

Abstract
Background
Familial partial lipodystrophy, Dunnigan variety is a rare autosomal dominant disorder caused by missense mutations in LMNA gene. Individuals are predisposed to insulin resistance and its complications including features of polycystic ovarian syndrome.
Case
A 27 year-old Hispanic female presented with oligomenorrhea and hirsutism. Examination revealed Cushingoid facies, significant hirsutism, acanthosis nigricans, and a lean body habitus. Metabolic testing identified diabetes mellitus, dyslipidemia, and steatohepatitis. A diagnosis of familial partial lipodystrophy, Dunnigan variety was confirmed by the detection of a heterozygous p.Arg482Trp (c.1444C>T) missense mutation in lamin A/C (LMNA) gene. Subsequently, seven female relatives were diagnosed with familial partial lipodystrophy, Dunnigan variety, four of whom had menstrual irregularities.
Conclusion
Familial partial lipodystrophy, Dunnigan variety can present with features similar to polycystic ovarian syndrome. Diagnosis is critical as the metabolic complications of the disorder have significant morbidity.
Introduction
Lipodystrophies are a heterogeneous group of acquired and inherited disorders characterized by selective loss of adipose tissue. While genetic lipodystrophies are rare, acquired lipodystrophy due to antiretroviral therapy in HIV-infected patients is increasingly recognized. The extent of fat loss in the different lipodystrophy syndromes varies, and may be either generalized or restricted to certain regions. Familial partial lipodystrophy, Dunnigan Variety (FPLD) is a rare autosomal dominant disorder, first described by Dunnigan et al in 1974 (1) with over 300 subsequent patients being reported. It is caused by heterozygous missense mutations in the lamin A/C (LMNA) gene, but the molecular basis of fat loss due to LMNA mutations is not clear.
Patients with FPLD have normal fat distribution durisng childhood, but following puberty, demonstrate progressive loss of adipose tissue from the extremities, variable loss from the body corpus, and often, excess fat deposition in the face, neck, and intra-abdominal regions (2). Fat loss leads to a characteristic muscular appearance, and other notable features include acanthosis nigricans and hepatomegaly. Metabolic abnormalities related to insulin resistance such as diabetes mellitus, hypertriglyceridemia and hepatic steatosis are commonly noted, especially in affected women. Not only is the diagnosis more apparent in women because of the unusual muscular prominence, but also the metabolic complications are more severe (3). Further, oligomenorrhea and hirsutism have been reported in female subjects, and thus is important to consider the diagnosis of FPLD in lean, muscular women who have polycystic ovarian syndrome (PCOS). We report a patient with typical features of PCOS who was subsequently diagnosed to have FPLD.
Case
A 27-year-old gravida 0 Hispanic female was referred to a gynecology clinic at a teaching institution for oligomenorrhea, hirsutism, and infertility. Gynecologic history revealed menarche at age 12, followed by regular cycles for 5 years. Thelarche was not identifiable as the patient described lack of breast development, and she could not recall time of pubarche. At approximately age 17, the patient described the onset of oligomenorrhea, which ensued for 9 years, after which she became amenorrheic. The patient also described a 12-year history of progressively worsening hirsutism, leading to a full beard which she shaved daily, and significant hair growth over her chest, abdomen, and back. The irregular, anovulatory cycles, together with hirsutism had resulted in the patient previously receiving a diagnosis of PCOS. In addition to these complaints, the patient was also concerned about her physical appearance, specifically her “double chin” and small breasts. Her sister, mother and maternal grandmother also had a similar body habitus, with the latter two also having diabetes.
Physical examination revealed a weight of 65.6 kg and height of 160.5 cm (body mass index of 25.5 kg/m2). She had marked loss of subcutaneous fat from the extremities leading to prominence of muscular contours and veins, with excess fat around the face, submental and dorsocervical region giving her a Cushingoid appearance (Fig. 1A and B). Breast tissue was nearly absent giving the appearance of Tanner stage 1 breasts with hirsute nipples. A male-pattern hair distribution was observed, with complete but light coverage of terminal hair on the abdomen, chest, back, upper arms, thighs, and buttocks (Ferriman-Gallwey score 39/48). She had extensive acanthosis nigricans of the neck, axilla, groin, and midline of chest, abdomen, and back, and was also found to have non-tender hepatomegaly. Vulvar examination revealed excess fat deposition. Ultrasonographic findings included a normal-appearing uterus and enlarged ovaries, with a total of more than 30 small follicles (measuring 2–9 mm in diameter), and ovarian volumes of 16.6 and 10.7 mL.
