

Abstract
A critical goal in cell biology is to develop a systems-level perspective of eukaryotic cell cycle controls. Among these controls, a complex signaling network (called ‘checkpoints’) arrests progression through the cell cycle when there is a threat to genomic integrity such as unreplicated or damaged DNA. Understanding the regulatory principles of cell cycle checkpoints is important because loss of checkpoint regulation may be a requisite step on the roadway to cancer. Mathematical modeling has proved to be a useful guide to cell cycle regulation by revealing the importance of bistability, hysteresis and time lags in governing cell cycle transitions and checkpoint mechanisms. In this report, we propose a mathematical model of the frog egg cell cycle including effects of unreplicated DNA on progression into mitosis. By a stepwise approach utilizing parameter estimation tools, we build a model that is grounded in fundamental behaviors of the cell cycle engine (hysteresis and time lags), includes new elements in the signaling network (Myt1 and Chk1 kinases), and fits a large and diverse body of data from the experimental literature. The model provides a validated framework upon which to build additional aspects of the cell cycle checkpoint signaling network, including those control signals in the mammalian cell cycle that are commonly mutated in cancer.
Keywords: checkpoint, hysteresis, Cdk, MPF, Cdc25, Wee1
Introduction
The ability of cells to arrest at checkpoints in response to damaged or unreplicated DNA is conserved among eukaryotes (Elledge, 1996), and loss of checkpoints results in genomic instability, which characterizes most human malignancies (Bartek and Lukas, 2003; Hanahan and Weinberg, 2000). Because the core cell cycle machinery as well as the checkpoint signaling networks that affect this machinery are highly conserved, information derived from studying checkpoints in one experimental system, such as the frog egg extract, should provide a framework of knowledge that can be extended to include more complex control circuits in mammalian cells. As details about the molecules that regulate checkpoints and their interactions with one another accumulate, it becomes increasingly difficult to predict how perturbations (such as a deletion or overexpression of a protein) will affect the entire system. Mathematical models provide a system-level view of molecular networks. Experimentally validated models identify critical interactions that control the system and assist in developing the next round of questions to test experimentally.
We and others have used mathematical modeling as a tool to organize a large body of experimental data and discover underlying regulatory principles governing entry into and exit from mitosis in cell-free egg extracts (Novak and Tyson, 1993). The Novák-Tyson model of the cell cycle is based on biochemical kinetics (to generate a set of rate equations for Cdks and their associated proteins) and modern dynamical systems theory (to analyze the solutions of these nonlinear differential equations). The wiring diagram for the model (Fig. 1) summarizes the regulation of cyclin B/Cdk1 by three feedback loops. In egg extracts, newly synthesized cyclin B associates with Cdk1 (present in excess) to form active mitosis-promoting factor (MPF) (Solomon et al., 1990). MPF is rapidly inhibited by phosphorylation on Tyr15 by the kinase, Wee1 (Mueller et al., 1995a). (A second MPF kinase, Myt1, was discovered after the Novák-Tyson model was published (Mueller et al., 1995b). Addition of Myt1 became a critical step in building the model of the DNA replication checkpoint described here.) MPF remains inactive until this phosphate group is removed by the phosphatase, Cdc25 (Gautier et al., 1991; Kumagai and Dunphy, 1991). In turn, active MPF phosphorylates and inhibits Wee1, in a mutually antagonistic feedback loop (Mueller et al., 1995a), and phosphorylates and activates Cdc25, in a positive feedback loop (Izumi and Maller, 1993; Kumagai and Dunphy, 1992). These feedback loops are responsible for the abrupt activation of MPF at the G2/M transition. Also important to this control system is a negative feedback loop with time delay in which active MPF indirectly activates the APC to target cyclin B for degradation via the ubiquitin-proteasome pathway (Lorca et al., 1998; Felix et al., 1990).
Figure 1. The Novák-Tyson model of the cell cycle engine in frog egg extracts.
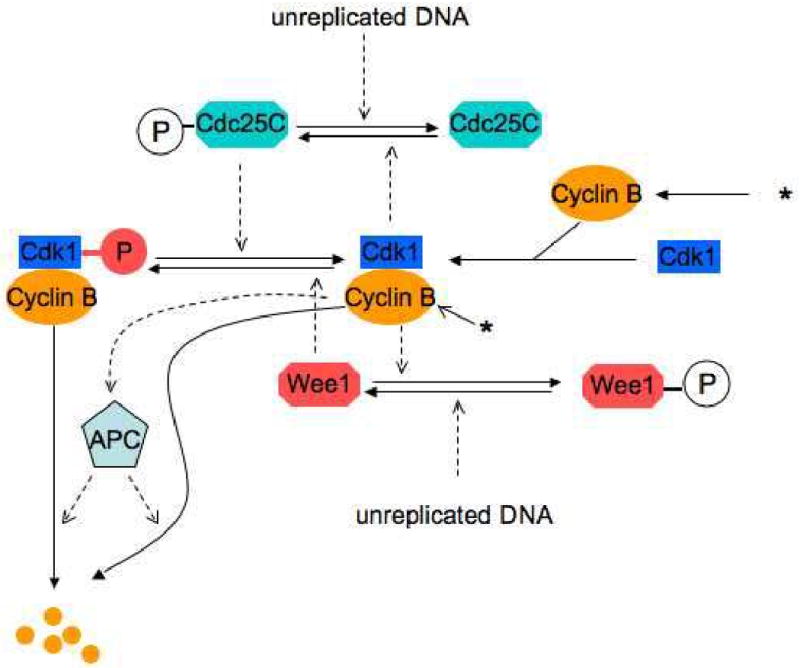
Positive feedback between Cdk1 and Cdc25 (Gautier et al., 1991; Izumi and Maller, 1993; Kumagai and Dunphy, 1991; Kumagai and Dunphy, 1992) and double-negative feedback between Cdk1 and Wee1 (Mueller et al., 1995a; Smythe and Newport, 1992) create the abrupt activation of Cdk1 at G2/M. Inactivation of Cdk1 results from negative feedback in which Cdk1 indirectly targets cyclin B for degradation through the Anaphase Promoting Complex (APC) (Lorca et al., 1998). Synthesis of cyclin is represented by an asterisk (*). Computer simulations of the model reproduce oscillations of Cdk1 (MPF) activity as they are observed in cell-free egg extracts (Novak and Tyson, 1993). Novák and Tyson proposed that unreplicated DNA blocks entry into mitosis by activating the phosphatases that oppose MPF's action on Cdc25 and Wee1.
The Novák-Tyson model made several predictions about the behavior of the underlying control system regulating entry into mitosis (Novak and Tyson, 1993), which we later validated experimentally (Sha et al., 2003). The first prediction we tested was that entry into mitosis (activation of MPF) and exit from mitosis (inactivation of MPF) are regulated by hysteresis. Novák and Tyson predicted that the phosphorylation reactions controlling the activity of MPF can persist in two alternative steady states: an interphase-arrested state with low MPF activity (because of inhibitory phosphorylation of Cdk1), and an M-phase arrested state with high MPF activity (because the inhibitory phosphate group has been removed). The system switches between the two states as the amount of cyclin B in the cell is manipulated (e.g. by altering its rate of synthesis or degradation). Such switch-like behavior is called hysteresis. Hysteresis is created by positive feedback in the underlying control mechanism. Goldbeter's model (Goldbeter, 1991), which lacks positive feedback, does not exhibit hysteresis.
In a series of experiments based on Solomon's protocol for MPF activation in frog egg extracts (Solomon et al., 1990), we confirmed that mitosis is governed by hysteresis and bistability. By strictly controlling the amount of cyclin B in the extract (using cycloheximide to block synthesis of endogenous cyclins and adding exogenous nondegradable cyclin at fixed concentrations), we determined that the activation threshold (amount of cyclin B required to activate MPF and drive entry into mitosis) was between 32 and 40 nM, whereas the inactivation threshold (amount of cyclin B required to keep an extract in mitosis) was between 16 and 24 nM (Sha et al., 2003). We showed that, for concentrations of cyclin B just above the activation threshold, the time lag for MPF activation gets very long (a phenomenon called ‘critical slowing down’). Thus, by pairing experimental and computational studies, we established a fundamental principle of cell cycle control in cell-free egg extracts. Pomerening et al. confirmed this principle in an independent study (Pomerening et al., 2003).
When a DNA replication checkpoint is engaged, stalled DNA replication forks trigger a signal transduction network that ultimately causes inactivation of MPF and arrests the cell cycle at the G2/M transition (Nyberg et al., 2002; Qin and Li, 2003; Sagata, 2002). In egg extracts supplemented with DNA, agents that lead to stalled replication forks (e.g. aphidicolin or UV-radiation) activate ATR (Hekmat-Nejad et al., 2000; Lupardus et al., 2002), a kinase that phosphorylates and activates Chk1 (Guo et al., 2000; Kumagai et al., 1998b). In frog eggs, Chk1 promotes cell cycle arrest by phosphorylating Cdc25C and Wee1. Phosphorylation of Cdc25C on S287 by Chk1 causes Cdc25C to bind 14-3-3 protein, sequestering Cdc25C in the cytosol (Kumagai et al., 1998a, b). Phosphorylation of Cdc25C on S287 is mutually exclusive with phosphorylation of Cdc25C on S285 by Cdks (Bulavin et al., 2003a, b). Phosphorylation of Wee1 by Chk1 causes 14-3-3 to bind to Wee1 and stimulates the activity of Wee1 towards Cdks (Lee et al., 2001).
