer). S-layers are capsule-like protein assemblages present on the surfaces of Gram-positive and Gram-negative bacteria as well as of Archaea. Among Gram-negative bacteria, 3 species (Aeromonas salmonicida, Campylobacter fetus, and Campylobacter rectus) have S-layers that have been characterized as having roles in pathogenesis. This review describes the current state of knowledge of the S-layers of C. fetus and C. rectus and how they allow the bacteria to productively interact with, resist, or evade the immune systems of their hosts.
C. fetus Disease in Animals
C. fetus is a small Gram-negative bacterial species. C. fetus cells are curved, S-shaped, or spiral rods, 0.2 to 0.8 μm in width and 0.5 to 5 μm in length. They are motile by means of a single polar unsheathed flagellum, and require a microaerophilic atmosphere (3% to 5% O2) for growth.
Two subspecies of C. fetus exist, C. fetus subsp. fetus and C. fetus subsp. venerealis, although the spectrum of disease caused by the 2 subspecies is significantly different. C. fetus subsp. venerealis infects the bovine reproductive tract and causes the sexually transmitted disease bovine venereal campylobacteriosis (BVC). BVC results in infertility and is a major problem for the cattle industry.2 A vaccine against BVC is available and results in a strong mucosal and systemic IgG response that both protects against new C. fetus subsp. venerealis infection and eradicates previous infection.3
C. fetus subsp. fetus also infects cattle, as well as sheep and other ungulates, but causes a different type of disease. C. fetus subsp. fetus infection of farm animals results in sporadic abortion but not infertility.4 C. fetus subsp. fetus is spread by ingestion of contaminated food and water rather than by sexual contact. Following ingestion, C. fetus subsp. fetus first colonizes the intestinal tract of the host, followed by a transient bacteremia that can seed extraintestinal sites including the placentas of pregnant animals, resulting in abortion.4
C. fetus Disease in Humans
Infection of humans is generally limited to the single subspecies C. fetus subsp. fetus, hereafter referred to as simply C. fetus. C. fetus disease is uncommon but its frequency is increasing and is certainly under-reported due to the fastidious growth characteristics of C. fetus.5 Disease nearly always occurs in persons with underlying immunocompromising conditions such as human immunodeficiency virus infection or hematological malignancies.5 C. fetus infection of humans is zoonotic and is probably acquired by ingestion of contaminated animal meat or animal products.5 Although most cases of C. fetus disease are sporadic, common source outbreaks have been traced to consumption of raw milk6,7 or raw calf's liver during alternative “nutritional therapy” for malignancies.8 Progression of C. fetus disease in humans is similar to that of C. fetus subsp. fetus infection of animals in that each probably involves primary intestinal colonization followed by dissemination.5 The ultimate manifestations of C. fetus infection in humans are many (including meningitis, pericarditis, cellulitis, and abortion), but each has a preceding systemic component. In fact, bacteremia is the most common detectable form of illness,9-12 and it is primarily the ability of C. fetus to disseminate through the bloodstream that allows it to cause disease.
Early Studies on the C. fetus S-Layer
Much of the early understanding of the C. fetus S-layer can be traced to the work of McCoy et al.13 They characterized a glycine extractable, variable surface antigen, called antigen [a], that was associated with antiphagocytic properties of C. fetus. They discovered that the presence of antigen [a] on the C. fetus cell surface blocked the agglutination of these cells with O antiserum (i.e., decreased the accessibility of the LPS O antigen epitopes required for agglutination) and therefore appeared to cover both LPS and the C. fetus cell surface.13 Next, these authors compared the proficiency with which wild-type C. fetus (strain 23D) and a spontaneous mutant (strain 23B) lacking antigen [a] were phagocytosed by macrophages. In the absence of immune serum, 23D cells were highly resistant to phagocytosis while 23B was internalized efficiently.13 However, in the presence of opsonizing (anti-antigen [a]) antiserum, both 23D and 23B were consumed by macrophages. Therefore, the presence of antigen [a] conferred antiphagocytic properties to C. fetus cells in the absence of specific opsonic antibodies.
One hypothesis for the propensity of C. fetus (relative to other Campylobacters) to cause disseminated disease was that C. fetus cells were resistant to the bactericidal effects of human serum. This was tested by examining the serum resistances of C. fetus, C. jejuni, and C. coli.14 When suspended in non-immune human serum, all C. jejuni and C. coli strains were killed readily by either antibody or complement. In contrast, all C. fetus strains were completely resistant to killing by either complement or antibody.14 C. fetus serum resistance was associated with proteins of apparent molecular masses of 100 to 125 kDa; lab-passaged strains that had spontaneously become serum sensitive had also lost these proteins.15 These proteins were identical to antigen [a] and were the sub-units of the S-layer (S-layer proteins, SLPs).
Further studies on serum resistance focused on the complement system, using wild-type strain 23D (possesses an S-layer; S+) and its spontaneous mutant 23B (lacks an S-layer; S−). These strains were assayed for their abilities to bind and be killed by components of the complement cascade.16 While S− strain 23B bound abundant C3, S+ strain 23D bound little C3 (Fig. 1 A). Furthermore, the small amount of C3 bound by 23D was in a degraded, non-functional form. The end result was a lack of C3b bound to the 23D cell surface, precluding the formation of the C5 convertase and the C5-9 membrane attack complex. Binding of C3b and downstream complement components to 23B resulted in significant killing (Fig. 1B); essentially no serum killing was observed for strain 23D. Therefore, the presence of the S-layer prevents components of the complement system from binding to the cell surface of C. fetus, with a consequent lack of complement-mediated cell lysis. The absence of C3b binding to the cell also precludes phagocytic killing of C. fetus using C3b as an opsonin.16
Figure 1.
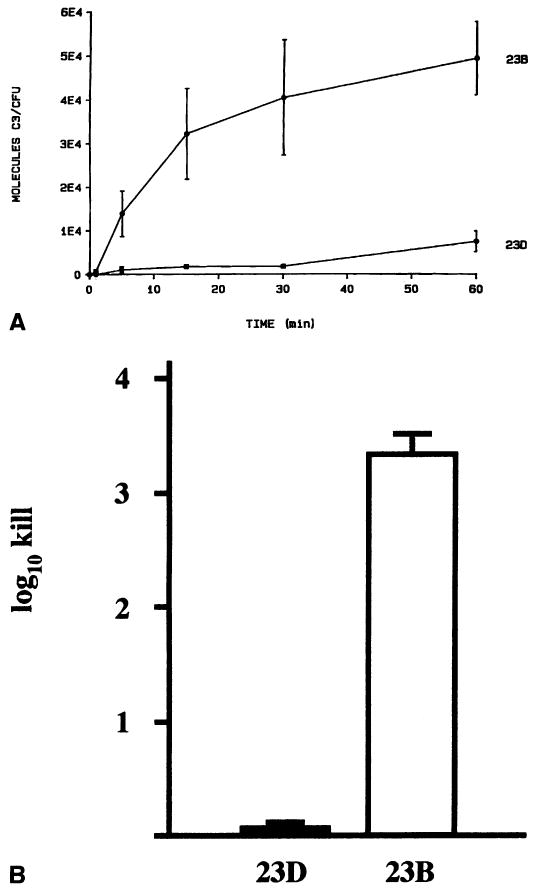
Role of the C. fetus S-layer in resisting complement-mediated killing. A. The C. fetus S-layer prevents the binding of 125I-labeled complement factor C3 in S+ strain 23D, but not in S− strain 23B. Reprinted from reference 16 with permission. B. Lack of complement factor C3b binding provides resistance to serum killing in S+ C. fetus strain 23D, but not in S− strain 23B. Data adapted from reference 47.