Figure 1.
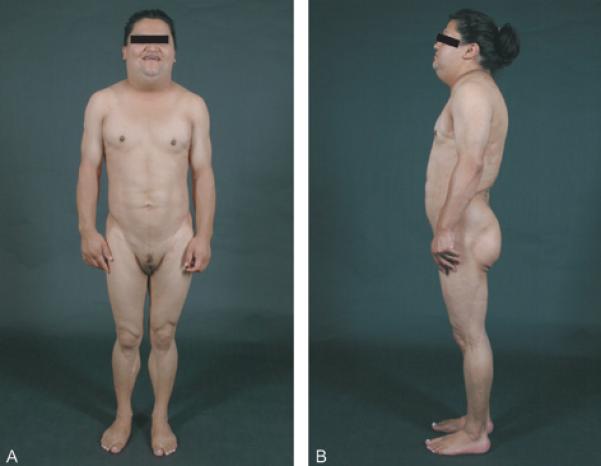

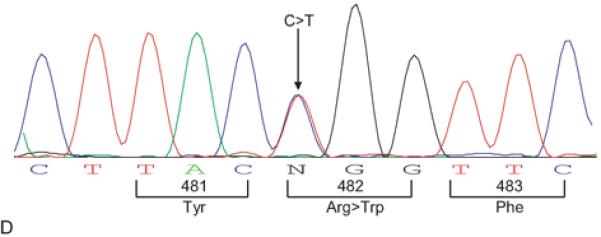
(A and B): Anterior and lateral views of the index patient showing marked loss of subcutaneous fat from the extremities, hips and anterior abdomen, with excess fat deposition around the face, and in the submental and dorsocervical regions. Also note the presence of acanthosis nigricans over the neck, poor breast development, and increased vulvar fat. (C): FPLD Pedigree with patient numbers corresponding to data in Table 1. Affected individuals are shown as filled black symbols, unaffected subjects as unfilled symbols, and deceased individuals are indicated by a diagonal line. Squares represent males and circles represent females. Arrow indicates the index subject. RR indicates a subject with the normal LMNA genotype (Arginine in position 482 for both alleles), while RW indicates patients with the heterozygous missense LMNA mutation (Arginine and Tryptophan in position 482). (D) Sequence electropherogram of LMNA exon 8, with arrow showing the site of heterozygous missense mutation, c.1444C>T, resulting in substitution of one of the normal alleles, Arginine by Tryptophan at position 482. The colors denote nucleic acids; blue=cytosine (C), green=adenine (A), red=thymine (T), black=guanine (G). N, possible missense mutation.
Initial laboratory evaluation revealed slightly increased total serum testosterone of 100 ng/dL (normal values, 11–59 ng/dL), but normal levels of serum thyroid stimulating hormone, prolactin, dehydroepiandrosterone sulphate, basal and ACTH stimulated 17- hydroxy progesterone, and 24 h urinary cortisol levels. Her fasting and post prandial plasma glucose levels after an oral glucose load were diagnostic of diabetes mellitus (151 and 298 mg/dL, respectively). She also had hypertriglyceridemia (158 mg/dL) and low levels of high density lipoprotein cholesterol (32 mg/dL). Liver function tests were abnormal with 2–3 fold elevation of serum alanine aminotransferase and aspartate aminotransferase (174 and 110 U/L, respectively), and right upper quadrant ultrasound revealed diffuse fatty infiltration of the liver.
In view of her characteristic physical appearance, history of similar affliction in other members of the family, and evidence of metabolic abnormalities related to insulin resistance, it was felt that her PCOS could be secondary to FPLD. She underwent further evaluation at University of Texas Southwestern Medical Center at Dallas along with other family members. Written informed consent was obtained. The protocol had been approved by the Institutional Review Board at UT Southwestern Medical Center. Measurement of skin fold thickness, and whole body magnetic resonance imaging confirmed loss of adipose tissue from the extremities with excess accumulation in the dorsocervical and intermuscular regions (Fig. 2). The patient's fasting leptin level was low at 2.3 ng/dL (less than 10th percentile of age and sex matched normal subjects). Sequencing of LMNA gene revealed a heterozygous missense mutation (c.1444C>T) leading to substitution of Arginine by Tryptophan at position 482 (R482W), confirming the diagnosis of FPLD (Fig. 1D). The same mutation was identified in other family members, including her mother, sister, maternal aunt, cousin, and 3 nieces (Fig. 1C). They had similar body fat distribution, but variable metabolic and menstrual irregularities as indicated in Table 1.
