

Abstract
c-kit, the transmembrane tyrosine kinase receptor for stem cell factor, is required for melanocyte and mast cell development, hematopoiesis, and differentiation of spermatogonial stem cells. We show here that in the heart, c-kit is expressed not only by cardiac stem cells but also by cardiomyocytes, commencing immediately after birth and terminating a few days later, coincident with the onset of cardiomyocyte terminal differentiation. To examine the function of c-kit in cardiomyocyte terminal differentiation, we used compound heterozygous mice carrying the W (null) and Wv (dominant negative) mutations of c-kit. In vivo, adult W/Wv cardiomyocytes are phenotypically indistinguishable from their wild-type counterparts. After acute pressure overload adult W/Wv cardiomyocytes reenter the cell cycle and proliferate, leading to left ventricular growth; furthermore in transgenic mice with cardiomyocyte-restricted overexpression of the dominant negative Wv mutant, pressure overload causes cardiomyocytes to reenter the cell cycle. In contrast, in wild-type mice left ventricular growth after pressure overload results mainly from cardiomyocyte hypertrophy. Importantly, W/Wv mice with pressure overload–induced cardiomyocyte hyperplasia had improved left ventricular function and survival. In W/Wv mice, c-kit dysfunction also resulted in an ≈14-fold decrease (P<0.01) in the number of c-kit+/GATA4+ cardiac progenitors. These findings identify novel functions for c-kit: promotion of cardiac stem cell differentiation and regulation of cardiomyocyte terminal differentiation.
Keywords: c-kit, cardiomyocyte, terminal differentiation, cardiac stem cells, pressure overload
The response of the mammalian heart to stresses such as pressure overload (PO) or injury is limited after birth by cardiomyocyte terminal differentiation.1,2 This restricts the adult heart to adaptation by growth through cardiomyocyte and left ventricular (LV) hypertrophy, a response associated with increased mortality.3 Recent discoveries of primitive cardiac stem cells (CSCs) and cardiomyocyte progenitors residing in the adult heart, however, have suggested that it may be possible to renew cardiomyocytes in the injured heart or to increase LV mass in response to PO by adding new cardiomyocytes.4,5 Of the different CSC populations, c-kit+ CSCs have the highest capacity to generate myocardial cells in vitro.6 Understanding how c-kit+ CSC differentiation is regulated should allow manipulation of these cells in situ.
c-kit is the transmembrane tyrosine kinase receptor for stem cell factor.7 Transphosphorylation of tyrosines in the intracellular portion of c-kit creates binding sites for Src homology 2 domain–containing proteins that recruit regulatory proteins. Phosphorylation of these proteins, which include phosphoinositide 3′-kinase, phospholipase Cγ, the Src family of tyrosine kinases, and p21ras GTP-activating protein,7,8 is crucial for c-kit signaling.7 Phenotypes resulting from germline loss-of-function mutations, as in WBB6F1/J-KitW/KitW-v (W/Wv) mice, have established a role for c-kit in mast cell (MC) and melanocyte development, hematopoiesis, and differentiation of spermatogonial stem cells.7,9,10 This led us to investigate the role of c-kit in CSC differentiation in W/Wv mice. The W allele, in which the transmembrane domain is deleted, is a null mutation that is not expressed at the cell surface. Wv is a missense mutation (T660M) in the kinase domain that reduces tyrosine kinase activity by >95%, thus preventing binding and phosphorylation of regulatory proteins.8 Heterozygous Wv mice have a more severe phenotype than heterozygous W mice: this dominant negative effect results from the formation of dysfunctional heterodimers.11 W/W mice have a perinatal lethal phenotype, but W/Wv mice survive.7,9 We have investigated the role of c-kit in the heart using W/Wv mice. Here we show that not only is c-kit required in vivo for c-kit+ CSC differentiation but it is also expressed in cardiomyocytes and regulates their terminal differentiation.
Materials and Methods
An expanded Materials and Methods is in the online data supplement, available at http://circres.ahajournals.org.
Mice with genetic c-kit dysfunction (W/Wv) and their congenic wild-type (WT) littermate controls were subjected to suprarenal aortic constriction (SAC) and euthanized at different intervals up to 2 weeks.
LV function was assessed by echocardiography and micromanometry, and cardiomyocyte proliferation was assessed by immunohistochemistry and flow cytometric analyses. Gene expression profiling was performed using microarrays followed by quantitative RT-PCR (supplemental Table I).