Although little molecular information about the DNA replication checkpoint was available at the time, Novák and Tyson predicted that unreplicated DNA enlarges the hysteresis loop governing mitosis. They placed the effects on the feedback loops involving MPF, Wee1 and Cdc25. Experimentally, we validated this prediction by showing that for a specific concentration of sperm nuclei (1200/μl), an increase in cyclin content from 40 to 100 nM was required to bypass a DNA replication checkpoint (Sha et al., 2003). This discovery of a relatively small quantitative difference (2.5 fold change in cyclin concentration) producing a striking change in physiologic behavior (bypassing cell cycle arrest) provides another example of the novel information gained by pairing experimental studies in egg extracts with mathematical modeling. As we identify points of control in the molecular network that are sensitive to small quantitative changes, we add new layers of understanding about how diseases develop (sometimes by subtle rather than gross changes in gene expression) and might be treated (by adjusting the cell cycle thermostat by degrees, not orders of magnitude). These preliminary studies provided insight into how a checkpoint signaling pathway affects the dynamical behavior of the cell-cycle engine.
To extend our understanding of cell cycle regulation, we address here the effect of unreplicated DNA on the core cell cycle engine, in particular, on the activating phosphatases (Cdc25) and the inhibitory kinases (Wee1 and Myt1) that engage in feedback loops with MPF. In order to preserve the fundamental properties of the control system, the model was constrained to reproduce the phenomena of hysteresis and critical slowing down that we demonstrated experimentally (Sha et al., 2003). Through an iterative process of parameter estimation aided by a newly developed parameter estimation tool (Zwolak et al., 2005a, b), we identified a set of parameter values that brings the model into good qualitative agreement with experiments in which the regulatory phosphatase and kinase (Cdc25 and Wee1) were manipulated in the absence and in the presence of unreplicated DNA. This model serves as a computational tool for investigating the underlying system dynamics of cell cycle checkpoints and predicting the effect of pharmacological and pathological perturbations to the system.
Results
Overall approach to building the model
In the original Novák-Tyson model (Novak and Tyson, 1993), it was assumed that unreplicated DNA upregulates the phosphatase(s) that oppose MPF (see Fig. 1) in the positive feedback loops whereby MPF activates Cdc25 and inhibits Wee1. Experimental studies that were published later suggested that the unreplicated DNA checkpoint operates by activating kinases (notably Chk1) that phosphorylate Cdc25 and Wee1 on sites distinct from the Cdk-phosphorylation sites. Phosphorylation of Cdc25 by Chk1 appears to inhibit the action of Cdc25, in part by promoting its binding to cytoskeletal 14-3-3 protein which retains Cdc25 in the cytoplasm. Cdc25 phosphorylation by Chk1 also precludes its phosphorylation by Cdk (Bulavin 2003a, b). Phosphorylation of Wee1 by Chk1 appears to activate Wee1 and oppose entry into mitosis by favoring the low activity, tyrosine-phosphorylated form of Cdk1. Furthermore, there is experimental evidence (to be described in detail later) that Myt1 kinase is active during normal mitotic cycles, when the checkpoint is not functioning, and that the main role of Wee1 is to transduce the checkpoint signal to MPF. These modifications of the Novák-Tyson scheme (Fig. 1) lead to a revised model (Fig. 2) that is the basis of the current study.
Figure 2. A modified Novák-Tyson model, including the effects of Myt1 and Chk1 kinases.
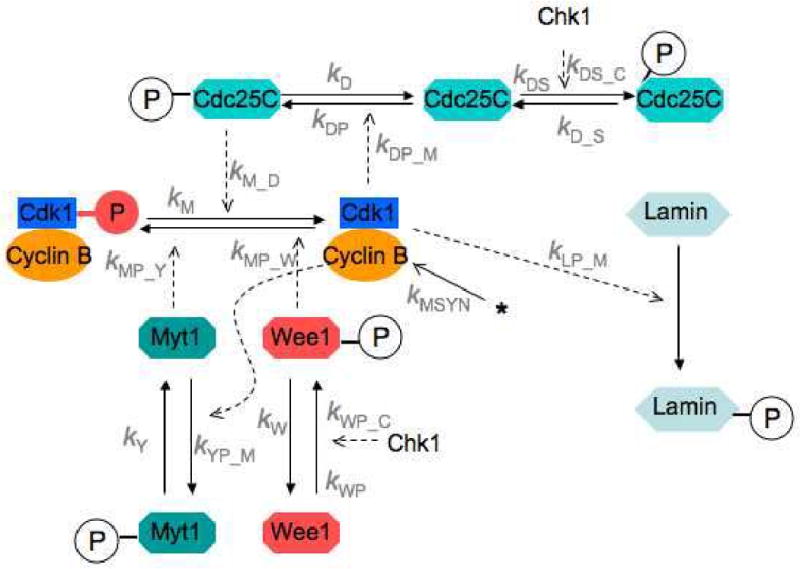
In the presence of unreplicated DNA, active Chk1 phosphorylates and activates Wee1 kinase, which phosphorylates and inactivates MPF. Active Chk1 also phosphorylates and deactivates Cdc25, thereby impeding progression into M phase. Nuclear lamin phosphorylation by MPF is an indicator, in the model, of preparation for nuclear envelope breakdown.
Our proposed model of the effects of Chk1 on the frog egg cell cycle (Fig. 2), involving reactions among 10 biochemical components (Table 1), can be described mathematically by 7 ordinary differential equations (ODEs) and 3 conservation relations (Table 2). The reaction rate terms on the right-hand-sides of these equations involve 18 unknown rate constants (Table 3). Our goal is to show that these rate constants can be estimated from relevant experimental data about the unreplicated DNA checkpoint in frog egg extracts, and then to test the model on data not previously used to estimate the rate constants. If we can do this successfully, we will have shown that the model in Fig. 2 is sufficient to account for all relevant experimental observations and can be used to predict new results with reasonable confidence.
Table 1. Components of the DNA replication checkpoint mechanism in frog eggs.
| MPF | Cdk1:cyclin B dimer, phosphorylated on Thr161 (active form) |
| preMPF | MPF phosphorylated on Thr161 and on Tyr15 (inactive form) |
| Myt1 | Myt1 (tyrosine kinase active during normal mitotic cycles) |
| Myt1P | Phosphorylated Myt1 (inactive form) |
| Cdc25 | Cdc25C (tyrosine phosphatase active during normal mitotic cycles) |
| Cdc25P | Cdc25C phosphorylated on Ser285 (active form) |
| Cdc25S | Cdc25C phosphorylated on Ser287 (inactive form) |
| Wee1 | Wee1 (tyrosine kinase activated by checkpoint signal) |
| Wee1P | Wee1 phosphorylated on Ser549 (active form) |
| Lamin | Protein required for nuclear envelope integrity |
Table 2. Computational model of the DNA replication checkpoint.
| 1. |
|
|
| 2. |
|
|
| 3. |
|
|
| 4. |
|
|
| 5. |
|
|
| 6. |
|
|
| 7. |
|
|
| 8. | YT = [Myt1]+[Myt1P] = constant | |
| 9. | WT = [Wee1] + [Wee1P] = constant | |
| 10. | DT = [Cdc25] + [Cdc25P] + [Cdc25S] = constant |
Table 3. The final, optimized set (Bfinal) of parameter values used in the model.
| Parameter (min-1) | Parameter (nM-1 min-1) | ||||
|---|---|---|---|---|---|
| Bini | Bfinal | Bini | Bfinal | ||
| kM | 0 | [0] | kDP_M | 0.64 | 0.45 |
| kMP_Y YT | 6.15 | 6.48 | kLP_M | 0.005 | 0.0042 |
| kMP_W WT | 0.08 | 0.075 | Parameter (nM min-1) | ||
| kM_D DT | 0.19 | 0.18 | Bini | Bfinal | |
| kD | 0.45 | 0.50 | kMSYN | 0.12 | 0.13 |
| kDP | 0.18 | 0.13 | Parameter (nM) | ||
| kW | 1 | [1] | kY/kYP_M | 0.034 | 0.035 |
| kWP | 0 | [0] | Parameter (dim' less) | ||
| kWP_C CT | 10 | [10] | Bini | Bfinal | |
| kDS | 3 | 5.38 | μNEB | 0.8 | 0.76 |
| kD_S | 1 | [1] | σNEB | 0.2 | 0.16 |
| kDS_C CT | 7 | 5.4 | |||
To this end, we first surveyed the experimental literature about MPF dynamics in frog egg extracts with and without unreplicated DNA and collected data that was sufficiently representative and quantitative to be useful in constraining the mathematical model (Table 4). We also built a general-purpose Parameter Estimation Toolkit (PET) that uses powerful and efficient optimization algorithms (both global and local) to fit ODE models to data of the type collected in Table 4. (Technical specifications of PET are outlined in ‘Models and Methods’.) Then we used PET to carry out a brute-force optimization of all 18 parameters of the full model from the full data set. This brute-force approach failed to find a parameter set that gave acceptable fits to the experimental data.
