

Abstract
Synthesis of a new class of phosphatidylcholine analogues derived from glyceric acid is reported for spectroscopic studies of phospholipases and conformation of phospholipid side-chains in biological membranes, using fluorescence resonance energy transfer (FRET) techniques.
Keywords: phosphatidylcholine analogue synthesis, glyceric acid ester, orthogonal O-protection, fluorescence resonance energy transfer (FRET)
Biologically active synthetic phospholipid compounds are required for structural and dynamic studies of biomembranes for establishment of structure-activity relationships with respect to phospholipid-phospholipid and phospholipid-protein interactions, as well as for mechanistic studies of phospholipid metabolizing enzymes.1–3 Specifically, with the discovery that phospholipases generate a number of physiologically important lipid metabolites, including second messengers that are involved in cell signaling,1 development of new synthetic methods for preparation of phospholipid derivatives became a key step in advancing membrane biochemistry. Phospholipid analogues incorporating spectroscopically active reporter groups have been shown to be valuable structural probes to study conformation and dynamics of phospholipids in aggregates (e.g. micelles, bilayers, and vesicles) as well as for development of spectroscopic assay systems of lipolytic enzymes,4–6 and for in vivo monitoring the fate of products generated by phospholipids metabolizing enzymes.7
As a part of our work in this area we focused on development of highly specific fluorescent phospholipase A2 substrates for kinetic studies and for detection of in vivo activity of the enzyme. Phospholipases A2 (PLA2, EC 3.1.1.4) comprise a large group of intracellular and secreted enzymes that catalyze the hydrolysis of the sn-2 ester function of glycerophospholipids to produce free fatty acids, such as arachidonic acid, and lysophospholipids.8 Both products are precursors of signaling molecules with a multitude of biological functions.8–13 Specifically, arachidonic acid is converted to eicosanoids that have been shown to be involved in immune response, inflammation, pain perception, and sleep regulation,8,9 while lysophospholipids are precursors of lipid mediators such as lysophosphatidic acid (LPA) and platelet activating factor (PAF). LPA is involved in cell proliferation, survival and migration, and PAF is particularly involved in inflammatory processes.8 Thus, mammalian secreted phospholipase A2’s (sPLA2s) have been implicated in a series of physiological and pathophysiological functions including lipid digestion, cell proliferation, neurosecretion, antibacterial defense, cancer, and inflammatory diseases.10 Kinetic characterization and mechanistic elucidation of sPLA2 enzymes should contribute to better understanding of their exact physiological role in these events, which remain to be elucidated.11 In addition, fluorescence-based phospholipase A2 assay methods should also be applicable for high-throughput screening of potential PLA2 inhibitors.4 Significantly, recent studies indicated therapeutic potential of phospholipase A2 inhibitors in cardiovascular disease,12 and it has also been suggested that select isoforms of the enzyme may be targets for drugs in the treatment of cancer.13
In the present communication we report the synthesis of a new series of phospholipid compounds derived from of glyceric acid (1), incorporating an “inverse ester” function at the sn-1-position preventing hydrolysis by PLA1, while keeping the sn-2-ester group intact for cleavage by phospholipase A2 enzymes (2).