Results
Expression and Function of c-kit in Cardiomyocytes
Using an antibody directed at a C-terminal epitope unique to c-kit, we found that c-kit is not detectible in fetal (embryonic day [E]15) or very early postnatal (day 0.5) cardiomyocytes but is expressed by cardiomyocytes from postnatal days 1 to 10 (Figure 1), a period coincident with terminal differentiation.2,3,12 At later times, c-kit expression is nearly undetectable (Figure 1). Photomicrographs in Figure 1 show cell surface and intracellular c-kit in postnatal cardiomyocytes; this dual cellular localization has been detected using some c-kit antibodies,4,13 but others recognize only cell surface c-kit.6
Figure 1.
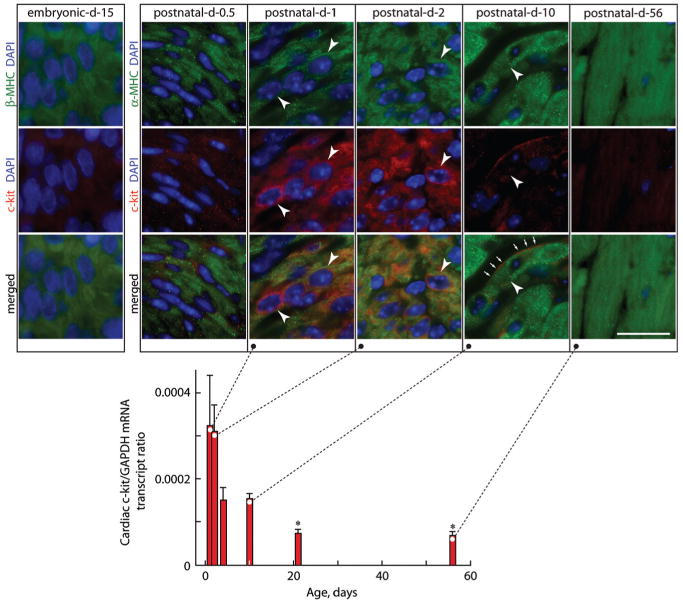
Expression of c-kit in mouse heart. Cardiac c-kit mRNA levels by quantitative RT-PCR are normalized to GAPDH mRNA levels in postnatal (1- to 10-day-old) and adult (21- and 56-day-old) mice. Levels of cardiac c-kit expression are high 1 day after birth, fall by postnatal day 4, and are barely evident by day 10. Values are means±SEM, and comparisons were made by ANOVA, followed by Tukey's test. *P<0.05 relative to day 1. Insets shows photomicrographs of LV sections from embryonic day 15 and postnatal day 0.5, 1, 2, 10, and 56 mice stained for α-MHC or β-MHC to identify cardiomyocytes and 4′,6′-diamidino-2-phenylindole (DAPI) to detect nuclei and c-kit, with dual staining or merged images as indicated. Arrowheads show some c-kit+ LV cardiomyocytes in postnatal hearts. Arrows show cell surface c-kit+. Scale bar=20 μm.
To investigate the role of c-kit in cardiomyocyte terminal differentiation, we used W/Wv mice.7,9 Eight-week-old W/Wv mice and their WT littermates have similar baseline mean proximal aortic blood pressures (Figure 2a) and similar left ventricles in terms of weight (LV-to-body weight ratio) (Figure 2b), morphology (LV wall thickness/diameter ratio), and isovolemic (±dP/dtmax), and ejection-phase function (rate-corrected velocity of circumferential shortening [VCFr]) (supplemental Table II). In addition, LV cardiomyocytes of adult animals of both genotypes were similar in cross-sectional area (Figure 2c) and predominantly binucleated (Figure 2d). Expression profiling of mRNA from adult W/Wv and WT cardiomyocytes (5 to 7 replicates per group) revealed that of >40 000 transcripts analyzed, only 8 unrelated genes were significantly (P<0.05) changed by ≥2-fold in W/Wv cardiomyocytes (supplemental Table III). This suggests that under basal conditions W/Wv and WT cardiomyocytes are phenotypically indistinguishable.
Figure 2.