Figure 2.
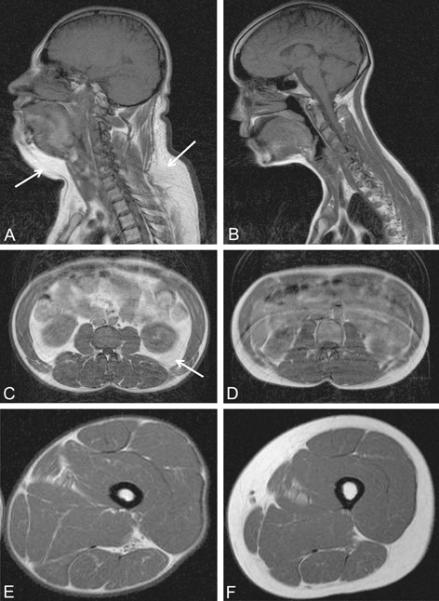
T1 weighted magnetic resonance images of the head and neck, abdomen and thigh from the index patient (A, C and E, respectively) and a normal 25-year old female subject (B, D and F, respectively). Compared to the normal subject, patient has increased fat in the dorsocervical, submental and intra-abdominal regions (as shown by arrows), but marked paucity of subcutaneous fat in the thigh and anterior abdomen.
Table 1.
Clinical characteristics, endocrine and metabolic variables in affected females.
| Patient | Current Age (y) |
Gravity and Parity |
Infertility | Menstrual Function |
Age at Menarche (y) |
Total † Testosterone (ng/dL) |
Ferriman- Gallwey Score |
Leptin ‡ (ng/dL) |
Fasting § insulin (μU/mL) |
Fasting glucose (mg/dL) |
Acanthosis Nigricans |
Oral GTT |
HDL (mg/dL) |
TG (mg/dL) |
Medications |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 57 | G4P4 | No | Amenorrhea** | 14 | ND | 0/48 | 1.4 | 17 | 257 | None | DM | 38 | 312 | Metformin |
| 2* | 28 | G0P0 | Yes | Amenorrhea | 12 | 100 | 39/48 | 1.6 | 31 | 151 | Significant | DM | 42.5 | 154 | Metformin |
| 3 | 24 | G4P2 | No | Oligomenorrhea | 13 | 54 | 18/48 | 2.7 | 52 | 95 | Moderate | NGT | 29 | 378 | No |
| 4 | 64 | G4P2 | No | Amenorrhea** | 14 | ND | 0/48 | 3.7 | 13 | 77 | None | IGT | 64 | 203 | No |
| 5 | 36 | G3P3 | No | Eumenorrhea | 13 | 42 | 5/48 | 9.0 | 29 | 103 | Mild | IGT | 46 | 448 | Thyroxine |
| 6 | 15 | G0P0 | NA | Eumenorrhea | 13 | ND | 0/48 | 5.2 | ND | 85 | None | ND | 73 | 84 | No |
| 7 | 14 | G0P0 | NA | Eumenorrhea | 12 | ND | 0/48 | 4.0 | ND | 80 | None | ND | 59 | 105 | No |
| 8 | 12 | G0P0 | NA | Prepubertal | NA | ND | 0/48 | 2.0 | ND | 82 | None | ND | 67 | 116 | No |
Abbreviations: GTT, glucose tolerance test; DM, diabetes mellitis; NGT, normal glucose tolerance; IGT, impaired glucose tolerance based on 2 hour glucose value (140–199 mg/dL); HDL, fasting high density lipoprotein; TG, fasting triglycerides; NA, not applicable; ND, not done;
normal range, 11–59 ng/dL;
normal range, 3.9–30.0 ng/dL;
normal range, 3–13 μU/mL.
Index case.
Menstrual cyclicity during late reproductive years.
Comment
The association of PCOS with obesity and insulin resistance is well known. However, not all women with PCOS are obese, and it is important to consider the diagnosis of lipodystrophy in lean PCOS subjects. The reported case highlights the importance of careful evaluation of body fat distribution by a thorough physical examination which can offer important clues to the diagnosis of this rare disorder. Recognition of these disorders of adipose tissue distribution is critical for the subsequent diagnosis of more serious metabolic abnormalities in patients and their family members. Only about 300 cases of FPLD have been described in the literature (2,4). To date, all pedigrees described have been of European origin, and this is the first case report of a Hispanic pedigree with the R482W missense mutation in the LMNA gene.