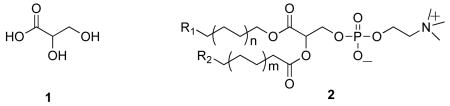
Phospholipid analogues such as compound 2 should also be applicable to detect the activity of PLA2 enzymes, because the change in fluorescence depends on specific cleavage of the ester linkage at the sn-2-position of the substrate. In designing the target structure we selected two photostable coumarin fluorophores with the requisite spectroscopic properties to provide a suitable donor-acceptor pair.14 In addition, the chain-terminal reporter groups are relatively small in size in order to minimize the impact on the properties of the hydrocarbon chains. Using the “inverse ester” strategy instead of relying on the sn-1-alkyl ether substitution to achieve PLA2 selectivity brings the compound closer to the naturally occurring glycerophospholipid structure.1 Furthermore, the synthetic sequence is designed in such a way that includes lysophospholipid intermediates, to allow incorporation of substituents with chain-terminal functional groups, including those that would not survive the reaction conditions involved in phosphorylation. Significantly, fluorescent lysophospholipid compounds have been shown to be important biologically active lipid analogues in their own right.1,15
The synthetic strategy, outlined in Scheme 1, is based upon two key elements. First, commercially available 2,3-O-isopropylidene-L-methyl glycerate 3 is used to provide the three-carbon skeleton to construct the target phospholipid compound. Specifically, we have discovered that while glyceric acid itself is rather intractable due to its poor solubility in organic solvents, base-catalyzed hydrolysis of the acetonide 3 readily affords the corresponding sodium glycerate in essentially quantitative yield. Treatment of the sodium salt with ion exchange resin provides the corresponding free acid, which in turn can be converted to the long-chain fluorescent ester 4 in condensation with the desired alcohol.16 The second principal element involves the orthogonal protection of the diol portion of the glyceric acid skeleton. Here we selected the phenoxyacetyl group to provide base-labile protection at the sn-3-position, since we have found that acylation of compound 5 using a combination of phenoxyacetyl chloride/collidine in CHCl3 proceeds with high regioselectivity at the primary hydroxyl group, the only byproduct being the diacyl derivative that is readily separated from the product 6 on column chromatography. On the other hand, for acid labile protection at the sn-2-alcohol function tetrahydropyranylation turned out to be the most suitable choice. Specifically, both introduction of the tetrahydropyranyl function as well as its cleavage later on in the sequence, can be carried out under mild acidic conditions, and most importantly without acyl group migration from the adjacent ester function. Although the protection step introduces a second chiral center, it does not complicate the synthesis because the intermediate can easily be carried through the sequence as a diastereoisomeric mixture up to the deprotection step.
Scheme 1.
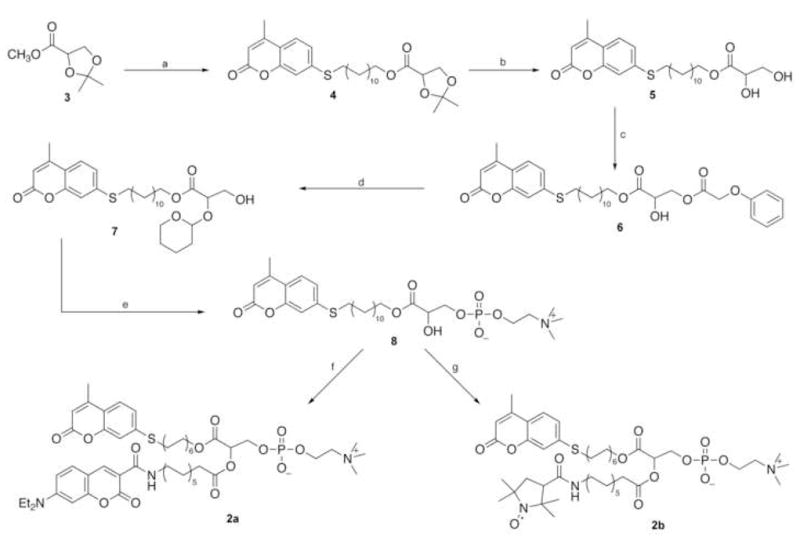
Reagents and conditions: (a) (i) 1.2 M NaOH, 80% aq. dioxane, (ii) Bio-Rad AG 50-X(H+), (iii) 12-(7′-mercapto-4′methylcoumarin)dodecanol-DCC-DMAP, CHCl3;(b) 0.4 M HCl, aq. dioxane, 2 h; (c) 1.4 equiv. C6H5OCH2COCl, 2,4,6-trimethylpyridine, CHCl3, 0°C, 3 h; (d) (i) DHP, PPTS, CH2Cl2, rt, 3 h, (ii) 5 equiv. tert.-butylamine, CHCl3-MeOH (1:1), 0°C, 14 h; (e) ethylene chlorophosphate, Et3N, benzene, (ii) (CH3)3N, MeCN, 65°C; (iii) 0.3 M HCl, rt, 6 h; (f) (i) FMOC-12-aminododecanoic acid-DCC-DMAP, CHCl3, (ii) DBU, rt, 1 h, (iii) p-nitrophenyl-7-diethylaminocoumarin-3-carboxylate, DMAP, CHCl3; (g) (i) and (ii) as in (f), then p-nitrophenyl-PROXYL-3-carboxylate, DMAP, CHCl3.