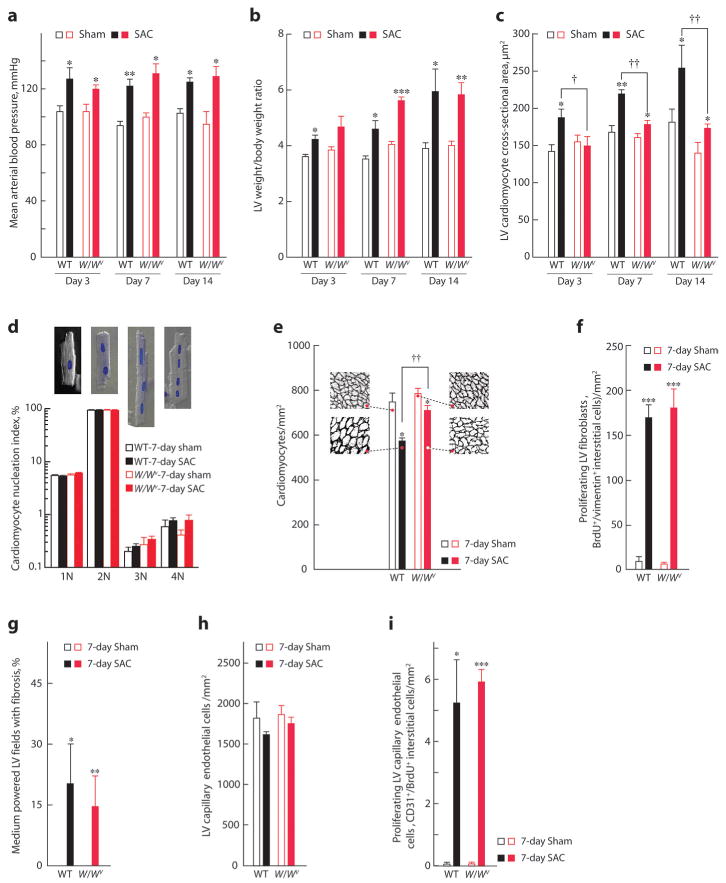
Hemodynamic and cardiac morphological changes after SAC in WT and W/Wv mice. Three to 7 days of SAC increased mean arterial blood pressure (a) and LV-to-body weight ratio (b) to a similar level in WT and W/Wv mice, compared with sham controls, but SAC-induced LV cardiomyocyte hypertrophy was markedly less in W/Wv mice (c). d, Percentage of cardiomyocytes from WT or W/Wv left ventricles after 7 days of SAC or sham with 1 (1N), 2 (2N), 3 (3N), or 4 (4N) nuclei per cells. Insets are examples of 1N to 4N cardiomyocytes in phase contrast with DAPI-stained nuclei. e, Cardiomyocyte density in 7-day SAC or sham-operated WT or W/Wv left ventricles. Insets are representative LV sections from each group, at the same magnification, stained with laminin to outline cardiomyocytes. Seven days of SAC produced genotype-independent changes in LV fibroblast proliferation (f), fibrosis (g), capillary EC density (h), and EC proliferation (i). Values are means±SEM (n=5 per group), and comparisons within each time-after-operation interval were made by ANOVA, followed by Tukey's test. *P<0.05, **P<0.01, ***P<0.001 for intragenotype comparisons; †P<0.05, ††P<0.01 for intergenotype comparisons.
We reasoned that a defect in cardiomyocyte terminal differentiation caused by c-kit dysfunction may be unmasked by a provocation such as PO.2 SAC for 3 to 14 days produced similar increases in mean proximal aortic blood pressures and LV-to-body weight ratios in WT and W/Wv mice (Figure 2a and 2b). In WT mice, LV cardiomyocyte cross-sectional areas increased 32% to 41% after 3 to 14 days of SAC (Figure 2c), consistent with hypertrophic growth. This growth resulted in an ≈23% decrease in cardiomyocyte density because as the cardiomyocytes enlarge there are fewer cardiomyocytes per millimeter squared (in 7-day SAC WT mice) (Figure 2e). In contrast, for an equivalent increase in LV growth in 7-day SAC W/Wv mice (Figure 2b), cardiomyocyte cross-sectional areas increased only by ≈10% (Figure 2c), resulting in an ≈9% decrease in LV cardiomyocyte density (Figure 2e; P<0.01). Thus, LV growth in SAC W/Wv mice is only partly mediated by cardiomyocyte hypertrophy. We found no differences in proliferating LV fibroblasts (Figure 2f) or fibrosis (Figure 2g). Also, LV capillary endothelial cell (EC) density did not decrease during SAC-induced LV growth in either W/Wv or WT mice (Figure 2h), suggesting equivalent increases in EC proliferation. This was confirmed by equivalent increases (>80-fold) in synthetic capillary ECs (CD31+/bromodeoxyuridine-positive [BrdUrd+] interstitial cells) (Figure 2i). Because there are no differential changes in other major LV cell types, increased LV weight in W/Wv SAC mice could be attributable to cardiomyocyte hyperplasia.
To assess the effect of PO on gene expression linked to cardiomyocyte proliferation, we carried out expression profiling on isolated cardiomyocytes from adult 7-day SAC WT and W/Wv mice (5 to 7 replicates per group). More than 150 genes were up- or downregulated ≥2-fold (P<0.05) in W/Wv SAC cardiomyocytes relative to WT SAC (supplemental Table IV). Upregulated genes in W/Wv SAC cardiomyocytes (confirmed by quantitative RT-PCR) are involved in transit through G1/S and G2/M phases and in cytokinesis (supplemental Table V).