Role of the C. fetus S-Layer in Virulence
The lack of serum killing correlated with the disseminating nature of C. fetus infections. The role of the S-layer in virulence was tested in an outbred HA/ICR mouse model of infection, using S+ and S− C. fetus strains inoculated either intraperitoneally or orally.17,18 Intraperitoneal inoculation of C. fetus resulted in death of the mice; however, the mortality due to the S+ strain was significantly higher than that of the S− strain. Oral inoculation with the S+ strain resulted in a high-grade and persistent bacteremia (without death); no bacteremia was observed for the S− strain.18 Passive immunization of the mice with anti-SLP antibodies provided protection against bacteremia with the S+ strain.17,18 Finally, reattachment of SLPs to the S− cells (see below) allowed them to express an S-layer and cause bacteremia in mice.18
The role of the S-layer in infection was subsequently tested in a sheep model,19 in which C. fetus infection can cause spontaneous abortion. Pregnant ewes were inoculated subcutaneously with S+ or S− C. fetus and the outcome on the pregnancy was determined. All of the sheep inoculated with the S+ strain excreted C. fetus in their feces; 10 of 11 fetuses aborted and C. fetus was recovered from the placentas and all fetal tissues.19 In contrast, none of the 7 ewes inoculated with the S− C. fetus strain either excreted C. fetus in their feces or aborted.19 However, if S− C. fetus cells were inoculated intraplacentally, abortion occurred. These results are consistent with the ability of the S-layer to cause dissemination through the bloodstream to systemic sites. Once disseminated, the S-layer is not required to cause abortion in sheep.
In humans, the role of serum killing due to antibody against the C. fetus S-layer was demonstrated in a case of C. fetus infection in a patient with acquired hypogammaglobulinemia.20 The case involved postoperative bacteremia with an S+ C. fetus strain, after which the organism became sequestered in a protected site within the patient's clavicle. Bacteremia did not return until 7 years later, when the patient acquired hypogammaglobulinemia. With the loss of specific anti-SLP antibodies (which can ultimately limit long-term infection by C. fetus), the latent infection with the S+ and serum-resistant C. fetus strain was reactivated, and the complement resistance due to the C. fetus S-layer allowed bacteremia to recur. In vitro experiments confirmed the inability of the patient's serum to kill the S+ C. fetus strain either by opsonizing S-layer antibodies or by complement. Consequently, lifelong antibiotic therapy was instituted in this patient to prevent further occurrence of bacteremia.20
In each of these hosts, C. fetus bacteremia is enabled by expression of its S-layer and its ability to resist serum killing. Protection against bacteremia is associated with the presence of opsonizing S-layer-specific antibody, since complement-dependent pathways are inefficient at killing C. fetus.
Structure of the C. fetus S-Layer
The C. fetus S-layer is composed of a regular monomolecular array of subunit S-layer proteins (SLPs) of various sizes, which are the immunodominant C. fetus antigens.21,22 Early electron micrographic studies showed that the S-layer could assume different appearances (Fig. 2A).21,23,24 The S-layer of C. fetus strain VC119 was tetragonal in appearance and was composed of 131 kDa subunits.21 Strain TK produced both hexagonal and tetragonal S-layers, resulting from the expression of either 97 kDa (hexagonal), or 127 or 149 kDa SLPs (tetragonal) (Fig. 2A).24 In addition to the differing S-layer structures observed when composed of different size SLP subunits, these S-layers have both unique and common antigenic epitopes.15,25-27
Figure 2.

The C. fetus S-layer viewed by electron microscopy. A. A freeze-etch preparation of the C. fetus cell surface allows visualization of the S-layer as either tetragonal (four-sided structures on the left side of the single C. fetus cell shown here) or hexagonal (six-sided structures on the right side of the cell) arrays, and demonstrates that even a single C. fetus cell can express multiple S-layer types. Reprinted from reference 24 with permission. B. An ultra-thin cross-section of C. fetus shows the S-layer as the ring-like structure (labeled “S” and indicated by an arrow) external to the bacterial outer membrane, the dark-staining, ruffled structure in this preparation. Reprinted from reference 21 with permission.
S-layers are attached to bacterial cell surfaces and are typically the outermost structure on these cells (Fig. 2B).28 In some cases the S-layer binds directly to membrane structures, in others binding is to protruding molecules such as lipopolysaccharide (LPS).28 Binding of the S-layer to C. fetus cells was investigated using SLPs that had been removed from the cell surface of S+ C. fetus by water extraction.29 Purified SLPs could be reattached to the surfaces of S− C. fetus only in the presence of divalent cations; neither monovalent nor trivalent cations supported binding. SLPs derived from serotype A C. fetus were able to bind the surfaces only of serotype A C. fetus, while serotype B SLPs bound only serotype B cells. Since the C. fetus serotyping scheme is based on LPS antigenicity, this suggested that, in C. fetus, SLPs bound directly to LPS in a serospecific manner. The LPS binding domain was localized to the amino terminal 50 kDa of the homologous SLPs.29 To determine the portion of LPS to which the SLPs attached, the binding of lectins to glycosyl moieties on C. fetus LPS was studied.30 While several lectins bound the LPS of S− serotype A C. fetus cells, the presence of the S-layer completely prevented lectin binding to S+ cells. This suggested that the S-layer totally masks LPS, and thus may be important in avoiding immune responses to LPS epitopes. Interestingly, although the S-layer appears to completely surround the C. fetus cell, it is not required for adherence of C. fetus to HEp-2 cells.31 Adherence instead appears to be mediated by an uncharacterized factor that remains surface-exposed even in the presence of the S-layer.
Structure of Slp Genes
Antigenic variation of C. fetus surface antigens occurs during infection,25,32,33 and this variation is due to the altered expression of C. fetus SLPs.25,27,34,35 Although antigenic variation occurs in many bacterial pathogens, variation of the C. fetus SLPs has unique aspects. C. fetus strains possess multiple (typically 7 to 9) SLP-encoding genes.36-38 The genetics of SLP-encoding genes and their variation has been best studied in C. fetus serotype A strain 23D, which has 8 genes that encode its SLPs.36 The alleles are designated sapA, sapA1, sapA2, etc. (through sapA7).