Thus, hydrolysis of methyl 2,3-O-isopropylidene-L-glycerate 3 with stoichiometric amount of 1.2 M NaOH in 80% aqueous 1,4-dioxane at room temperature for 2 h yielded the sodium salt of the protected glyceric acid as a white powder (97%). The compound was isolated from the reaction mixture by freeze-drying. The product was treated with 30 mL Bio-Rad AG50-X (H+) ion exchange resin in dioxane for 15 min at room temperature to obtain the carboxylic acid as a colorless oil, which was converted to the fluorescent ester 4 in reaction with 12-(7′-mercapto-4′-methylcoumarin)dodecanol-DCC-DMAP in chloroform at room temperature for 6 h. The resulting ester 4 was purified by silica gel chromatography (CHCl3-EtOAc 9.5:0.5), and freeze-dried from benzene as a white solid (88%). Acid-catalyzed hydrolysis of the isopropylidene function with 0.4 M HCl in 97% aq. dioxane at room temperature for 2 h, followed by freeze-drying and subsequent chromatography (CHCl3-EtOAc 5:1) led to the pure diol 5 in 78% yield.
Regioselective acylation at the primary hydroxyl group of compound 5 was accomplished using 1.4 equiv. phenoxyacetyl chloride in the presence of 2.0 equiv 2,4,6-trimethyl pyridine in chloroform at 0°C for 3 h. The product 6 was purified by silica gel chromatography using chloroform-ethyl acetate (5:1) as eluant, then freeze-dried from benzene to give a colorless oil that turned to a white waxy solid on standing (68%). The high-field 1H-NMR spectrum of compound 6 shows baseline resonance in the δ 5.00–5.09 region, confirming that the secondary hydroxyl group remained intact in the isolated product.17 Next, the tetrahydropyranyl function was elaborated at the sn-2-glycerol position using fivefold excess of 3,4-dihydro-2H-pyran with pyridinium p-toluenesulfonate catalysis in CH2Cl2 at room temperature for 3 h, in 96% yield, and base-catalyzed chemoselective cleavage of the phenoxyacetyl ester function was accomplished using 5 equiv. tert.-butylamine in chloroform-methanol (1:1) at 0°C for 14 h. The product 7 was purified by column chromatography on silica gel using a stepwise gradient of CHCl3-EtOAc (9:1 followed by 6:1) to obtain the primary alcohol 7 in 97% yield.