Table 4. Sources of data used for parameter estimation in the model.
| # | Data type | Source | Fig # here | Parameter(s) estimated |
|---|---|---|---|---|
| 1a | Cyclin thresholds | J. Moore (unpublished) | 3 | M0, MY, MD |
| 1b | MPF time lags | Sha et al 2003, Fig. 6D | 3 | kM_DDT |
| 2 | Cdc25 phos'n and dephos'n | Kumagai & Dunphy 1992 Fig 10 | 4 | kD, kDP, kDP_M |
| 3 | MPF phos'n and dephos'n | Kumagai & Dunphy 1995 Figs 3, 4 | 5 | kM_DDT, kM, kMP_YYT |
| 4a | NEB Time series: Wee1-depletion | Lee et al 2001, Fig 5 | 6 | KLP_M, KDS_S, KMSYN, μNEB, σNEB |
| 4b | NEB Time series: Mock-depletion | Lee et al 2001, Fig 5 | 6 | kMP_WWT, kW |
| 5 | NEB Time series: Cdc25-depletion and add-back | Kumagai et al 1998, Fig 6 | 7 | Test 1 |
| 6 | MPF kinase assay | Sha et al 2003 Fig 6 | 8 | Test 2 |
As an alternative to brute force, we were successful in optimizing the model by taking small, logical steps from the central network of MPF activation to the complex effects of unreplicated DNA on frog egg extracts supplemented with recombinant proteins. By addressing a few experiments at a time, we were able to build up the model with a few new components at each step, and to use PET at each step to find a set of parameter values that fit the data in the new experiments while retaining good fits to the data in previous steps.
By the end of this process we had estimates of 14 of the 18 parameters in the full model (see Table 3), and the fully parameterized model provided a good fit to all of the experimental data in our collection (Table 4). The quality of the fit can be judged informally by comparing simulated curves with data points in Figures 3-8. To explain how we reached our goal and how the rate constants in the model are constrained by the data available, we describe in some detail the steps taken in the optimization procedure.
Figure 3. Hysteresis and critical slowing down in the MPF control system.
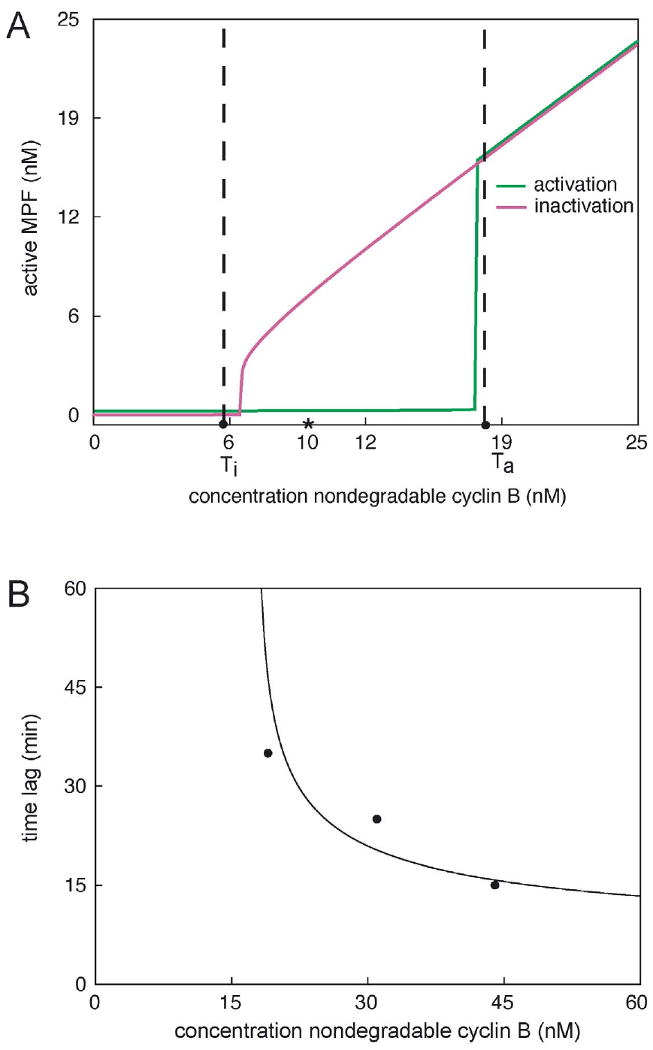
(A) Unpublished data of J. Moore indicate a cyclin between 15 and 20 nM (set at 18 nM in this figure) and a cyclin threshold (Ti) for MPF inactivation between 5 and 7 nM (set at 6 nM here). Moore's extracts showed definitive bistability (stable interphase or M-phase states) at 10 nM cyclin B (asterisk). The theoretical MPF-response curves (red and green lines) were simulated by the model in Table 2 and the parameter set Bfinal in Table 3. (B) Moore (unpublished) also observed increasing time lags (data points) for entry into mitosis (nuclear envelope breakdown) for cyclin B concentrations slightly above Ta. The simulated lag times (solid line) were computed from the full model and the final parameter set.
Figure 8. Simulations of critical slowing down in frog egg extracts.
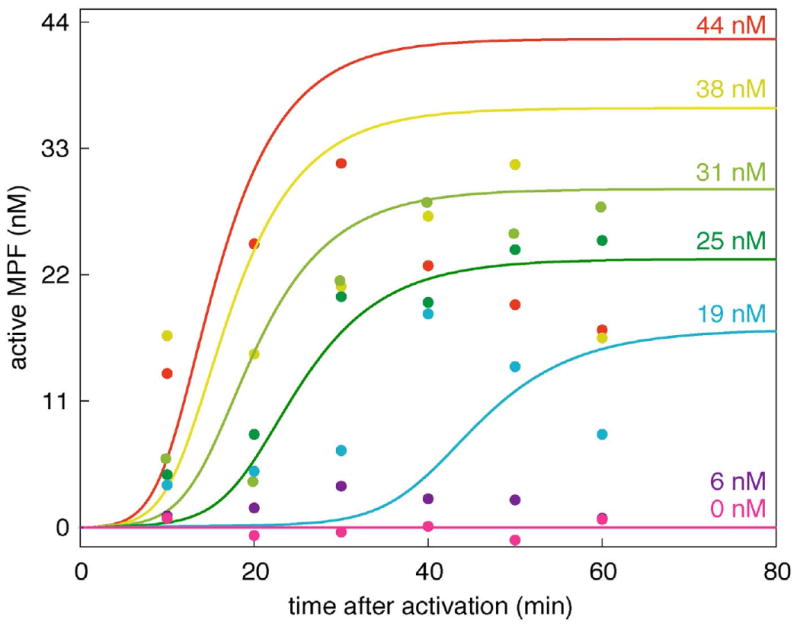
Simulations (lines) are compared to experimental measurements (data points) of MPF activity vs. time at several cyclin concentrations approaching the activation threshold from above. The data points are from (Sha et al., 2003) as well as unpublished data (Sha and Sible) collected during the same experiment at 25 and 38 nM cyclin. The theoretical curves were computed from the full model (Table 2) and the final parameter set (Table 3).
Step 0: Initial assumptions
In all the experiments that we shall be considering, cyclin B degradation is negligible, either because the extracts contain only non-degradable cyclin B, or because MPF activity is measured only up to the early stages of mitosis (nuclear envelope breakdown), before the APC is activated. Hence, we assume throughout our calculations that kMDEG = 0. Furthermore, the Novák-Tyson model is most conducive to bistability when kM ≪ kM_D DT. Because bistability of MPF activity in frog egg extracts has been demonstrated (Pomerening et al., 2003; Sha et al., 2003), we assume that kM = 0.
Step 1: A simplified model of MPF dynamics with bistability and time lags
Our first step is to derive a simple model (a single differential equation for MPF activity) that is sufficient to describe crucial features of the MPF control system, including cyclin thresholds for MPF activation and inactivation and the dependence of lag-time for MPF activation on cyclin concentration. In the ‘Models and Methods’ section, we show how, under appropriate conditions and suitable assumptions, the first 5 differential equations of Table 3 can be subsumed in a single equation for M(t) = concentration of the active form of MPF (as a function of time),
| (1) |
In this equation, the ‘total’ concentrations, YT = [Myt1]+[Myt1P], DT = [Cdc25]+[Cdc25P], are assumed to be constants. In all experiments to be modeled in this step, kMSYN = kMDEG = 0, and so the total MPF concentration in the extract, MT = [MPF]+[preMPF], is also a constant. In Eq. (1) we have introduced some ‘derived’ parameters:
| (2) |
M0, MDMY have units nM (nanomolar), whereas ρ is a dimensionless ratio of rate constants. We will estimate the parameters in Eq. (1) by fitting the cyclin threshold data of Jonathan Moore (unpublished) and time lag data of Sha et al. (2003).