Given these findings, we sought immunohistochemical evidence for cardiomyocyte cell cycle reentry. Adult cardiomyocytes are normally refractory to cell cycle reentry.1,2,12 It is therefore remarkable that in W/Wv mice, after 7 and 14 days of SAC, Ki67+ (expressed in G1, S, G2, and early mitosis) (Figure 3a) and BrdUrd+ (specific to S phase) (Figure 3b through 3d) nuclei were readily observed in large rod-shaped LV cardiomyocytes (identified by α-myosin heavy chain [α-MHC] expression) with mature sarcomeric organization. After 7 days of SAC, W/Wv Ki67+ or BrdUrd+ cardiomyocyte nuclei increased to ≈15 nuclei/mm2 (Figure 3e and 3f). In vivo BrdUrd labeling identifies all cells that enter the S phase during the labeling period. Moreover, this nuclear label exists in cells even after they exit the cell cycle and is therefore an integrative measurement. Thus, in long-term in vivo BrdUrd-labeling studies, BrdUrd+ cells exceed those proliferating cells that are identified by Ki67 immunofluorescence, which labels cells that were proliferating at the moment of euthanasia. Our terminal BrdUrd-labeling interval was short (a single intraperitoneal BrdUrd injection given 12 hours before euthanasia) and hence identified only those cardiomyocytes that reentered the cell cycle in the 12-hour interval before euthanasia. The short labeling interval likely explains why BrdUrd+ cardiomyocytes were similar in frequency to those that were Ki67+. In W/Wv SAC mice, nuclear length was 20% to 25% of cardiomyocyte length (data not shown), and most cells were binucleated (Figure 2d). Thus, a nucleus in cross-section may not be visible in up to 50% of cardiomyocytes viewed. Allowing for this underestimation, it is evident that as many as 30 cardiomyocytes/mm2 (that is, ≈4% of the LV cardiomyocytes [calculated from Figure 2e and 3f]) had reentered the cell cycle in 7-day SAC W/Wv mice. In the absence of SAC, no BrdUrd+ or Ki67+ cardiomyocytes were observed in multiple LV sections of WT or W/Wv mice. Similarly, no BrdUrd+ and only 2 Ki67+ cardiomyocytes/mm2 were observed in 3- to 14-day SAC-WT mice. The absence of BrdUrd+ cardiomyocytes in WT hearts is likely attributable to the short interval of in vivo BrdUrd labeling (see above) and does not contradict the potential of low level cardiomyocyte renewal in WT hearts subjected to PO.5
Figure 3.
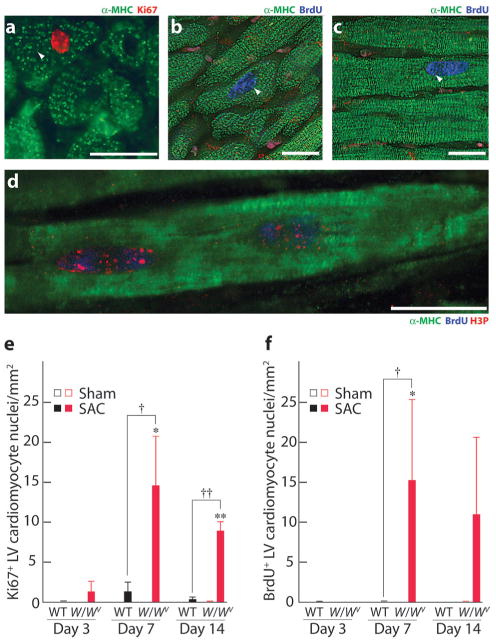
Cardiomyocytes proliferate in W/Wv mice in response to PO. a, Photomicrograph of a 7-day SAC W/Wv LV section stained for α-MHC to identify cardiomyocytes and Ki67 (arrow) to identify G1/M phase nuclei. b through d, Fourteen-day SAC W/Wv LV sections stained for α-MHC and BrdUrd to identify S phase nuclei (arrows). d, Confocal image of a cardiomyocyte from a 7-day SAC W/Wv LV stained for α-MHC, BrdUrd, and H3P (marker of mitosis). Scale bars=20 μm. SAC-induced changes in Ki67+ (e) and BrdUrd+ (f) cardiomyocytes in WT and W/Wv left ventricles. Values are means±SEM (n=5 per group), and comparisons within each time-after-operation interval were made using ANOVA followed by Tukey's test. *P<0.05, **P<0.01 for intragenotype comparisons; †P<0.05, ††P<0.01 for intergenotype comparisons.