Approximately 20–33% of patients with FPLD develop hirsutism, menstrual abnormalities, and polycystic-appearing ovaries (3). These signs of androgen excess likely result from both the decrease in sex-hormone binding globulins (SHBG) production from the liver caused by hyperinsulinemia, and the direct effect of insulin on theca cells leading to androgen production. Additionally, it can be hypothesized that a greater reduction in SHBG and a higher production of testosterone occurs with an increase in visceral fat deposition, as has been observed in patients with central obesity (5). The exact etiology of insulin resistance and hyperinsulinemia leading to these abnormalities in FPLD is not clear, but is likely the consequence of ectopic fat accumulation in non-adipose tissues. Due to the limitation in sc fat storage depots, dietary fat is redirected to non adipose tissue stores such as the liver and muscle leading to lipotoxicity from steatosis. However, it is also not clear why some patients with FPLD develop the PCOS-phenotype while others do not. Potential causative factors include the regional differences in fat deposition, the quantity of excess intra-abdominal fat, and the severity of insulin resistance, leading to hyperandrogenemia. Undoubtedly, as in patients with other causes for PCOS, multiple factors play a role in the development of metabolic and endocrine dysfunction, including variable expressivity of the genetic mutation, age, and lifestyle.
The cornerstone of management of FPLD patients is the prevention of excess fat accumulation in non adipose tissues. Lifestyle factors including low fat diet and regular exercise is vital. In patients affected by the PCOS- phenotype, improvements in insulin sensitivity lead to decreases in circulating androgens and resumption of menses. Traditional pharmacologic therapy for diabetes and dyslipidemia is also often necessary, but optimal control is generally difficult to achieve due to marked insulin resistance. Replacement therapy with human recombinant leptin offers much promise in the treatment of patients with generalized lipodystrophy, but its role in FPLD patients remains to be defined. A recent open-label trial involving 6 patients with FPLD over a period of 4 years demonstrated a significant improvement in triglycerides, fasting glucose, and insulin sensitivity following replacement with recombinant leptin (8), but larger, controlled studies are needed.
Awareness of FPLD is important for the gynecologist as female patients may initially present with features similar to lean PCOS. Accurate diagnosis is critical to both treatment and counseling. More detailed studies on the risk factors for PCOS in FPLD cohorts are likely to shed much information on the relationship between fat deposition, insulin resistance, and hyperandrogenemia.
Acknowledgments
Supported by the National Institutes of Health, grant RO1-DK54387.
Footnotes
Financial Disclosure: The authors did not report any potential conflicts of interest.
References
- 1.Dunnigan MG, Cochrane M, Kelly A, Scott JW. Familial lipoatrophic diabetes with dominant transmission. Q J Med. 1974;49:33–48. [PubMed] [Google Scholar]
- 2.Garg A. Acquired and Inherited Lipodystrophies. New England Journal of Medicine. 2004;350:1220–43. doi: 10.1056/NEJMra025261. [DOI] [PubMed] [Google Scholar]
- 3.Garg A. Gender Differences in the Prevalence of Metabolic Complications in Familial Partial Lipodystrophy (Dunnigan Variety) J of Clin Endoc & Metab. 2000;86:1776–81. doi: 10.1210/jcem.85.5.6605. [DOI] [PubMed] [Google Scholar]
- 4.Speckman RA, Garg A, Du F, Bennett L, Veile R, Arioglu E, et al. Mutational and Haplotype Analyses of Families with Familial Partial Lipodystrophy (Dunnigan Variety) Reveal Recurrent Missense Mutations in the Globular C-Terminal Domain of Lamin A/C. Am J Hum Genet. 2000;66:1192–98. doi: 10.1086/302836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pasquali R. Obesity and Androgens: Facts and Perspectives. Fertil Steril. 2006;85:1319–36. doi: 10.1016/j.fertnstert.2005.10.054. [DOI] [PubMed] [Google Scholar]
- 6.Park JY, Javor ED, Cochran EK, DePaoli AM, Gorden P. Long-Term Efficacy of Leptin Replacement in Patients with Dunnigan-type Familial Partial Lipodystrophy. Metab Clin Exp. 2007;56:508–16. doi: 10.1016/j.metabol.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]