The cyclin threshold experiments of Moore imply that Eq. (1) must have three steady-state solutions for MPF activity for MT in the range: 6 nM < MT < 18 nM. Bistability was also observed by Sha et al. (2003) and by Pomerening et al. (2003), although the range they observed was at larger values of total cyclin, presumably because their preparations of cyclin protein were not as active as Moore's. The steady state solutions of Eq. (1) satisfy the algebraic equation
| (3) |
Equation (3) is a cubic equation in M, and it may have three real positive roots, 0 < M1 < M2 < M3 < MT, for a proper choice of parameters MT, M0, MD, MY and ρ. In general, it would be very difficult to find the real roots of Eq. (3) as functions of all the parameters. However, we assume, quite reasonably, that kDP ≪ kDP_M MT and kY ≪ kYP_M MT (i.e., that MD ≪ MT and MY ≪ MT). Given these assumptions, the three real roots of Eq. (3) are:
| (4) |
provided that
| (5) |
The facts that M1 ≈ 0 and M3 ≈ MT (as expected) are evidence that the assumptions we have made are reasonable. Recall that Moore found bistability in the range 6 nM < MT < 18 nM, implying that ρM0 ≈ 18 nM and MY ≈ 0.5 nM.
This analysis shows us how to choose MY and ρM0 in Eq. (1) to get a bistable response in MPF activity with thresholds close to the values observed by Moore. Next, we have to choose MD and the time-scale factor, kM_DDt, so that solutions of Eq. (1) exhibit the correct temporal profile (i.e., lag time ≈ 15-30 min and rise time ≈ 15 min). A few simulations of Eq. (1) demonstrate that the parameter set B1 (see Table 5) might provide a reasonable starting point for automatic parameter estimation by PET. Indeed, from this starting point, PET quickly finds an optimal parameter set (B1opt in Table 5) that gives a very good fit to the data in Table 4, lines 1a and b.
Table 5. Table of parameter values used during construction of the model.
| Parameter* | Units | B1 | B1opt | B2 | B2opt | B3 | B3opt |
|---|---|---|---|---|---|---|---|
| M0 | nM | 20 | 25 | 25 | (6.7) | - | - |
| MY | nM | 1 | 0.42 | 0.42 | 0.094 | 0.094 | 0.034 |
| MD | nM | 0.02 | 0.029 | 0.029 | (0.12) | - | - |
| ρ | dim' less | 1 | [1] | 1 | (11) | - | - |
| kMP_Y YT | min-1 | (1) | (0.39) | (0.39) | 2.11 | 2.11 | 6.15 |
| kM | min-1 | 0 | [0] | 0 | [0] | 0 | [0] |
| kM_D DT | min-1 | 1 | 0.39 | 0.39 | 0.19 | 0.19 | 0.19 |
| kD | min-1 | - | - | 0.25 | 0.49 | 0.49 | 0.45 |
| kDP | min-1 | - | - | 0.0003 | 0.009 | 0.09 | 0.18 |
| kDP_M | nM-1 min-1 | - | - | 0.01 | 0.073 | 0.73 | 0.64 |
| kDS | min-1 | - | - | - | - | 10 | [10] |
| kD_S | min-1 | - | - | - | - | 1 | [1] |
Values in square brackets [ ] are fixed during optimization. Values in parentheses ( ) are derived from the estimated and fixed parameters according to these definitions:
Parameter set B1opt is defined in terms of ‘derived’ parameters: ρ, M0, MY and MD. We can easily translate these values back to the fundamental rate constants in Eqs. (1-3), whose estimated values are also recorded in Table 5. Notice that we are not able to estimate kY, kD or kDP because we have not yet provided any data to set the time scales for the phosphorylation and dephosphorylation of Cdc25 and Myt1.
Step 2: Adding the rate equation for Cdc25
In a series of elegant experiments, Kumagai and Dunphy (1992;1995) measured directly the rates of phosphorylation and dephosphorylation of Cdc25 and MPF in frog egg extracts prepared in interphase (with MPF activity small) or in M phase (with MPF activity large). These measurements provide an independent test of the rate constants estimated in parameter set B1opt (Table 5) and allow us to estimate kD and kDP. Because Myt1 is a membrane-bound enzyme and hence very difficult to study biochemically in cell extracts, there are no data on the rates of phosphorylation and dephosphorylation of Myt1. Hence, we will not be able to estimate values for kY and kYP_M separately, but can only estimate their ratio MY = kY / kYP_M.
We used PET to fit the Kumagai-Dunphy data (Figs. 4 and 5) to the following two-variable model,
Figure 4. Rates of phosphorylation and dephosphorylation of Cdc25.
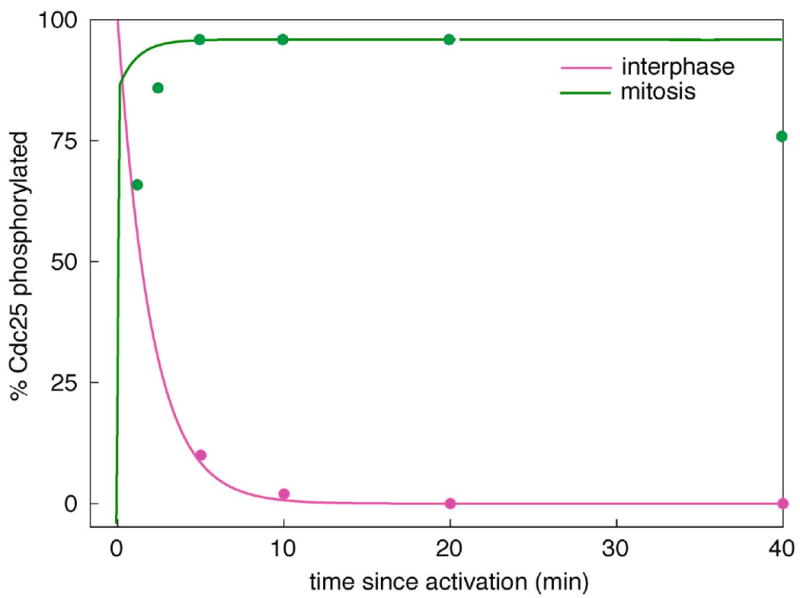
Simulations (lines) are compared to experimental data (circles) estimated from Kumagai et al. 1992, Figure 10. The phosphorylation of Cdc25 during M phase was monitored by gel-shift of endogenous Cdc25, and the dephosphorylation of Cdc25 during interphase was monitored by loss of 32P from radioactively phosphorylated, exogenous Cdc25 added to an extract. The theoretical curves were computed from the full model (Table 2) and the final parameter set (Table 3). Assumptions: [MPF] = 100 nM in M phase; = 0 in interphase.
Figure 5. Rates of phosphorylation and dephosphorylation of MPF.
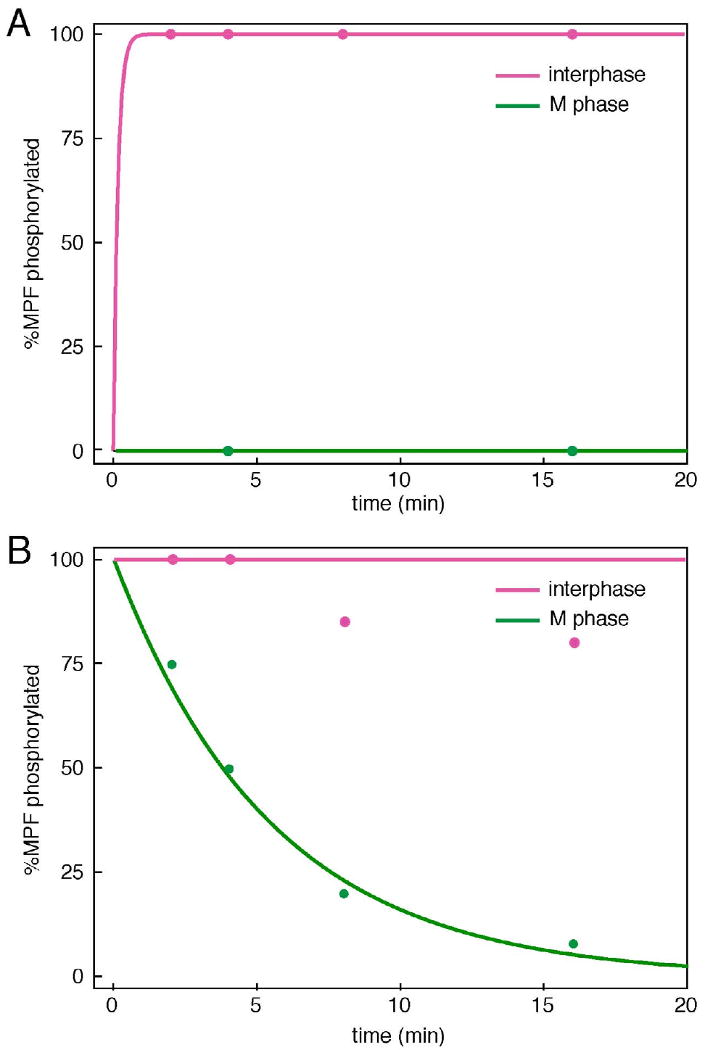
Simulations (lines) are compared to experimental data estimated from Kumagai & Dunphy 1995, Figures 3 and 4. (A) Phosphorylation of MPF during interphase and M-phase was monitored by gel-shift of endogenous MPF. (B) Dephosphorylation of MPF during interphase and M-phase was monitored by loss of 32P from radioactively phosphorylated, exogenous Cdc25 added to an extract. The theoretical curves were computed from the full model (Table 2) and the final parameter set (Table 3).