Serotype B strains have a parallel and homologous family of SLPs.39 Hybridization studies on 23D chromosomal DNA using probes specific for different parts of the sapA gene revealed diversity within the different alleles.40 Probes derived from the 5′ end of the sapA gene hybridized strongly to all 8 alleles. However, probes for sections of sapA increasingly 3′ in the gene hybridized with progressively fewer bands. This showed that the different sapA genes had both conserved and variable regions. The 5′ ends of each gene were highly similar, yet the remainder of each gene was increasingly divergent. The significant differences in the 3′ ends of the genes are reflected in antigenic differences in the encoded proteins.
To date, the DNA sequences of a total of 5 SLP-encoding genes have been determined.35,39-42 Three of these (sapA, sapA1, and sapA2) are from serotype A strains, and 2 are from serotype B strains (sapB and sapB2). Sequence analysis of the remaining 5 alleles from type A strain 23D (sapA3 through sapA7) is currently in progress. Comparison of the gene sequences reveals several features (Fig. 3). First, each sapA homolog encodes a complete protein, although only some are associated with an active promoter (sapA promoter).36 C. fetus cells have a single promoter that is responsible for expression of sapA genes,36 and its position relative to sapA genes determines which allele is expressed (Figs. 4 and 5). Deletion of this promoter can occur during laboratory passage of C. fetus strains (e.g., in spontaneous S− mutant strain 23B) and results in a strain that is unable to synthesize SLPs, cannot present an S-layer on it surface, and therefore is sensitive to killing by components of the immune system. Second, 626 bp regions encompassing the 5′ ends of each gene are identical. The homology begins 74 bp upstream of the open reading frame (ORF) and extends 552 bp into each ORF. The nucleotide identities within the 5′ end of the ORFs yield SapA proteins with identical amino termini. This 184 amino acid amino-terminal region constitutes the LPS binding domain of the SLPs and is conserved among families of type A and type B SLPs, respectively.39 Third, the 3′ ends of each sapA gene are divergent, sharing only limited nucleotide identity among the various alleles. This is reflected in a large amount of amino acid variability among the predicted sapA proteins (26% to 44% similarity); these differences result in significant antigenic differences in the different sapA proteins. Mapping studies showed that all of the sapA alleles are clustered within a region of the C. fetus chromosome of less than 60 kb.37,40 (Tu et al., unpublished data). These features, along with phylogenetic reconstructions of sapA genes (Tu et al., unpublished data), suggest that the C. fetus sapA gene family originally arose by the introduction of a single sapA progenitor gene into C. fetus. Successive tandem duplications of this gene or its derivatives resulted in the currently existing sapA gene complement. Presumably, positive selection for SLP antigenic variation resulted in antigenic drift of the duplicated sapA genes, mediated by amino acid substitutions, insertions, and/or deletions. In contrast, the highly conserved 5′ ends of these genes were maintained for functional (LPS binding) and mechanistic (SLP variation) purposes (see below).
Figure 3.

Schematic representations of 4 SLP-encoding genes from type A and type B C. fetus strains. The 626 nucleotide 5′ conserved regions of each gene are indicated by large black (type A) or white (type B) rectangles. Stippled or hatched boxes represent the divergent 3′ regions of each type A gene. Solid and broken arrows denote expressed and non-expressed genes, respectively, aa, amino acids. Reprinted from reference 22 with permission.
Figure 4.

Structure and inversion of the 6.2 kb sap invertible region. Bold arrows indicate genes within the sapA invertibie region. Bent arrows represent the divergent sapA and putative sapCDEF promoters. Hatched lines denote the 626 bp 5′ conserved regions of the flanking sapA genes, designated here arbitrarily as sapAx and sapAy.
Figure 5.
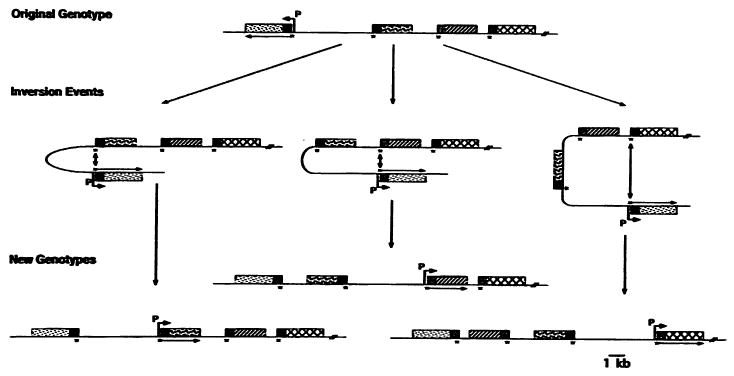
Complex rearrangement of sapA genes. Extensions of the simple inversions shown in Figure 4 allow rearrangement of both the 6.2 kb invertible region and one or more flanking sapA alleles. The black boxes represent the 626 bp 5′ conserved regions of each sapA gene; other types of shading indicate variable regions of each allele. Bent arrows show the locations of the sapA promoter, associated with the expression of the adjacent sapA homolog (straight arrow). Reprinted from reference 43 with permission.