Cell cycle progression from G0/G1 into G2/M is expected to result in increased ploidy (DNA synthesis without nuclear division) if the nuclei arrest in G2. PO (for 7 days) increased the ratio of 2N to ≥4N cardiomyocyte nuclei in W/Wv mice (Figure 4). Along with increased DNA synthesis in W/Wv SAC cardiomyocytes, these results indicate that c-kit dysfunction accelerates cardiomyocyte nuclear division (karyokinesis), resulting in a left-shifted ploidy profile (Figure 4). This is supported by an increase in phosphorylated histone 3–positive (H3P+) (0±0 versus 1.1 ±0.07 H3P+ cardiomyocytes/mm2 in 7-day sham and 7-day SAC W/Wv left ventricles, respectively; n=5 per group; P<0.01) (Figure 3d) and aurora B+ cardiomyocytes in 7-day SAC-W/Wv left ventricles (0±0 versus 10.4±3.1 aurora B+ cardiomyocytes/mm2 in 7-day sham and 7-day SAC W/Wv left ventricles, respectively; n=5 per group; P<0.05) (Figure 5). H3P is associated with chromosomal condensation that accompanies the onset of mitosis, and aurora B, a regulator of kinetochore-microtubule attachment, is crucial for mitosis. Adjacent cardiomyocyte nuclei with intervening aurora B immunofluorescence (Figure 5) provide immunohistochemical evidence for karyokinesis in W/Wv SAC cardiomyocytes.
Figure 4.
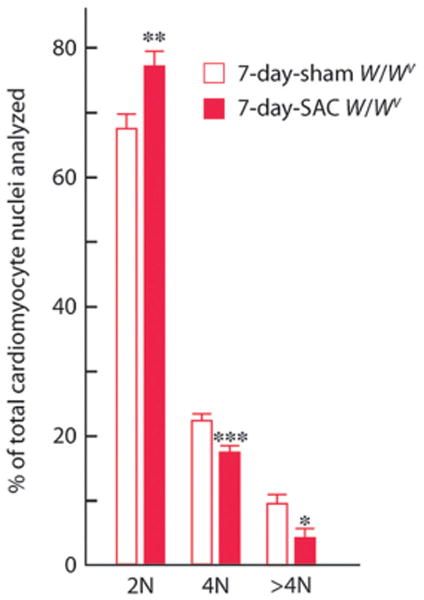
Effect of PO on DNA content (ploidy) in W/Wv cardiomyocyte nuclei. Ventricular cardiomyocytes from each heart were dispersed by enzymatic digestion. The nuclei were isolated and stained with propidium iodide for determination of DNA content by flow cytometric analyses. The results, expressed as 2N, 4N, and >4N, are means±SEM (n=6 to 7 per group). Comparisons were made by t tests. *P<0.05, **P<0.01, ***P<0.001.
Figure 5.
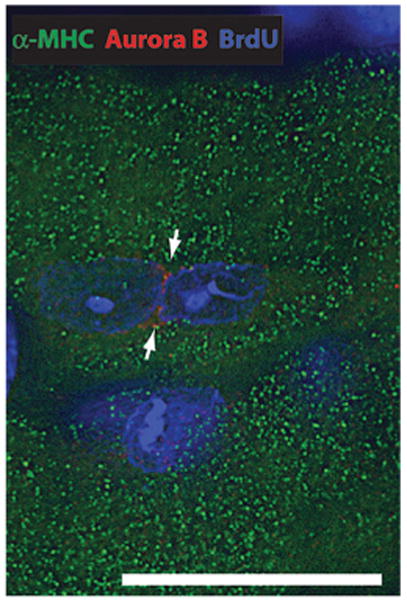
Karyokinesis in a 7-day SAC W/Wv LV cardiomyocyte. Photomicrograph (0.5 μm thick; using digital deconvolution) of a cardiomyocyte from a W/Wv SAC LV section stained with α-MHC (to identify the cardiomyocyte), DAPI (to identify nuclei), and aurora B. The arrows show the localization of aurora B between 2 nuclei to illustrate in vivo karyokinesis. Scale bar=20 μm.
We next asked whether cardiomyocyte karyokinesis is uncoupled from cytokinesis in W/Wv SAC mice. Such uncoupling should be readily apparent by the finding of cardiomyocyte multinucleation.14 Enzymatic dissociation of heart muscle in W/Wv mice and cytochemical assessment of the number of nuclei per cardiomyocyte revealed that SAC does not promote multinucleation (Figure 2d). Thus, the finding of karyokinesis in W/Wv SAC left ventricles indicates cardiomyocyte division. To test this, we enzymatically dispersed cardiac cells from 7-day SAC or sham W/Wv mice and counted ventricular cardiomyocytes. Consistent with cytokinesis, PO increased the total number of ventricular cardiomyocytes in W/Wv mice by ≈38% (2.68±0.3×106 total ventricular cardiomyocytes in 7-day sham W/Wv hearts versus 3.71±0.3×106 total ventricular cardiomyocytes in 7-day SAC W/Wv hearts; n=7 per group; P<0.05).