| (6) |
| (7) |
To simulate an extract arrested in interphase or M-phase, we assumed that [MPF]initial = 0 or 100 nM, respectively. For a starting point we used set B1opt plus some rough guesses for kD, kDP_M and kDP. From this starting point (parameter set B2 in Table 5), PET estimated a set of parameters (B2opt) that provides a good fit for the experimental data in Table 4, rows 1-3.
Nonetheless, there are some discrepancies with the measurements of Kumagai & Dunphy. Their observed rate of Cdc25 phosphorylation in an M-phase extract is about five-fold slower than what the model requires in order to fit other data in Table 4. In addition, their observed rate of Cdc25 phosphorylation in an interphase extract is ten-times faster than what we would expect from kDP ≈ 0.01 min-1. Also, they observed different rates of Cdc25P dephosphorylation in interphase and M phase extracts, suggesting that the phosphatase acting on Cdc25P is regulated in some fashion. Our model has no such regulatory signal. We estimate the rate constant for Cdc25P dephosphorylation (kD) from the interphase extract.
Step 3: Adding the mechanism of the unreplicated-DNA checkpoint
Next, the model was expanded to include relevant features of the DNA replication checkpoint (Fig. 2), in order to simulate the experimental studies of Lee et al. (2001) and Kumagai et al. (1998a) (rows 4 and 5 of Table 4). In these experiments, the extracts are constantly synthesizing endogenous cyclin B, so we added cyclin synthesis (kMSYN) to the model. Degradation of cyclin B is negligible during the time frame of these experiments, so we maintain kMDEG = 0. Two forms of Wee1, were added, an inactive (unphosphorylated) form and an active form that has been phosphorylated by Chk1. A third form of Cdc25, one that is phosphorylated on Ser287 by Chk1, was also introduced. We assumed that phosphorylation of Cdc25 on Ser285 by MPF and on Ser287 by Chk1 are mutually exclusive, based on the work of Bulavin et al. (Bulavin et al., 2003a, b). More recent studies indicate that Cdc25 can be dually phosphorylated (Margolis et al., 2006), but since the conversion from dual (Ser285 and Ser287) to mono phosphorylated (Ser287) forms of Cdc25 is rapid, we leave the ‘exclusivity’ assumption in place.
Step 3A: Estimating the background level of phosphorylation of Ser287
The phosphorylation of Cdc25 on Ser287 by Chk1 is quantified by rate constants kDS, kD_S and kDS_C. Even in the absence of the checkpoint signal ([Chk1] = 0), Cdc25 is significantly phosphorylated on Ser287 (see (Hutchins et al., 2002; Margolis et al., 2006)). According to the model, the steady state fraction of Cdc25 phosphorylated on Ser287 is kDS/(kDS+kD_S) when [Chk1] = 0. As a starting value, we choose kDS/kD_S = 10. (We have no data on the absolute rates of phosphorylation or dephosphorylation of Cdc25 on Ser287; only their ratio is relevant to the experimental data on hand. We assume kDS = 10 min-1 and kD_S = 1 min-1. Later we found, by trial-and-error, that kDS = 3 min-1 is a better starting value.) Because [Cdc25] (i.e., the form not phosphorylated on either Ser285 or Ser287) is now ten-fold smaller in an untreated interphase extract, we increase the values of kDP and kDP_M by ten-fold, to compensate.
Using B3 as a starting point, we let PET fit the new model, including Cdc25 phosphorylation on Ser287, to the data in Table 4, rows 1-3. The results of this fitting procedure are not shown, but the preliminarily optimized parameter values are given in set B3opt in Table 5.
Step 3B: Establishing the role of Wee1 in the unreplicated-DNA checkpoint
The roles of Wee1 during mitotic cycles of frog extracts with or without unreplicated DNA were investigated by Lee et al. (2001). In their experiments, the extracts were prepared with sperm nuclei (typically 1000 nuclei per μl), with or without aphidicolin (APH), an inhibitor of DNA synthesis in the sperm nuclei. Progress into mitosis was monitored as % NEB (nuclear envelope breakdown) of the sperm nuclei. Entry into mitosis is defined as the time when NEB = 50%. In an untreated extract (with nuclei and without APH), the nuclei enter mitosis about 110 min after the extract is prepared (t = 0). In an extracted treated with APH, entry into mitosis is delayed about 80 min (see Fig. 6). If Wee1 is removed from an untreated extract by immunodepletion, there is no acceleration or delay of mitosis (Fig. 6), strongly suggesting that Wee1 plays no effective role in determining entry into mitosis during the unperturbed mitotic cycle. On the other hand, if Wee1 is depleted from a treated extract (checkpoint-induced), then the delay of entry into mitosis is knocked in half (Fig. 6). Hence, it would appear that Wee1 is involved in inhibiting MPF in response to unreplicated DNA but is not involved in inhibiting MPF during unperturbed cycles. It is for this reason that we constructed our model (Fig. 2) with Myt1 as the tyrosine kinase that inhibits MPF during normal mitotic cycles and Wee1 as the tyrosine kinase that relays the signal from unreplicated DNA (via Chk1).
Figure 6. Time course of entry into mitosis: effects of Wee1 depletion and unreplicated DNA.
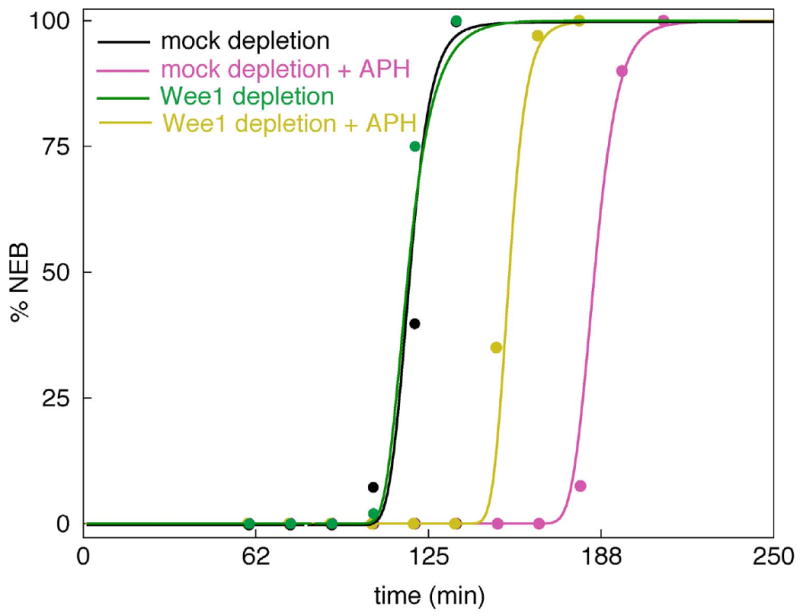
In the experiments of Lee et al. (2001) Wee1-depleted and mock-depleted extracts were monitored for nuclear envelope breakdown (NEB, circles) in the presence and absence of aphidicolin. The theoretical curves were computed from the full model (Table 2) and the final parameter set (Table 3).
Before we can fit the model to NEB measurements, we must relate MPF activity to breakdown of the nuclear envelope. We assume that MPF phosphorylates lamin proteins and that NEB is a steeply sigmoidal function of the fractional phosphorylation of the lamin pool. For details, see ‘Models and Methods’ section.
We fit the model to the curves in Fig. 6 as follows. The mock-depletion curve is the control. It achieves 50% NEB about 110 min after the extract is stimulated with Ca2+. By choosing kMSYN = 0.12 nM min-1, we find that MPF activates at about 100 min and 50% NEB occurs 5-10 min later if we choose: kLP_M = 0.005 nM-1 min-1, μNEB = 0.8 and σNEB = 0.2. These initial estimates are recorded in parameter set Bini in Table 3. Because NEB occurs at the same time in Wee1-depleted extracts as in mock-depleted extracts (in the absence of APH), Wee1 must be in its inactive (unphosphorylated) form in the absence of a checkpoint signal ([Chk1]=0), suggesting that kWP = 0.