Molecular Mechanism of SLP Variation
C. fetus is able to rapidly vary the expression of its SLPs, resulting in a continual changing of the antigenic type of S-layer expressed on the cell surface. Consequently, the cell is able to avoid or delay a potentially clearing antibody response. SLP antigenic variation is known to occur in vivo during experimental infection of sheep and cattle,19,25 and studies investigating SLP antigenic variation showed that all events resulting in the expression of alternate SLP types were accompanied by genomic rearrangements within the sapA locus.25,35 The genomic rearrangements involved the 626 bp conserved regions at the 5′ of each sapA gene and resulted from DMA inversions, including a 6.2 kb segment of DNA containing the sapA promoter.35,36
Antigenic variation results from high frequency base pairing and homologous recombination between the 626 nucleotide regions of identity at the 5′ ends of pairs of sapA genes. Since the sapA genes are oriented in inverse relation to one another, inversion of the intervening DNA segment occurs (Fig. 4). When this occurs, the sapA promoter is moved to a location upstream of another sapA homolog to promote its expression. Inversion can occur between adjacent sapA genes (Fig. 4), resulting in the reversal of only the minimal invertible region (containing the sapA promoter). However, more complex rearrangements can occur as well, such that the inverting DNA segment also contains one or more sapA genes (Fig. 5). Since the minimal invertible region is 6.2 kb and each of the 8 sapA genes is 3 to 4 kb in length, it is apparent that the size of the inverting DNA can be quite large (perhaps as much as 40 to 50 kb). Furthermore, all of the inversion events appear to occur at roughly similar frequencies (∼10−4), suggesting the extreme efficiency of the inversion system.43
Since these inversions are reciprocal events (unlike gene conversion) mediated by homologous recombination, the original sapA gene remains intact and available for future recombination and expression. Furthermore, C. fetus recA protein is required for efficient homologous recombination, and variation in the expression of sapA genes is undetectable in a C. fetus recA mutant under laboratory conditions.44 However, an unexpected result occurred when a C. fetus recA mutant expressing a 97 kDa SLP was used to experimentally infect sheep.19 One of 16 isolates recovered from the placenta of an infected ewe had switched from the expression of the 97 kDa SLP in the inoculated strain to a 127 kDa SLP, despite the stability of the recA mutation.45 The most likely explanation for this result is that there is strong selection for antigenic variation of C. fetus SLPs during infection, and that there are both recA-dependent (high efficiency) and recA-independent (low efficiency) recombination pathways that can result in sapA recombination. The recA-independent recombination pathway(s) involved in low-frequency sapA rearrangements have yet to be identified. Whichever pathway is involved, multiple crossover points within the 5′ conserved regions of sapA genes can be used during recombination, and no specific sequences within the conserved regions are evident.46
Secretion of SLPs
To ascertain whether genes required for DNA inversion were located within this invertible segment of DNA, the invertible region was cloned from strains of both serotype A (strain 23D) and serotype B (strain 84-107) and their DNA sequences determined.47 In each case, the invertible regions were essentially identical; the invertible region of type A strain 23D is described as follows (Fig. 4). The 74 bp at each end of the 6.2 kb invertible region were inverted repeats of each other, and represented the 5′ ends of the 626 bp conserved DNA boxes that are required for sapA DNA inversion. Adjacent to one of the 74 bp conserved sequences is the outward facing sapA promoter that is responsible for the expression of sap homologs. This promoter is highly active,36,47 as expected due to the high abundance of SLPs required to form the S-layer. The sapA promoter is not known to be regulated by environmental conditions, although due to the extreme energy requirements of SLP synthesis, the production of SLPs is typically tightly regulated.28 The distance between the transcriptional initiation site and the ATG start codons of the sapA genes is 114 bp, longer than the typical bacterial mRNA. This 114 bp untranslated region (UTR) of sapA mRNA is predicted to assume stable secondary structure (ΔG = − 23.4 kcal/mol). These data suggest that this region might be involved in post-transcriptional regulation of sapA expression, although this has not yet been tested experimentally.
The remainder of the 6.2 kb invertible region is composed of a probable operon of 4 genes designated sapCDEF,47 transcribed in a direction opposite that of the sapA promoter (Fig. 4). While the function of the sapC gene has yet to be determined, the sapDEF genes encode proteins that constitute a type I transport apparatus. Such systems have been shown to promote the secretion of a variety of bacterial toxins (hemolysins and leukotoxins), proteases, and other proteins (including SLPs) from Gram-negative bacteria. Type I protein transporters represent a mechanism for the secretion of proteins from the bacterial cytoplasm directly to the extracellular space,47 and are composed of 3 proteins homologous to C. fetus SapDEF that form a pore spanning the cytoplasmic and outer membranes. Proteins are translocated in a single step, without a periplasmic intermediate, using a poorly understood C-terminal secretion signal. To investigate the role of the putative type I secretion apparatus in C. fetus SLP transport, a sapD mutant was contructed. This sapD mutant was unable to transport SLPs to the cell surface and consequently was S− and serum-sensitive.47 As predicted by homology, SapDEF therefore constitute the transporter required for SLP secretion.
Summary: C. fetus S-Layer
The C. fetus S-layer is critical for its virulence, and C. fetus has devoted a significant segment of its chromosome (more than 5%) to the factors required for full expression and function of the S-layer. SLP-encoding genes are clustered in this region and their expression varies by means of complex DNA inversion events involving a single sapA promoter. These events are accomplished primarily by recA-mediated homologous recombination using conserved segments found at the 5′ ends of each sapA gene, although low frequency recA-independent inversions can be detected given the appropriate in vivo selective pressures. A type I secretion system, relatively uncommon for SLPs, is encoded within the sap invertible region, and is responsible for the efficient export of SLPs to the cell surface for assembly into an S-layer. During infection, the S-layer allows C. fetus to disseminate through the bloodstream because of its ability to resist complement-mediated lysis or phagocytosis. Antigenic variation of SLPs delays an opsonic and potentially protective antibody response against C. fetus.
C. Rectus in Periodontal Disease
C. rectus (from the Latin “rectus,” meaning “straight”), despite being closely related to C. fetus, causes an entirely different type of disease. C. rectus has been implicated in the etiology of inflammatory periodontitis.48-55 It is believed that colonization of periodontal tissue by C. rectus (among other organisms such as Porphyromonas gingivalis and Actinobacillus actino-mycetemcomitans) leads to the induction of proinflammatory cytokines such as interleukin (IL)-1 and tumor necrosis factor-(TNF) α. The resulting chronic inflammation results in the destruction of periodontal tissue with subsequent tooth loss. Periodontal disease, therefore, results from a combination of the actions of a pathogen (colonization/infection) and the host (inflammation). Evidence suggests that the C. rectus S-layer plays a role in determining the balance between these 2 opposing responses.
C. rectus S-Layer Genetics
The C. rectus S-layer has a characteristic hexagonal structure similar to many other bacteria.56-58 Two S-layer encoding genes have been characterized from different C. rectus strains, designated as crs and Slp.59,60 crs (strain 314) and slp (strain ATCC 33238) are homologous proteins of 1361 amino acids each (∼145 kDa), with 89% identical amino acids overall. Similarity is especially high in the amino-terminal 552 amino acids of crs and slp (99% identical). The proteins were most similar to SLPs of C. fetus, although the overall similarity was weak (<20% amino acid identity).59,60 Somewhat greater similarity to C. fetus SLPs was evident in the N-terminal 110 amino acids of crs and slp (37% identity/56% similarity). Since this is the region of C. fetus SLPs that is responsible for LPS binding, it was speculated that it is also the N-terminus of C. rectus SLPs that binds LPS.59 Unlike C. fetus strains, each of which have multiple copies of SLP-encoding genes, there is no evidence of multiple SLP genes in C. rectus. In C. rectus strain 314, crs is transcribed from a monocistronic gene in single copy in the chromosome.60 Although antigenic differences have been noted in SLPs from different C. rectus strains,61,62 antigenic variation within a single strain has not been observed.60,63
Neither of the C. rectus SLPs contain amino-terminal signal sequences.59,60 Consequently, it appears that C. rectus SLPs are transported to the cell surface by a type I secretion system that has not yet been characterized. Precedence for SLP secretion by type I transporters exists in C. fetus (described above), Caulobacter crescentus, and Serratia marcescens.41,64,65
Resistance to Complement-Mediated Killing and Phagocytosis
Limited information exists regarding the relationship of the C. rectus S-layer and immune evasion. As with C. fetus and other bacterial infections, complement is expected to play an important role in resisting C. rectus disease. Consequently, Okuda et al. investigated the complement-dependent bactericidal activity against C. rectus.66 C. rectus ATCC 33238 was exposed to guinea pig complement, either in the presence or absence of S-layer specific antibodies, and survival was assayed at timepoints of 1 to 24 hours. In the absence of S-layer-specific antibodies, C. rectus was totally resistant to complement-mediated cell lysis.66 Some killing was noted in cells exposed to 5% anti-C. rectus rabbit antiserum and monoclonal antibody against the C. rectus S-layer. However, no killing of C. rectus was noted in control samples lacking S-layer specific antibodies, suggesting that S+ C. rectus were resistant to killing by the alternative complement pathway.