Phosphorylation of the substituted glycerol 7 at the sn-3-position was carried out using 2 equiv. 2-chloro-2-oxo-1,3,2-dioxaphospholane and triethylamine in sodium-dried benzene at room temperature for 4 h. The phosphorylated intermediate was treated with 5 mL anhydrous trimethylamine in acetonitrile at 65°C (in a pressure bottle) for 24 h to afford the crude product that separated from the reaction mixture on cooling. This intermediate was purified by silica gel chromatography (CHCl3-CH3OH-H2O 65:25:4, 74% yield), and treated with a 0.3 M HCl in a biphasic system of chloroform-water (4:0.1) for 2 h. Freeze-drying from benzene yielded the crude fluorescent lysophospholipid 8, which was purified by chromatography (CHCl3-CH3OH-H2O 65:25:4) and isolated as a white solid (57%). Next, the compound 8 was acylated with 2.5 fold excess of FMOC-12-aminododecanoic acid-DCC-DMAP in CHCl3 in the presence of glass beads at 25oC, with sonication, for 5 h.18 Purification by silica gel chromatography, first with CHCl3/MeOH (4:1), followed by CHCl3-MeOH-H2O (65:25:4) as eluant gave the FMOC-protected phosphatidylcholine derivative (60%). Base-catalyzed elimination of the FMOC protecting group using 4 equiv. DBU in CHCl3 at room temperature for 1 h, followed by treatment with p-nitrophenyl-7-diethylaminocoumarin-3-carboxylate in the presence of catalytic amount of DMAP for 4 h in the same reaction mixture,18 afforded the target phospholipid 2a. Purification by silica gel chromatography using the same solvent systems as above yielded the fluorescent product 2a (66% from 8) as a yellow solid.19
With the availability of compound 8 we were able to extend the scope of reporter groups to include a nitroxide spin label introduced at the sn-2-ester group’s chain-terminal 2b. Conversion of compound 8 in the same sequence of reactions as outlined for 2a afforded the double-labeled phosphatidylcholine derivative 2b in 52% overall yield.
Both compounds 2a and 2b were completely hydrolyzed by bee-venom phospholipase A2, yielding the corresponding lysophospholipid 8 (eq.1).
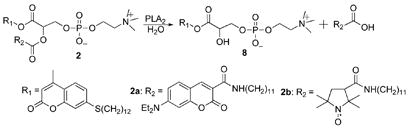 |
(1) |
Specifically, hydrolysis of 2a occurs with decrease in the fluorescence emission of the acceptor at 462 nm, and increase in the emission of the donor at 397 nm due to loss of fluorescence resonance energy transfer. On the other hand, cleavage of the sn-2-ester linkage of 2b leads to enhanced fluorescence at 397 nm via de-quenching, due to release of the paramagnetically labeled fatty acid from the molecule.20
In conclusion, the synthesis here reported provides a facile and efficient method for the preparation of a new class of phospholipid analogues. The method should be applicable for the preparation of a wide range of functionalized phospholipid derivatives not only for development of new real-time spectrophotometric assays of phospholipases and high-throughput screening of phospholipase inhibitors, but also for the design and development of new membrane probes for the study of conformation and interaction of phospholipids in monolayers, bilayers and micelles. Work toward these goals is underway in our laboratory.
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of Health, grant 2S06 GM/HD48680 for financial support.
Footnotes
Supplementary Data. Characterization of the synthetic compounds by spectroscopic methods including IR, 1H, 13C NMR, HRMS, and elemental analyses are given in the Electronic Supplementary Information. The supplementary data is available online with the paper in Science Direct.
References and notes
- 1.McPhail L. In: Biochemistry of Lipids, Lipoproteins and Membranes. 4. Vance DE, Vance JE, editors. Elsevier Science; Amsterdam: 2002. pp. 315–340. [Google Scholar]