Step 3C: Establishing the role of Ser287-phosphorylation of Cdc25 by Chk1
Next, we consider the Wee1-depleted extract in the presence of APH ([Chk1]=1). Because Wee1 is absent, the 30 min delay of NEB is due solely to the phosphorylation of Cdc25 on Ser287 by Chk1. By adjusting kDS_C to the value 7 min-1, we get a 40 min delay of NEB in the Wee1-depleted, APH-treated extract.
Step 3D: Estimating the effect of Chk1-phosphorylated Wee1 in inhibiting MPF
Finally, we used the mock-depleted, APH-treated extracts to measure the role of Wee1 in the unreplicated DNA checkpoint. First, we assumed that kWP_C = 10 min-1 and kW = 1 min-1, so that Wee1 would become 90% phosphorylated (active) when the checkpoint is invoked. Granted this modest assumption, we quickly found that kMP_W WT = 0.08 min-1 is consistent with the 75 min delay experienced by the mock-depleted, APH-treated extract (Fig. 6).
Step 3E: Optimizing the parameter set
All of these estimations and assumptions on rate constant values are collected in parameter set Bini in Table 3. We reduced kDS from 10 min-1 to 3 min-1 and let kDS be a free parameter, to be estimated from the data. Bini is now taken as a starting set for a final optimization of the full model with respect to all the data in Table 2, rows 1-4. The final, optimal parameter set is Bfinal in Table 3. The fit of the full model to the data simultaneously is illustrated in Figs. 3-6.
We have reserved some of the data in Table 2 (rows 5,6) to test the parameterized model.
Test 1a: Adding back wild-type Cdc25 to Cdc25-depleted extracts
Kumagai et al. (1998a) did an interesting experiment in which they depleted an extract of endogenous Cdc25 and then added back a known amount of exogenously synthesized, wild-type Cdc25 (10 μg/ml = 140 nM). The add-back extract experienced NEB about 15 min earlier than the control-depleted extract (see Fig. 7), which has the normal complement of endogenous Cdc25. We suppose that the advance of NEB can be attributed to a higher concentration of total Cdc25 in the add-back extract relative to the control extract. Letting δ be the ratio of total Cdc25 in the add-back extract to total Cdc25 in the control extract (i.e., δ = (140 nM)/DT), we can ask the parameter estimator to find δ that optimizes the fit of the model to the measured curves. The result, δ = 1.3, implies that DT = 108 nM. This is a prediction of the model.
Figure 7. Time course of entry into mitosis: effects of exogenous Cdc25 and unreplicated DNA.
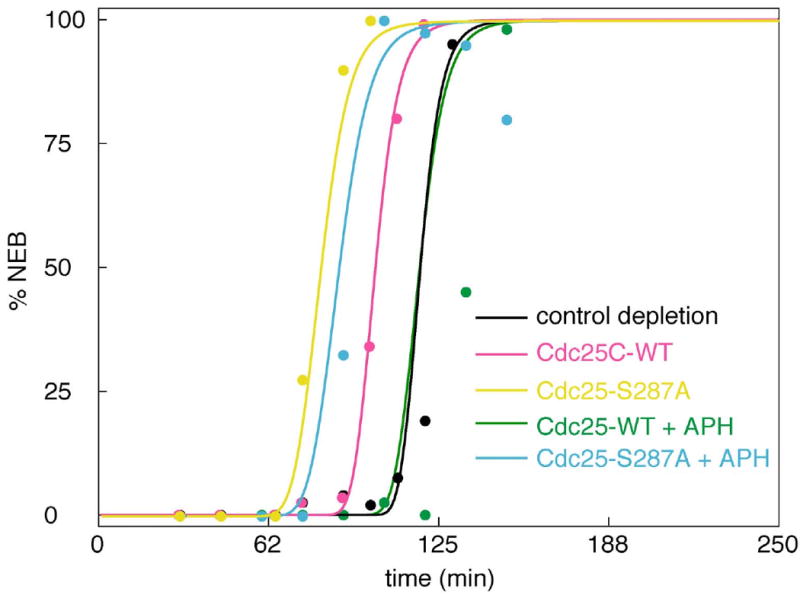
In the experiments of Kumagai et al. (1998a) extracts were immunodepleted of endogenous Cdc25 and exogenous, recombinant Cdc25 was added back, either wild type (WT) or non-phosphorylable by Chk1 (S287A). The theoretical curves were computed from the full model (Table 2) and the final parameter set (Table 3).
Next, Kumagai et al. (1998a) measured the delay of NEB in the Cdc25-WT add-back extract treated with APH (green data points in Fig. 7). These data were not used to estimate the parameters in Bfinal, so they are a valid test of the model. In the model simulations, NEB occurs 17 min earlier than in the experiments.
Test 1b: Adding back Cdc25 that cannot be phosphorylated on Ser287
Finally, Kumagai et al. (1998a) supplemented Cdc25-depleted extracts with an exogenous Cdc25 that cannot be phosphorylated on Ser287 (Cdc25-S287A). These extracts, with or without APH, entered mitosis earlier than control extracts. Based on the final parameter set in Table 3, the model simulations agree nicely with the experimental data (Fig. 7).
Test 2: Simulation of MPF time courses close to the cyclin threshold for MPF activation
To test predictions of the Novák-Tyson model, Sha et al. (2003) measured MPF activity as a function of time for a series of extracts supplemented with decreasing amounts of non-degradable cyclin B. We used a small part of this data set (just 3 points, in Fig. 3 inset) to estimate the magnitude of kM_D DT. Now we use the entire data set as a test of the full model and final, optimized parameter set. The simulated time courses fitted well to the observed time courses (Fig. 8), without any further adjustment of parameter values.
Discussion
Our model (Table 2) of the DNA replication checkpoint in early embryonic frog cells consists of 7 ODEs for the protein interaction network, involving 19 parameters (rate constants, binding constants, etc.). Starting from a robust model of the regulation of MPF (mitosis promoting factor) activity (Novak and Tyson, 2003), we represented the effect of unreplicated DNA as an increase in the activity of the checkpoint kinase Chk1, which phosphorylates and activates the MPF inhibitor Wee1 (Lee et al., 2001) and phosphorylates and inactivates the MPF activator Cdc25. By a stepwise application of our parameter estimation tool (PET), we identified a set of parameter values (Bfinal in Table 3) that fits a large body of experimental data (>110 individual data points), mostly from the published literature. Of the 18 parameters in the model, 14 are constrained by the data and can be estimated with some confidence. Four rate constants are assigned reasonable, nominal values because we have no experimental data that would constrain their values.
This model preserves the key features of hysteresis and bistability in the cell cycle engine. The model also distinguishes the role of Wee1 and Myt1 kinases and suggests that further experimentation is needed to likewise distinguish the relative contributions of their opposing Cdc25A and Cdc25C phosphatases.
Although the MPF kinases, Wee1 and Myt1, were cloned from frog eggs during the same year by the same research group (Mueller et al., 1995a, b), most subsequent experimentation has been performed on Wee1 exclusively, and modelers have either grouped Wee1 and Myt1 as a single enzymatic activity or ignored Myt1 entirely. Part of the reason that Myt1 is less studied is that it is a membrane-associated protein, and therefore, much less easy to manipulate experimentally (e.g., to remove by immunodepletion or to add purified, recombinant protein.) Nonetheless, studies by Lee et al. (2001), in which Wee1 is immunodepleted from frog egg extracts, indicate that Wee1 is not required for timing mitotic entry in unperturbed cell cycles. In early versions of the DNA replication checkpoint model, where Wee1 was the only kinase, simulations of Wee1 depletion caused the extract to lose bistability and enter mitosis prematurely due to the lack of inhibitory phosphorylation on MPF. Our current model corrects this error by adding Myt1 as the major MPF kinase during the normal cell cycle. Basal activity of Wee1 is low, but becomes activated during a DNA replication checkpoint due to its phosphorylation by Chk1. The model fits existing data well (Fig. 7) but requires experimental validation in studies in which Myt1 levels are manipulated directly in frog egg extracts. These studies are underway in our lab and the experimental data will inform the next iteration of the DNA replication checkpoint model.
In addition to validating the treatment of Myt1 and Wee1 in the model, the next version should expand “outward” to describe additional features of the DNA replication control network. Cdc25A needs to be added to the model and its role and regulation distinguished from that of Cdc25C. Studies with intact frog embryos indicate that Cdc25A may be the more important of the two phosphatases in driving the rapid cleavage cell cycle in frog eggs (Kim et al., 1999; Shimuta et al., 2002), and we have confirmed (in unpublished studies) that Cdc25A is translated during the first cycle in the cell-free frog egg extract system. Furthermore, when phosphorylated by checkpoint kinases in response to damaged or unreplicated DNA, Cdc25A is degraded (Sagata, 2002), whereas Cdc25C is reversibly bound to the cytoskeleton via 14-3-3 proteins (Kumagai et al., 1998a). Thus, a combination of modeling and experimental studies is needed to uncover dynamical differences in the regulation of Cdc25A and Cdc25C during both normal cell cycles and when checkpoints are engaged.