Because of the importance of leukocytes in periodontal lesions,67 these same authors also tested the ability of leukocytes to kill C. rectus.66 Exposure of C. rectus to either guinea pig intraperitoneal or human peripheral leukocytes resulted in phagocytic killing, but only in the presence of antibodies against the S-layer (either 5% anti-C. rectus rabbit antiserum or monoclonal antibody against the C. rectus S-layer). Control antibody preparations (anti-LPS antibodies, pre-immune rabbit serum, or control mouse ascites) did not promote phagocytic killing by leukocytes. Thus, the C. rectus S-layer appeared to resist phagocytic killing in the absence of S-layer-specific antibody.66
An important point to note about these studies is that all of these experiments were done using only S+ cells. While the results point to the involvement of the S-layer in resistance to complement-mediated killing and phagocytosis, confirmation of these results awaits the comparison of the resistances of isogenic S+ and S− C. rectus strains.
Induction of Cytokines by C. rectus
Inflammation is a characteristic of periodontitis, and cytokines TNFα, IL-1, IL-6, and IL-8 have been implicated in this process.68-74 To investigate the potential role of the C. rectus S-layer in inflammation, Wang et al. constructed an isogenic crs mutant (S−) of strain 314 that lacks an S-layer.60 In cell binding assays, both S+ and S− C. rectus adhered to HEp-2 cells, although the binding of the wild-type (S+) 314 strain consistently bound slightly better. This is in contrast to earlier studies, which found that the binding of spontaneous S− bacteria to fibroblasts was better than that of S+ bacteria.63 The reason for the discrepancy between these results is unclear, although it could be related to differences in the bacterial strains and host cells used. Also unclear is the biological relevance of the differences in binding between S+ and S− C. rectus, given the small magnitude of the binding difference.
Once bound to HEp-2 cells, both S+ and S− C. rectus induced the expression of the proinflammatory cytokines IL-1α, IL-1β, IL-6, IL-8, and TNFα.75 The induction of IL-1α and IL-1β was rapid but transient, returning to baseline levels by 2 hours (IL-1α) or by 6 hours (IL-1β) post-exposure. The amounts of IL-1α and IL-1β induced by S+ and S− C. rectus were not significantly different. In contrast, IL-6, IL-8, and TNFα were strongly induced at later timepoints; their levels remained high throughout the remainder of the experiment. At some timepoints, the amounts of IL-6, IL-8, and TNFα produced in response to S− C. rectus were significantly higher than those for S+ C. rectus. These results suggested 2 concepts. First, exposure of C. rectus to epithelial cells induced the production of proinflammatory cytokines. In vivo, this would be expected to result in inflammation that might release factors required for the survival of C. rectus at the site of infection, as well as lead to periodontal tissue degradation. Second, the presence of the S-layer appeared to diminish the production of some of the proinflammatory cytokines. Therefore, the S-layer may play a role in modulating the host inflammatory response to a level that is most favorable for C. rectus survival. These seemingly paradoxical outcomes suggest that the interaction of C. rectus with periodontal tissue may be complex, and that neither rampant inflammation nor a total lack of inflammation is most beneficial for C. rectus colonization. Rather, optimal colonization would result from the release (by inflammation) of sufficient growth factors to promote C. rectus survival, balanced by S-layer-mediated limitation of the exposure of C. rectus to toxic molecules released during the inflammatory process.
Summary
Like many bacterial pathogens, Campylobacter species have evolved means by which they are able to resist the immune responses of their hosts. C. fetus and C. rectus both achieve this by expression of S-layers. The C. fetus S-layer endows these organisms with the ability to prevent binding of complement to the cell surface and thereby avoid complement-dependent lysis. While opsonic S-layer-directed antibodies can still be effective in killing C. fetus, SLPs are subject to high frequency antigenic variation. Antigenic variation results from efficient, but complex recombination events involving multiple SLP-encoding genes such that different antigenic S-layer variants are presented to the immune system in an effort to thwart an opsonic antibody response. In addition to preventing complement-mediated killing, the C. rectus S-layer may modulate the host immune response by down-regulating the production of proinflammatory cytokines, promoting the persistence of C. rectus within periodontal tissue. Despite recent advances in the understanding of the role of the C. rectus S-layer in periodontal disease, several questions remain regarding the C. rectus S-layer. For example, is the C. rectus S-layer a ubiquitous (and presumably essential) structure found in virtually all natural C. rectus isolates, as is the S-layer of C. fetus? What domains of C. rectus SLPs are required for their various functions (e.g., cell binding, S-layer formation)? To what cellular structures does the C. rectus S-layer bind? Are C. rectus SLPs secreted by a type I transporter, as suggested by the lack of N-terminal signal sequences? Are there interstrain differences in the efficiency by which C. rectus SLPs inhibit (or induce) inflammation in epthelial tissue? Such a finding might suggest that the presence of some C. rectus strains can lead to greater or lesser periodontal damage. Does the C. rectus S-layer influence the induction of cytokines by other cell types, such as phagocytic cells recruited to inflamed periodontal tissue? Although the C. rectus S-layer is thought to confer resistance to complement-mediated killing, the availability of isogenic S-layer-deficient C. rectus now allows this to be tested in a more thorough manner. Finally, is it possible that because of its S-layer-dependent complement resistance that C. rectus is able to disseminate via the bloodstream to other bodily sites, influencing previously unknown disease processes? Answers to these questions are likely to illuminate understanding of a bacterial species that may have profound importance on periodontal health.