- 2.(a) Lee AG. Biochim Biophys Acta. 2003;1612:1–40. doi: 10.1016/s0005-2736(03)00056-7. [DOI] [PubMed] [Google Scholar]; (b) Carrillo-Tripp M, Feller SE. Biochemistry. 2005;44:10164–10169. doi: 10.1021/bi050822e. [DOI] [PubMed] [Google Scholar]; (c) Wang P, Blank DH, Spencer TA. J Org Chem. 2004;69:2693–2702. doi: 10.1021/jo030329d. [DOI] [PubMed] [Google Scholar]
- 3.Berg OG, Gelb MH, Tsai MD, Jain MK. Chem Rev. 2001;101:2613–3653. doi: 10.1021/cr990139w. [DOI] [PubMed] [Google Scholar]
- 4.(a) Fuji M, Watanabe F, Fuji Y, Hashizume H, Okuno T, Shirahase K, Teshirogi I, Ohtani M. J Org Chem. 1997;62:6804–6809. [Google Scholar]; (b) Reynolds LJ, Hughes LL, Yu L, Dennis EA. Anal Biochem. 1994;217:25–32. doi: 10.1006/abio.1994.1079. [DOI] [PubMed] [Google Scholar]
- 5.(a) Wichmann O, Schultz C. Chem Commun. 2001:2500–2501. doi: 10.1039/b107670c. [DOI] [PubMed] [Google Scholar]; (b) Mukherjee S, Raghuraman H, Dasgupta S, Chattopadhyay A. Chem Phys Lipids. 2004;127:91–101. doi: 10.1016/j.chemphyslip.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 6.Feng L, Manabe K, Shope JC, Widmer S, DeWald DB, Prestwich GD. Chem Biol. 2002;9:795–803. doi: 10.1016/s1074-5521(02)00168-0. [DOI] [PubMed] [Google Scholar]
- 7.Farber SA, Pack M, Ho SY, Johnson ID, Wagner DS, Dosch R, Mullins MC, Hendrickson HS, Hendrickson EK, Halpern ME. Science. 2001;292:1385–1388. doi: 10.1126/science.1060418. [DOI] [PubMed] [Google Scholar]
- 8.Schaloske RH, Dennis EA. Biochim Biophys Acta. 2006;1761:1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 9.Funk CD. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 10.Valentin E, Ghomashchi F, Gelb MH, Ladzunski M, Lambeau G. J Biol Chem. 2000;275:7492–7496. doi: 10.1074/jbc.275.11.7492. [DOI] [PubMed] [Google Scholar]
- 11.Kudo I, Murakami M. Prostaglandins Other Lipid Mediat. 2002;68–69:3–58. doi: 10.1016/s0090-6980(02)00020-5. [DOI] [PubMed] [Google Scholar]
- 12.White MC, McHowat J. Cardiovascular Hematological Agents. Med Chem. 2007;5:91–95. doi: 10.2174/187152507779315859. [DOI] [PubMed] [Google Scholar]
- 13.Cummings BS. Biochem Pharmacol. 2007;74:949–959. doi: 10.1016/j.bcp.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 14.The fluorescence emission peak of the donor, 7-mercapto-4-methylcoumarin group (λem = 397 nm) shows substantial overlap with the excitation spectrum of the acceptor, 7-dimethyl-aminocoumarin (λexc = 405 nm), and its emission can be followed at λem = 462 nm. These spectral parameters have been recorded in aqueous buffer at pH 8, in the presence of Triton X-100, conditions that have been used for assaying phospholipase A2 enzymes.3
- 15.(a) Ishii I, Fukushima N, Ye X, Chun J. Ann Rev Biochem. 2004;73:321–340. doi: 10.1146/annurev.biochem.73.011303.073731. [DOI] [PubMed] [Google Scholar]; (b) Xu Y. Biochim Biophys Acta. 2002;1582:81–88. doi: 10.1016/s1388-1981(02)00140-3. [DOI] [PubMed] [Google Scholar]
- 16.To best of our knowledge this is the first synthetic method for preparation of glyceric acid esters.
- 17.Roodsari FS, Wu D, Pum GS, Hajdu J. J Org Chem. 1999;64:7727–7737. [Google Scholar]
- 18.Rosseto R, Hajdu J. Tetrahedron Lett. 2005;46:2941–2944. [Google Scholar]
- 19.The new compounds were characterized by spectroscopic methods including IR, 1H, 13C NMR, HRMS, and elemental analysis. In addition, compound 2a and 2b were completely hydrolyzed by bee-venom phospholipase A2 yielding the corresponding sn-2-lysophospholipid 8 as a single phosphate positive spot on thin layer chromatography, with the same Rf as the authentic reference compound. For experimental details of the assay system used cf. Balet C, Clingman KA, Hajdu J. Biochem Biophys Res Commun. 1988;150:561–567. doi: 10.1016/0006-291x(88)90430-5.
- 20.Ranganathan R, Tcacenco CM, Rosseto R, Hajdu J. Biophys Chem. 2006;122:79–89. doi: 10.1016/j.bpc.2006.02.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.