When the DNA replication checkpoint model is expanded outward to the event that initiates cell cycle arrest (damaged DNA or stalled DNA replication forks), then other behaviors and properties of the network should be explored. These include the quantitative effects of the total DNA content and the number of DNA lesions or stalled replication forks, as both total DNA and damaged DNA seem to have separate, and possibly additive, effects on the strength of a checkpoint signal. Furthermore, a comprehensive model should reproduce checkpoint adaptation, the phenomenon where the cell cycle resumes after checkpoint arrest, even if the lesion has not been repaired. For frog egg extracts, the model will need to be expanded to include the proteins claspin and Plx1, which interact with Chk1 during checkpoint adaptation (Yoo et al., 2004).
The DNA replication checkpoint model provided an excellent test case for our recently developed PET (Zwolak et al., 2005a, b). Our initial strategy to load the entire model into PET and fit all of the experiment data simultaneously proved unsuccessful. The set of parameter values that was returned did not maintain bistability in the activity of MPF and many of the individual parameter values appeared to be orders of magnitude away from expected values. When no quick fixes were identified, we adopted the more conservative approach to utilizing PET in a step-wise fashion, adding experiments one at a time, beginning with those that demonstrated the fundamental properties of the hysteresis and critical slowing down in the cell cycle engine. Each time new data were added, new parameters values were originally set by hand to fit the data, then PET was run and a new set of parameter values was generated. If the new set reproduced the fundamental behaviors of the system, it was then compared to the previous set to see which was a better fit for the growing body of experimental data points. Thus our approach used PET to refine rather than define parameter values and relied on the user's knowledge of the biological system to guide the order of data input and validate the output from PET. Optimally, parameter estimation software should minimize the need for user input and expertise, but at present, we offer our approach as one that makes efficient use of PET for fitting a medium-to-large numerical model to a large and diverse body of experimental data.
Our construction of the DNA replication checkpoint model also illustrates the value of another tool to modelers: the published experimental literature. Aside from our own experimental data (published in 2003) in which we demonstrated hysteresis and critical slowing down during entry into mitosis (Sha et al., 2003), all data used for parameter estimation was generated in the laboratory of William Dunphy by Akiko Kumagai and others, and published between 1992 and 2001. The experiments were not designed with the intent of informing a mathematical model, and the conclusions drawn from them in the original articles were largely qualitative in nature. Nonetheless, the experiments were skillfully performed and quantitative data could be extracted from the various Western blots and NEB time course in order to inform the model and estimate parameter values. We offer our thanks to Kumagai and Dunphy for their skillful and timeless experiments and suggest to other modelers to invest time reading and mining the primary literature, including older papers, as a rich source of information for building numerical models of complex biological systems.
Models and Methods
Derivation and analysis of the simplified model in Eq. (1)
The simplified model describes the activation and inactivation of cyclin B/Cdk1 by dephosphorylation and phosphorylation (respectively) of the Tyr15 residue of Cdk1. Cdc25 is the phosphatase (as in the Novák-Tyson model) and Myt1 replaces Wee1 as the inhibitory kinase (based on data of Lee et al. (2001)). Both Cdc25 and Myt1 are phosphorylated by Cdk1, leading to their activation and inactivation, respectively. We express these interactions in mathematical terms by introducing the time-dependent variables: M(t) = concentration of MPF (as a function of time), DP(t) = concentration of the phosphorylated (active) form of Cdc25, and Y(t) = concentration of the unphosphorylated (active) form of Myt1. In terms of these variables, the kinetic rate equations for the core interactions of MPF, Cdc25 and Myt1 are assumed to be
| (1) |
| (2) |
| (3) |
In these equations: MT, DT and YT are the (assumed constant) total concentrations of cyclin B, Cdc25 and Myt1 in the reaction system; kM, kD, kDP and kY are first-order rate constants (units = time-1); and kMP_Y, kM_D, kDP_M and kYP_M are second-order rate constants (units = concentration-1 time-1). For the time being, we assume that Cdc25 and Myt1 come quickly to steady state levels as functions of active MPF (later we will relax this assumption). The steady-state solutions of Eqs. (2) and (3) are
With DP and Y given by these equations, Eq. (1) for MPF activity becomes
| (4) |
where ρ = (kMP_YYT)/(kM_DDT) and D0 = kM/kM_D.
Simulating nuclear envelope breakdown (NEB)
To relate the model to key experimental studies of the unreplicated-DNA checkpoint (see Figs. 7 and 8) we must relate MPF activity (output of the model) to % nuclear envelope breakdown (NEB, the experimental observable). Because NEB occurs as a result of phosphorylation by MPF of lamin proteins in the nuclear membrane (Peter et al., 1990), we add to the model a differential equation (#7 in Table 2) for the fraction of lamin proteins that remain unphosphorylated during a time course of MPF activation, [MPF](t). We assume that NEB is an increasing sigmoidal function of [LaminP] = 1 − [Lamin] = fractional phosphorylation of lamin pool. Any sigmoidal function will do. We choose to use the error function, erf(x), which is related to the integral of a Gaussian function with mean μ and standard deviation σ. We choose μ = 0.8 and σ = 0.2, and define
This function has the properties we desire: it ranges from 0 at [LaminP] = 0 to 1 at [LaminP] = 1. And it has a sigmoidal shape: NEB takes on the values 0.1, 0.5 and 0.9 at [LaminP] ≈ 0.55, 0.75 and 0.95, respectively.
Parameter Estimation Toolkit (PET)
PET (Shaffer et al., in press) is a graphical user interface (GUI) for estimating rate constants of a molecular reaction network by fitting simulations to experimental data. It is designed to help theoretical biologists to build regulatory network models and to compare their models to experimental results.
PET allows the user to set up multiple in silico “experiments” and then to scan quickly through alternative parameter values and view simulations of the experimental setups. In silico “experiments” are specified in PET by changes to model parameters and initial conditions that mimic or parallel the protocol of the wet-lab experiments. Experimental data can be input into PET, so the user can see plots, similar to those in this paper, that directly compare experimental data to model simulations.
For automatic parameter estimation PET uses two algorithms, ODRPACK95 (Zwolak et al., 2007) and VTDirect (He, 2007; He et al., 2007), that adjust parameters to fit model simulations to experimental data. ODRPACK95 is a local optimization algorithm based on orthogonal distance regression and Levenberg-Marquardt minimization. Starting from an initial guess, ODRPACK95 moves through parameter space in the direction that provides the fastest improvement of the model's fit to the experimental data, stopping when no further significant improvement can be made. VTDirect is a global optimization algorithm based on the method of dividing rectangles. The user specifies a range for the values of each parameter, and then VTDirect methodically divides the parameter space into parallelepipeds, sampling each region at its center and comparing the model to experimental data at those parameter values. Regions that provide good fits are subdivided further, as are very large regions that have not yet been much explored. VTDirect stops after a maximum number of simulations.
Acknowledgments
This work was funded by grants from the National Institutes of Health (GM076112) to JCS and the Defense Advanced Research Project Agency (F30602-01-2-0572) to JJT. We thank Amit Dravid and Ian Auckland for their preliminary work in building the model.
Abbreviations
- Cdk
cyclin-dependent kinase
- MPF
mitosis-promoting factor
- NEB
nuclear envelope breakdown
- ODE
ordinary differential equation
- PET
Parameter Estimation Tools
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Jason Zwolak, Email: jzwolak@vt.edu.
Nassiba Adjerid, Email: nadjerid@vt.edu.
Elife Z. Bagci, Email: ezbagci@vt.edu.
John J. Tyson, Email: tyson@vt.edu.
Jill C. Sible, Email: siblej@vt.edu.