Kühn et al15 recently showed that periostin, an extracellular matrix component, induces cell cycle reentry in adult cardiomyocytes. Although periostin mRNA expression was not different between WT and W/Wv left ventricles (data not shown), c-kit dysfunction could, by inhibiting MC development and hematopoiesis, alter the extracellular matrix in a manner that lowers the threshold for cardiomyocyte cell cycle reentry. To investigate the loss-of-function effects associated directly with c-kit activity in cardiomyocytes, transgenic mice were generated with cardiac-restricted expression of the dominant negative c-kit mutant Wv using an α-MHC promoter coupled to the Wv coding sequence (Figure 6a).11 In the highest-expressing line (α-MHC/Wv-Tg-2.1) LV Wv transgene expression was ≈12-fold and ≈62-fold higher than c-kit expression in 2-day and 13-week-old mice, respectively (Figure 6b). c-kit immunofluorescence was nearly undetectable in adult WT cardiomyocytes (Figure 6c) but was readily observed in adult α-MHC/Wv-Tg-2.1 LV cardiomyocytes (Figure 6d). Because LV Wv expression was much higher than endogenous c-kit expression in these transgenic mice and c-kit mRNA levels were similar to those in WT littermates (Figure 6b), it is likely that c-kit–like immunofluorescence in adult α-MHC/Wv-Tg-2.1 LV cardiomyocytes is mainly attributable to Wv overexpression. We did not observe BrdUrd+, H3P+, and aurora B+ cardiomyocytes in 13-week-old normotensive α-MHC/Wv-Tg-2.1 left ventricles. However, after 7 days of SAC, BrdUrd+ (0±0 and 9.7± 1.5 α-MHC+/BrdUrd+ LV cardiomyocytes/mm2 in α-MHC/Wv-Tg-2.1 sham and 7-day SAC mice, respectively; n=5 per group; P<0.01), H3P+ (0±0 and 7.8±2.9 α-MHC+/H3P+ LV cardiomyocytes/mm2 in α-MHC/Wv-Tg-2.1 sham and 7-day SAC mice, respectively; n=5 per group; P<0.05), and aurora B+ cardiomyocytes (0±0 and 16.9±6.6 α-MHC+/aurora B+ LV cardiomyocytes/mm2 in α-MHC/Wv-Tg-2.1 sham and 7-day SAC mice, respectively; n=5 per group; P<0.05) were readily apparent in 13-week-old α-MHC/Wv-Tg-2.1 left ventricles. These findings suggest that cardiomyocyte-restricted c-kit inhibition is sufficient for PO-induced cell cycle reentry in adult cardiomyocytes.
Figure 6.
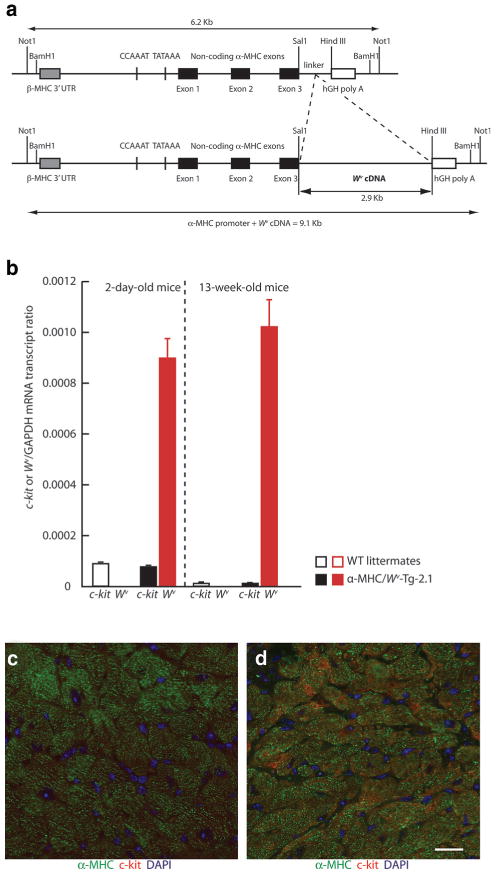
c-kit and Wv expression in α-MHC/Wv-Tg-2.1 transgenic mice. a, Structure of the α-MHC/Wv transgene. b, Relative expression of c-kit and the Wv transgene in the LV of 2-day postnatal and adult α-MHC/Wv-Tg-2.1 transgenic mice and their age-matched WT littermates. c-kit-like immunofluorescence in cardiomyocytes from 13-week-old WT (c) and α-MHC/Wv-Tg-2.1 transgenic (d) mice.