References
- 1.Finlay BB, Falkow S. Common themes in microbial pathogenicity revisited. Microbiol Mol Biol Rev. 1997;61:136–169. doi: 10.1128/mmbr.61.2.136-169.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hum S. Bovine venereal Campylobacteriosis. In: Newell DG, Ketley JM, Feldman RA, editors. Campylobacters, Helicobacters, and Related Organisms. New York: Plenum Press; 1996. pp. 355–358. [Google Scholar]
- 3.Corbeil LB. Immunization and diagnosis in bovine reproductive tract infections. Adv Vet Med. 1999;41:217–239. doi: 10.1016/s0065-3519(99)80018-4. [DOI] [PubMed] [Google Scholar]
- 4.Thompson SA, Blaser MJ. Pathogenesis of Campylobacter fetus infections. In: Nachamkin I, Blaser MJ, editors. Campylobacter. Washington, DC: ASM Press; 2000. pp. 321–347. [Google Scholar]
- 5.Blaser MJ. Campylobacter fetus — emerging infection and model system for bacterial pathogenesis at mucosal surfaces. Clin Infect Dis. 1998;27:256–258. doi: 10.1086/514655. [DOI] [PubMed] [Google Scholar]
- 6.Taylor PR, Weinstein WM, Bryner JH. Campylobacter fetus infection in human subjects: association with raw milk. Am J Med. 1979;66:779–783. doi: 10.1016/0002-9343(79)91116-1. [DOI] [PubMed] [Google Scholar]
- 7.Klein BS, Vergeront JM, Blaser MJ, et al. Campylobacter infection associated with raw milk. An outbreak of gastroenteritis due to Campylobacter jejuni and thermotolerant Campylobacter fetus subsp fetus. JAMA. 1986;255:361–364. doi: 10.1001/jama.255.3.361. [DOI] [PubMed] [Google Scholar]
- 8.Ginsberg MM, Thompson MA, Peter CR, Ramras DG, Chin J. Campylobacter sepsis associated with “nutritional therapy” in California. Morbid Mortal Weekly Rep. 1981;30:294–295. [PubMed] [Google Scholar]
- 9.Verresen L, Vrolix M, Verhaegen J, Lins R. Campylobacter bacteremia. Report of 2 cases and review of the literature. Acta Clin Belg. 1985;40:99–104. doi: 10.1080/22953337.1985.11719061. [DOI] [PubMed] [Google Scholar]
- 10.Rettig PJ. Campylobacter infections in human beings. J Pediatr. 1979;94:855–864. doi: 10.1016/s0022-3476(79)80202-4. [DOI] [PubMed] [Google Scholar]
- 11.Morrison VA, Lloyd BK, Chia JKS, Tuazon CU. Cardiovascular and bacteremic manifestations of Campylobacter fetus infection: Case report and review. Rev Infect Dis. 1990;12:387–392. doi: 10.1093/clinids/12.3.387. [DOI] [PubMed] [Google Scholar]
- 12.Guerrant RL, Lahita RG, Winn WC, Jr, Roberts RB. Campylobacteriosis in man: Pathogenic mechanisms and review of 91 bloodstream infections. Am J Med. 1978;65:584–592. doi: 10.1016/0002-9343(78)90845-8. [DOI] [PubMed] [Google Scholar]
- 13.McCoy EC, Doyle D, Burda K, Corbeil LB, Winter AJ. Superficial antigens of Campylobacter (Vibrio) fetus: Characterization of antiphagocytic component. Infect Immun. 1975;11:517–525. doi: 10.1128/iai.11.3.517-525.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blaser MJ, Smith PF, Kohler PF. Susceptibility of Campylobacter isolates to the bactericidal activity of human serum. J Infect Dis. 1985;151:227–235. doi: 10.1093/infdis/151.2.227. [DOI] [PubMed] [Google Scholar]
- 15.Blaser MJ, Smith PF, Hopkins JA, Heinzer I, Bryner JH, Wang WL. Pathogenesis of Campylobacter fetus infections: Serum resistance associated with high-molecular-weight surface proteins. J Infect Dis. 1987;155:696–706. doi: 10.1093/infdis/155.4.696. [DOI] [PubMed] [Google Scholar]
- 16.Blaser MJ, Smith PF, Repine JE, Joiner KA. Pathogenesis of Campylobacter fetus infections. Failure of encapsulated Campylobacter fetus to bind C3b explains serum and phagocytosis resistance. J Clin Invest. 1988;81:1434–1444. doi: 10.1172/JCI113474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blaser MJ, Pei Z. Pathogenesis of Campylobacter fetus infections: Critical role of high-molecular-weight S-layer proteins in virulence. J Infect Dis. 1993;167:372–377. doi: 10.1093/infdis/167.2.372. [DOI] [PubMed] [Google Scholar]
- 18.Pei Z, Blaser MJ. Pathogenesis of Campylobacter fetus infections. Role of surface array proteins in virulence in a mouse model. J Clin Invest. 1990;85:1036–1043. doi: 10.1172/JCI114533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grogono-Thomas R, Dworkin J, Blaser MJ, Newell DG. Roles of the surface layer proteins of Campylobacter fetus subsp. fetus in bovine abortion. Infect Immun. 2000;68:1687–1691. doi: 10.1128/iai.68.3.1687-1691.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neuzil KM, Wang E, Haas DW, Blaser MJ. Persistence of Campylobacter fetus bacteremia associated with absence of opsonizing antibodies. J Clin Microbiol. 1994;32:1718–1720. doi: 10.1128/jcm.32.7.1718-1720.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dubreuil JD, Logan SM, Cubbage S, et al. Structural and biochemical analyses of a surface array protein of Campylobacter fetus. J Bacteriol. 1988;170:4165–4173. doi: 10.1128/jb.170.9.4165-4173.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dworkin J, Blaser MJ. Molecular mechanisms of Campylobacter fetus surface layer protein expression. Mol Microbiol. 1997;26:433–440. doi: 10.1046/j.1365-2958.1997.6151958.x. [DOI] [PubMed] [Google Scholar]
- 23.Fujimoto S, Umeda A, Takade A, Murata K, Amako K. Hexagonal surface layer of Campylobacter fetus isolated from humans. Infect Immun. 1989;57:2563–2565. doi: 10.1128/iai.57.8.2563-2565.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujimoto S, Takade A, Amako K, Blaser MJ. Correlation between molecular size of the surface array protein and morphology and antigenicity of the Campylobacter fetus S-layer. Infect Immun. 1991;59:2017–2022. doi: 10.1128/iai.59.6.2017-2022.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garcia MM, Lutze-Wallace CL, Denes AS, Eaglesome MD, Hoist E, Blaser MJ. Protein shift and antigenic variation in the S-layer of Campylobacter fetus subsp. venerealis during bovine infection accompanied by genomic rearrangement of sapA homologs. J Bacteriol. 1995;177:1976–1980. doi: 10.1128/jb.177.8.1976-1980.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pei Z, Ellison RT, Lewis RV, Blaser MJ. Purification and characterization of a family of high molecular weight surface-array proteins from Campylobacter fetus. J Biol Chem. 1988;263:6416–6420. [PubMed] [Google Scholar]