References
- Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- Bulavin DV, Demidenko ZN, Phillips C, Moody SA, Fornace JAJ. Phosphorylation of Xenopus Cdc25C at Ser285 interferes with ability to activate a DNA damage replication checkpoint in pre-midblastula embryos. Cell Cycle. 2003a;2:263–266. [PubMed] [Google Scholar]
- Bulavin DV, Higashimoto Y, Demidenko ZN, Meek S, Graves P, Phillips C, Zhao H, Moody SA, Piwnica-Worms H, Fornace JAJ. Dual phosphorylation controls Cdc25 phosphatases and mitotic entry. Nat Cell Biol. 2003b;5:545–551. doi: 10.1038/ncb994. [DOI] [PubMed] [Google Scholar]
- Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science. 1996;274:1664–1671. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- Felix MA, Labbe JC, Doree M, Hunt T, Karsenti E. Triggering of cyclin degradation in interphase extracts of amphibian eggs by cdc2 kinase. Nature. 1990;346:379–382. doi: 10.1038/346379a0. [DOI] [PubMed] [Google Scholar]
- Gautier J, Solomon MJ, Booher RN, Bazan JF, Kirschner MW. cdc25 is a specific tyrosine phosphatase that directly activates p34cdc2. Cell. 1991;67:197–211. doi: 10.1016/0092-8674(91)90583-k. [DOI] [PubMed] [Google Scholar]
- Goldbeter A. A minimal cascade model for the mitotic oscilllator involving cyclin and cdc2 kinase. Proc Natl Acad Sci USA. 1991;88:9107–9111. doi: 10.1073/pnas.88.20.9107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Kumagai A, Wang SX, Dunphy WG. Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev. 2000;14:2745–2756. doi: 10.1101/gad.842500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- He J. Vol Ph.D. Department of Computer Science, Virginia Polytechnic Insitutute and State University; Blacksburg, VA: 2007. Design and evaluation of a data-distributed massively parallel implementation of a global optimization algorithm DIRECT. [Google Scholar]
- He J, Watson LT, Sosonkina M. Algorithm XXX: VTDIRECT95: Serial and parallel codes for the global optimization algorithm DIRECT. Virginia Polytechnic Institute and State University; Blacksburg, VA: 2007. [Google Scholar]
- Hekmat-Nejad M, You Z, Yee MC, Newport JW, Cimprich KA. Xenopus ATR is a replication-dependent chromatin-binding protein required for the DNA replication checkpoint. Curr Biol. 2000;10:1565–1573. doi: 10.1016/s0960-9822(00)00855-1. [DOI] [PubMed] [Google Scholar]
- Hutchins JR, Dikovskaya D, Clarke PR. Dephosphorylation of the inhibitory phosphorylation site S287 in Xenopus Cdc25C by protein phosphatase-2A is inhibited by 14-3-3 binding. FEBS Lett. 2002;528:267–71. doi: 10.1016/s0014-5793(02)03327-6. [DOI] [PubMed] [Google Scholar]
- Izumi T, Maller JL. Elimination of cdc2 phosphorylation sites in the cdc25 phosphatase blocks initiation of M-phase. Mol Biol Cell. 1993;4:1337–1350. doi: 10.1091/mbc.4.12.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Li C, Maller J. A maternal form of the phosphatase Cdc25A regulates early embryonic cell cycles in Xenopus laevis. Devel Biol. 1999;212:381–391. doi: 10.1006/dbio.1999.9361. [DOI] [PubMed] [Google Scholar]
- Kumagai A, Dunphy WG. The cdc25 protein controls tyrosine dephosphorylation of the cdc2 protein in a cell-free system. Cell. 1991;64:903–914. doi: 10.1016/0092-8674(91)90315-p. [DOI] [PubMed] [Google Scholar]
- Kumagai A, Dunphy WG. Regulation of the cdc25 protein during the cell cycle in Xenopus extracts. Cell. 1992;70:139–151. doi: 10.1016/0092-8674(92)90540-s. [DOI] [PubMed] [Google Scholar]
- Kumagai A, Dunphy WG. Control of the Cdc2/Cyclin B complex in Xenopus egg extracts arrested at a G2/M checkpoint with DNA synthesis inhibitors. Mol Biol Cell. 1995;6:199–213. doi: 10.1091/mbc.6.2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Yakowec PS, Dunphy WG. 14-3-3 proteins act as negative regulators of the mitotic inducer Cdc25 in Xenopus egg extracts. Mol Biol Cell. 1998a;9:345–354. doi: 10.1091/mbc.9.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Guo Z, Emami KH, Wang SX, Dunphy WG. The Xenopus Chk1 protein kinase mediates a caffeine-sensitive pathway of checkpoint control in cell-free extracts. J Cell Biol. 1998b;142:1559–1569. doi: 10.1083/jcb.142.6.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kumagai A, Dunphy WG. Positive regulation of Wee1 by Chk1 and 14-3-3 proteins. Mol Biol Cell. 2001;12:551–563. doi: 10.1091/mbc.12.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorca T, Castro A, Martinez AM, Vigneron S, Morin N, Sigrist S, Lehner C, Doree M, Labbe JC. Fizzy is required for activation of the APC/cyclosome in Xenopus egg extracts. EMBO J. 1998;17:3565–3575. doi: 10.1093/emboj/17.13.3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupardus PJ, Byun T, Yee MC, Hekmat-Nejad M, Cimprich KA. A requirement for replication in inactivation of the ATR-dependent DNA damage checkpoint. Genes Dev. 2002;16:2327–2332. doi: 10.1101/gad.1013502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis SS, Perry JA, Weitzel DH, Freel CD, Yoshida M, Haystead TA, Kornbluth S. A role for PP1 in the Cdc2/Cyclin B-mediated positive feedback activation of Cdc25. Mol Biol Cell. 2006;17:1779–89. doi: 10.1091/mbc.E05-08-0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller PR, Coleman TR, Dunphy WG. Cell cycle regulation of a Xenopus Wee1-like kinase. Mol Biol Cell. 1995a;6:119–134. doi: 10.1091/mbc.6.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller PR, Coleman TR, Kumagai A, Dunphy WG. Myt1: a membrane-associated inhibitory kinase that phosphorylates Cdc2 on both threonine-14 and tyrosine-15. Science. 1995b;270:86–90. doi: 10.1126/science.270.5233.86. [DOI] [PubMed] [Google Scholar]
- Novak B, Tyson JJ. Numerical analysis of a comprehensive model of M-phase control in Xenopus oocyte extracts and intact embryos. J Cell Sci. 1993;106:1153–1168. doi: 10.1242/jcs.106.4.1153. [DOI] [PubMed] [Google Scholar]
- Nyberg KA, Michelson RJ, Putnam CW, Weinert TA. Toward maintaining the genome: DNA damage and replication checkpoints. Ann Rev Genet. 2002;36:617–656. doi: 10.1146/annurev.genet.36.060402.113540. [DOI] [PubMed] [Google Scholar]
- Peter M, Nakagawa J, Doree M, Labbe JC, Nigg EA. In vitro disassembly of the nuclear lamina and M phase-specific phosphorylation of lamins by cdc2 kinase. Cell. 1990;61:591–602. doi: 10.1016/0092-8674(90)90471-p. [DOI] [PubMed] [Google Scholar]
- Pomerening JR, Sontag ED, Ferrell JEJ. Buidling a cell cycle oscillator: hysteresis and bistability in activation of Cdc2. Nat Cell Biol. 2003;5:346–351. doi: 10.1038/ncb954. [DOI] [PubMed] [Google Scholar]
- Qin J, Li L. Molecular anatomy of the DNA damage and DNA replication checkpoints. Radiation Res. 2003;159:139–148. doi: 10.1667/0033-7587(2003)159[0139:maotdd]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Sagata N. Untangling checkpoints. Science. 2002;298:1905–1907. doi: 10.1126/science.1079225. [DOI] [PubMed] [Google Scholar]
- Sha W, Moore J, Chen K, Lassaletta AD, Yi CS, Tyson JJ, Sible JC. Hysteresis drives cell-cycle transitions in Xenopus laevis egg extracts. Proc Natl Acad Sci U S A. 2003;100:975–80. doi: 10.1073/pnas.0235349100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer CA, Zwolak JW, Randhawa R, Tyson JJ. Modeling molecular regulatory networks with JigCell and PET. In: Maley I, editor. Systems Biology. Human Press in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimuta K, Nakajo N, Uto K, Hayano Y, Okazaki K, Sagata N. Chk1 is activated transiently and targets Cdc25A for degradation at the Xenopus midblastula transition. EMBO J. 2002;21:3694–3703. doi: 10.1093/emboj/cdf357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smythe C, Newport JW. Coupling of mitosis to the completion of S phase in Xenopus occurs via modulation of the tyrosine kinase that phosphorylates p34cdc2. Cell. 1992;68:787–797. doi: 10.1016/0092-8674(92)90153-4. [DOI] [PubMed] [Google Scholar]
- Solomon MJ, Glotzer M, Lee TH, Phillippe M, Kirschner MW. Cyclin activation of p34cdc2. Cell. 1990;63:1013–1024. doi: 10.1016/0092-8674(90)90504-8. [DOI] [PubMed] [Google Scholar]
- Yoo HY, Kumagai A, Shevchenko A, Dunphy WG. Adaptation of a DNA replication checkpoint response depends upon inactivation of claspin by the polo-like kinase. Cell. 2004;117:575–588. doi: 10.1016/s0092-8674(04)00417-9. [DOI] [PubMed] [Google Scholar]
- Zwolak JW, Tyson JJ, Watson LT. Parameter estimation from a mathematical model. J Comput Biol. 2005a;12:48–63. doi: 10.1089/cmb.2005.12.48. [DOI] [PubMed] [Google Scholar]
- Zwolak JW, Tyson JJ, Watson LT. Globally optimized parameters for a model of mitotic control in frog egg extracts. IEE Proc -Syst Biol. 2005b;152:81–92. doi: 10.1049/ip-syb:20045032. [DOI] [PubMed] [Google Scholar]
- Zwolak JW, Boggs PT, Watson LT. ODRPACK95: A weighted orthogonal distance regression code with bound constraints. ACM Trans Math Software. 2007;33 [Google Scholar]