Improved LV Function in W/Wv Mice After PO
Echocardiography demonstrated an early significant increase in LV wall thickness at 3 days after SAC in WT mice, whereas in W/Wv SAC mice, there was a trend toward an increase in LV wall thickness at day 7, which achieved significance by day 14 (supplemental Table II). In a similar fashion, LV wall thickness-to-diameter ratio increased significantly early on day 3 in WT SAC mice, whereas W/Wv SAC mice demonstrate a trend toward an increase in LV concentric hypertrophy at days 7 and 14, but these changes did not achieve significance. Although there were no differences in peak ±dP/dtmax at any time, LV VCFr was significantly increased after 7 days of SAC in W/Wv but not in WT mice. Also, a positive correlation between VCFr and the number of proliferating cardiomyocytes in W/Wv SAC left ventricles (P<0.01, Figure 7a) indicates that improvements in LV contractility are related to the cardiomyocyte hyperplastic response. Early mortality increases after acute PO, and hypercontractile cardiac function reduces this mortality.16 In the first 7 days of SAC, 41% of the WT mice, but no W/Wv mice, died (Figure 7b), suggesting that improved cardiac contractility after PO could improve survival in W/Wv SAC mice.
Figure 7.
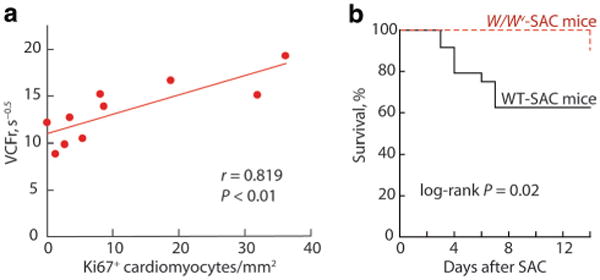
Effects of c-kit dysfunction on PO-induced changes in LV function and survival. a, In W/Wv mice subjected to 7 or 14 days of SAC, LV Ki67+ LV cardiomyocyte numbers were positively correlated with ejection-phase LV systolic function (VCFr). b, Actuarial survival in WT SAC and W/Wv SAC mice (n=24 for WT mice and n=21 for W/Wv mice).
c-kit dysfunction inhibits inflammatory MC development and hematopoiesis,7,9 which could in turn influence the LV inflammatory response to PO and hence contractility. We examined the extent of this abnormality in W/Wv SAC left ventricles and its effect on LV function. We sought leukocyte L1 protein immunofluorescence, which identifies macrophages, neutrophils, and monocytes,17 to study potential infiltration of inflammatory cells in WT and W/Wv left ventricles after 7 days of SAC. Interstitial inflammatory cells numbers were not different between WT and W/Wv left ventricles (0.17±0.11, 0.28±0.07, 0.17±0.1, and 0.22±0.14 LV L1+ interstitial cells/mm2 in WT sham, WT SAC, W/Wv sham, and W/Wv SAC mice, respectively; n=5 per group). In WT mice, LV MC numbers were not changed after 7 days of SAC (2.2±0.64 and 1.8±0.37 MCs/mm2 in WT-sham and WT SAC left ventricles, respectively; n=5 per group). However, as expected, MCs could not be detected in the LV of W/Wv SAC mice. Cromolyn, a MC stabilizer, blocks MC-dependent phenomena.18 Cromolyn (60 mg/kg per day) did not alter the response to 7 days of SAC; ejection-phase LV function VCFr was not different in vehicle- and cromolyn-treated WT sham and SAC mice (10.7±0.6, 12.6±1.2, 9.0±1.5, and 10.1 ± 1.1 sec−0.5 in vehicle-treated WT sham and SAC mice and cromolyn-treated WT sham and SAC mice, respectively; n=10 to 12 per group). Also, BrdUrd+ and Ki67+ cardiomyocytes were not observed in cromolyn-treated WT SAC mice (data not shown). Together, these findings do not support an inflammatory mechanism or MC involvement in influencing LV function in W/Wv and WT mice.
c-kit Dysfunction Inhibits CSC Differentiation
Using genetic-fate mapping, Hsieh et al5 provided evidence that stem or precursor cells add cardiomyocytes to the heart after long-term PO. We thus asked whether c-kit dysfunction alters the levels and/or differentiation state of c-kit+ CSCs. c-kit+ LV interstitial cells (≈20 μm diameter) were relatively infrequent (≈10 cells/mm2) but equally abundant in WT and W/Wv mice (Figure 8a), suggesting that c-kit activity is not essential for maintaining their numbers in the heart. To explore the origin of these cells, we used expression of CD45 to discriminate between c-kit+ LV interstitial cells of hematopoietic cell lineage (CD45+) and those (CD45−) resident in heart.19 Absence of vascular EC growth factor receptor-2 (VEGFR2) expression was additionally used to indicate that c-kit+/CD45− CSCs were not of bone marrow origin.13 We studied the colocalization of c-kit and CD45, or c-kit and VEGFR2, in c-kit+/α-MHC− small LV interstitial cells. Fewer than 2.5% of c-kit+ LV interstitial cells were CD45+ and <1% were VEGFR2+ in WT and W/Wv left ventricles, suggesting that the majority of c-kit+ LV interstitial cells were resident CSCs not derived from the bone marrow.