- 27.Wang E, Garcia MM, Blake MS, Pei Z, Blaser MJ. Shift in S-layer protein expression responsible for antigenic variation in Campylobacter fetus. J Bacteriol. 1993;175:4979–4984. doi: 10.1128/jb.175.16.4979-4984.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sára M, Sleytr UB. S-Layer proteins. J Bacteriol. 2000;182:859–868. doi: 10.1128/jb.182.4.859-868.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang LY, Pei Z, Fujimoto S, Blaser MJ. Reattachment of surface array proteins to Campylobacter fetus cells. J Bacteriol. 1992;174:1258–1267. doi: 10.1128/jb.174.4.1258-1267.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fogg GC, Yang LY, Wang E, Blaser MJ. Surface array proteins of Campylobacter fetus block lectin-mediated binding to type A lipopolysaccharide. Infect Immun. 1990;58:2738–2744. doi: 10.1128/iai.58.9.2738-2744.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Graham LL, MacDonald KL. The Campylobacter fetus S-layer is not essential for initial interaction with HEp-2 cells. Can J Microbiol. 1998;44:244–250. [PubMed] [Google Scholar]
- 32.Schurig GD, Hall CE, Burda K, Corbeil LB, Duncan JR, Winter AJ. Persistent genital tract infection with Vibrio fetus intestinalis associated with serotypic alteration of the infecting strain. Am J Vet Res. 1973;34:1399–1403. [PubMed] [Google Scholar]
- 33.Corbeil LB, Schurig GGD, Bier PJ, Winter AJ. Bovine venereal vibriosis: Antigenic variation of the bacterium during infection. Infect Immun. 1975;11:240–244. doi: 10.1128/iai.11.2.240-244.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dubreuil JD, Kostrzynska M, Austin JW, Trust TJ. Antigenic differences among Campylobacter fetus S-layer proteins. J Bacteriol. 1990;172:5035–5043. doi: 10.1128/jb.172.9.5035-5043.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tummuru MKR, Blaser MJ. Rearrangement of sapA homologs with conserved and variable regions in Campylobacter fetus. Proc Natl Acad Sci (USA) 1993;90:7265–7269. doi: 10.1073/pnas.90.15.7265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tummuru MKR, Blaser MJ. Characterization of the Campylobacter fetus sapA promoter: Evidence that the sapA promoter is deleted in spontaneous mutant strains. J Bacteriol. 1992;174:5916–5922. doi: 10.1128/jb.174.18.5916-5922.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fujita M, Fujimoto S, Morooka T, Amako K. Analysis of strains of Campylobacter fetus by pulsed-field gel electrophoresis. J Clin Microbiol. 1995;33:1676–1678. doi: 10.1128/jcm.33.6.1676-1678.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fujita M, Amako K. Localization of the sapA gene on a physical map of Campylobacter fetus chromosomal DNA. Arch Microbiol. 1994;162:375–380. doi: 10.1007/BF00282100. [DOI] [PubMed] [Google Scholar]
- 39.Dworkin J, Tummuru MKR, Blaser MJ. Segmental conservation of sapA sequences in type B Campylobacter fetus cells. J Biol Chem. 1995;270:15093–15101. doi: 10.1074/jbc.270.25.15093. [DOI] [PubMed] [Google Scholar]
- 40.Dworkin J, Tummuru MKR, Blaser MJ. A lipopolysaccharide-binding domain of the Campylobacter fetus S-layer protein resides within the conserved N terminus of a family of silent and divergent homologs. J Bacteriol. 1995;177:1734–1741. doi: 10.1128/jb.177.7.1734-1741.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blaser MJ, Gotschlich EC. Surface array protein of Campylobacter fetus. Cloning and gene structure. J Biol Chem. 1990;265:14529–14535. [PubMed] [Google Scholar]
- 42.Casadémont I, Chevrier D, Guesdon JL. Cloning of a sapB homologue (sapB2) encoding a putative 112-kDa Campylobacter fetus S-layer protein and its use for identification and molecular genotyping. FEMS Immunol Med Microbiol. 1998;21:269–281. doi: 10.1111/j.1574-695X.1998.tb01174.x. [DOI] [PubMed] [Google Scholar]
- 43.Dworkin J, Blaser MJ. Nested DNA inversion as a paradigm of programmed gene rearrangement. Proc Natl Acad Sci (USA) 1997;94:985–990. doi: 10.1073/pnas.94.3.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dworkin J, Shedd OL, Blaser MJ. Nested DNA inversion of Campylobacter fetus S-layer genes is recA dependent. J Bacteriol. 1997;179:7523–7529. doi: 10.1128/jb.179.23.7523-7529.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ray KC, Tu ZC, Grogono-Thomas R, Newell DG, Thompson SA, Blaser MJ. Campylobacter fetus sap inversion occurs in the absence of RecA function. Infect Immun. 2000;68:5663–5667. doi: 10.1128/iai.68.10.5663-5667.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tu ZC, Ray KC, Thompson SA, Blaser MJ. Campylobacter fetus uses multiple loci for DNA inversion within the 5′ conserved regions of sap homologs. J Bacteriol. 2001;183:6654–6661. doi: 10.1128/JB.183.22.6654-6661.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thompson SA, Shedd OL, Ray KC, Beins MH, Jorgensen JP, Blaser MJ. Campylobacter fetus surface layer proteins are transported by a type I secretion system. J Bacteriol. 1998;180:6450–6458. doi: 10.1128/jb.180.24.6450-6458.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dzink JL, Tanner ACR, Haffajee AD, Socransky SS. Gram-negative species associated with active destructive periodontal lesions. J Clin Periodontol. 1985;12:648–659. doi: 10.1111/j.1600-051x.1985.tb00936.x. [DOI] [PubMed] [Google Scholar]
- 49.Tanner ACR, Dzink JL, Ebersole JL, Socransky SS. Wolinella recta, Campylobacter concisus, Bacteroides gracilis, and Eikenella corrodens from periodontal lesions. J Periodont Res. 1987;22:327–330. doi: 10.1111/j.1600-0765.1987.tb01593.x. [DOI] [PubMed] [Google Scholar]
- 50.Listgarten MA, Lai CH, Young V. Microbial composition and pattern of antibiotic resistance in subgingival microbial samples from patients with refractory periodontitis. J Periodontol. 1993;64:155–161. doi: 10.1902/jop.1993.64.3.155. [DOI] [PubMed] [Google Scholar]
- 51.Rams TE, Feik D, Slots J. Campylobacter rectus in human periodontitis. Oral Microbiol Immunol. 1993;8:230–235. doi: 10.1111/j.1399-302x.1993.tb00565.x. [DOI] [PubMed] [Google Scholar]
- 52.Gmür R, Guggenheim B. Interdental supragingival plaque — a natural habitat of Actinobacillus actinomycetemcomitans, Bacteroides forsythus, Campylobacter rectus, and Prevotella nigrescens. J Dent Res. 1994;73:1421–1428. doi: 10.1177/00220345940730080501. [DOI] [PubMed] [Google Scholar]
- 53.López NJ, Mellado JC, Giglio MS, Leighton GX. Occurrence of certain bacterial species and morphotypes in juvenile periodontitis in Chile. J Periodontol. 1995;66:559–567. doi: 10.1902/jop.1995.66.7.559. [DOI] [PubMed] [Google Scholar]
- 54.Lai CH, Oshima K, Slots J, Listgarten MA. Wolinella recta in adult gingivitis and periodontitis. J Periodont Res. 1992;27:8–14. doi: 10.1111/j.1600-0765.1992.tb02079.x. [DOI] [PubMed] [Google Scholar]
- 55.van Steenbergen TJM, van der Velden U, Abbas F, de Graaff J. Microbiological and clinical monitoring of non-localized juvenile periodontitis in young adults: A report of 11 cases. J Periodontol. 1993;64:40–47. doi: 10.1902/jop.1993.64.1.40. [DOI] [PubMed] [Google Scholar]
- 56.Lai CH, Listgarten MA, Tanner ACR, Socransky SS. Ultrastructures of Bacteroides gracilis, Campylobacter concisus, Wolinella recta, and Eikenella corrodens, all from humans with periodontal disease. Int J Syst Bacterid. 1981;31:465–475. [Google Scholar]
- 57.Kerosuo E, Haapasalo M, Lounatmaa K. Ultrastructural relationship of cell envelope layers in Wolinella recta. Scand J Dent Res. 1989;97:54–59. doi: 10.1111/j.1600-0722.1989.tb01430.x. [DOI] [PubMed] [Google Scholar]
- 58.Dokland T, Olsen I, Farrants G, Johansen BV. Three-dimensional structure of the surface layer of Wolinella recta. Oral Microbiol Immunol. 1990;5:162–165. doi: 10.1111/j.1399-302x.1990.tb00415.x. [DOI] [PubMed] [Google Scholar]
- 59.Miyamoto M, Maeda H, Kitanaka M, Kokeguchi S, Takashiba S, Murayama Y. The S-layer protein from Campylobacter rectus: Sequence determination and function of the recombinant protein. FEMS Microbiol Lett. 1998;166:275–281. doi: 10.1111/j.1574-6968.1998.tb13901.x. [DOI] [PubMed] [Google Scholar]
- 60.Wang B, Kraig E, Kolodrubetz D. A new member of the S-layer protein family: Characterization of the crs gene from Campylobacter rectus. Infect Immun. 1998;66:1521–1526. doi: 10.1128/iai.66.4.1521-1526.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kobayashi Y, Ohta H, Kokeguchi S, et al. Antigenic properties of Campylobacter rectus (Wolinella recta) major S-layer proteins. FEMS Microbiol Lett. 1993;108:275–280. doi: 10.1111/j.1574-6968.1993.tb06115.x. [DOI] [PubMed] [Google Scholar]
- 62.Nitta H, Holt SC, Ebersole JL. Purification and characterization of Campylobacter rectus surface layer proteins. Infect Immun. 1997;65:478–483. doi: 10.1128/iai.65.2.478-483.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Borinski R, Holt SC. Surface characteristics of Wolinella recta ATCC 33238 and human clinical isolates: Correlation of structure with function. Infect Immun. 1990;58:2770–2776. doi: 10.1128/iai.58.9.2770-2776.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Awram P, Smit J. The Caulobacter crescentus paracrystalline S-layer protein is secreted by an ABC transporter (type I) secretion apparatus. J Bacteriol. 1998;180:3062–3069. doi: 10.1128/jb.180.12.3062-3069.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kawai E, Akatsuka H, Idei A, Shibatani T, Omori K. Serratia marcescens S-layer protein is secreted extracellularly via an ATP-binding cassette exporter, the Lip system. Mol Microbiol. 1998;27:941–952. doi: 10.1046/j.1365-2958.1998.00739.x. [DOI] [PubMed] [Google Scholar]
- 66.Okuda K, Kigure T, Yamada S, et al. Role for the S-layer of Campylobacter rectus ATCC33238 in complement mediated killing and phagocytic killing by leukocytes from guinea pig and human peripheral blood. Oral Dis. 1997;3:113–120. doi: 10.1111/j.1601-0825.1997.tb00022.x. [DOI] [PubMed] [Google Scholar]
- 67.Van Dyke TE, Horoszewicz HU, Cianciola LJ, Genco RJ. Neutrophil chemotaxis dysfunction in human periodontitis. Infect Immun. 1980;27:124–132. doi: 10.1128/iai.27.1.124-132.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Assuma R, Oates T, Cochran D, Amar S, Graves DT. IL-1 and TNF antagonists inhibit the inflammatory response and bone loss in experimental periodontitis. J Immunol. 1998;160:403–409. [PubMed] [Google Scholar]
- 69.Hönig J, Rordorf-Adam C, Siegmund C, Wiedemann W, Erard F. Increased interleukin-1 beta (IL-1 β) concentration in gingival tissue from periodontitis patients. J Periodont Res. 1989;24:362–367. doi: 10.1111/j.1600-0765.1989.tb00883.x. [DOI] [PubMed] [Google Scholar]
- 70.Roberts FA, Hockett RD, Jr, Bucy RP, Michalek SM. Quantitative assessment of inflammatory cytokine gene expression in chronic adult periodontitis. Oral Microbiol Immunol. 1997;12:336–344. doi: 10.1111/j.1399-302x.1997.tb00735.x. [DOI] [PubMed] [Google Scholar]
- 71.Roberts FA, McCaffery KA, Michalek SM. Profile of cytokine mRNA expression in chronic adult periodontitis. J Dent Res. 1997;76:1833–1839. doi: 10.1177/00220345970760120501. [DOI] [PubMed] [Google Scholar]
- 72.Stashenko P, Fujiyoshi P, Obernesser MS, Prostak L, Haffajee AD, Socransky SS. Levels of interleukin 1 β in tissue from sites of active periodontal disease. J Clin Periodontol. 1991;18:548–554. doi: 10.1111/j.1600-051x.1991.tb00088.x. [DOI] [PubMed] [Google Scholar]
- 73.Stashenko P, Jandinski JJ, Fujiyoshi P, Rynar J, Socransky SS. Tissue levels of bone resorptive cytokines in periodontal disease. J Periodontol. 1991;62:504–509. doi: 10.1902/jop.1991.62.8.504. [DOI] [PubMed] [Google Scholar]
- 74.Dongari-Bagtzoglou AI, Ebersole JL. Production of inflammatory mediators and cytokines by human gingival fibroblasts following bacterial challenge. J Periodont Res. 1996;31:90–98. doi: 10.1111/j.1600-0765.1996.tb00469.x. [DOI] [PubMed] [Google Scholar]
- 75.Wang B, Kraig E, Kolodrubetz D. Use of defined mutants to assess the role of the Campylobacter rectus S-layer in bacterium-epithelial cell interactions. Infect Immun. 2000;68:1465–1473. doi: 10.1128/iai.68.3.1465-1473.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]