Figure 8.
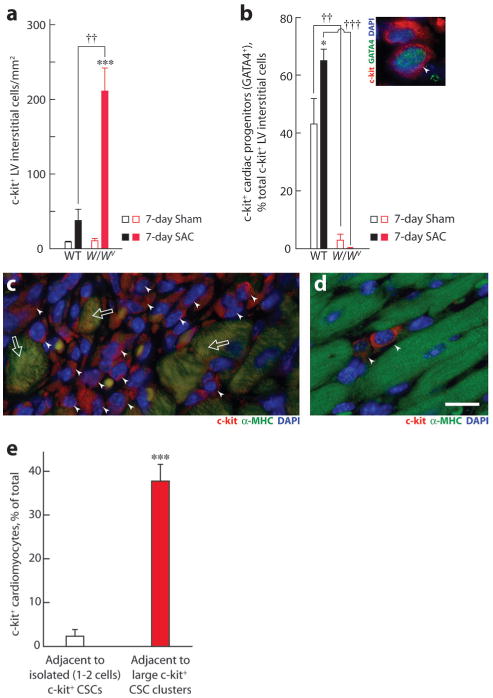
c-kit+ LV interstitial cells and cardiac progenitors (GATA4+) in WT and W/Wv left ventricles. Effect of 7 days of SAC on total c-kit+ interstitial cells (a) and the percentage of these cells that were also GATA4+ in WT and W/Wv left ventricles (b). Inset shows photomicrograph showing 2 adjacent cells in a WT LV, stained with c-kit and GATA4 to identify c-kit+ cardiac progenitors (arrowhead). Nuclei were stained using DAPI. Comparisons were made using ANOVA, followed by Tukey's test (n=5 hearts per group). c, Photomicrograph showing a large cluster of c-kit+ interstitial cells (arrowheads) and some c-kit+/α-MHC+ cardiomyocytes (arrows) in the LV. d, Photomicrograph showing isolated c-kit+ LV interstitial cells in the LV of a 7-day sham WT mouse. Magnification was the same in c and d. Scale bar=20 μm. e, Number of c-kit+ cardiomyocytes adjacent to large clusters of c-kit+ LV interstitial cells (filled bar) vs isolated (1 to 2) c-kit+ cells (open bar). Twenty-six clusters and 17 isolated c-kit+ LV interstitial cells from multiple 7-day SAC left ventricles were compared using a t test. Values are means±SEM. *P<0.05, ***P<0.001 for intragenotype comparisons; ††P<0.01, †††P<0.01 for intergenotype comparisons.
As c-kit+ CSCs differentiate into cardiac progenitors, they begin to express the transcription factor GATA4.4,20 Under basal conditions, the W/Wv left ventricles exhibited an ≈14-fold reduction (P<0.01) in the number of GATA4+/c-kit+ cardiac progenitors (Figure 8b), indicating that c-kit is critical for in vivo differentiation of these CSCs. Acute PO (7 days of SAC) increased GATA4+/c-kit+ cardiac progenitors in the WT left ventricles by ≈50% (P<0.05) (Figure 8b), but total numbers of LV c-kit+ interstitial cells were not significantly increased (Figure 8a). These findings suggest that stem or precursor cell-based addition of cardiomyocytes to the LV after long-term PO5 could be initiated by an early PO-dependent increase in c-kit+ CSC differentiation. However, the absence of synthetic cardiomyocytes in 7- or 14-day SAC WT mice may be attributable to the acute nature of our in vivo studies, which may not allow sufficient time for c-kit+ CSCs to proliferate (and thus be labeled by a short terminal 12-hour BrdUrd pulse), differentiate into cardiac progenitors, and then develop into mature integrated cardiomyocytes.
In contrast to WT mice, PO produced an >10-fold increase in c-kit+ interstitial cells in W/Wv left ventricles (Figure 8a); these cells were found in large niches (Figure 8c) and <1% were differentiated (ie, GATA4+/c-kit+) (Figure 8b), whereas those in WT SAC left ventricles were found as isolated cells (1 to 2 cells) (Figure 8d), but ≈65% were differentiated (Figure 8b). One possibility for this differential increase in c-kit+ cell numbers is that stem cells have a higher mitotic potential than their more differentiated daughters.21 As a result, PO may have disproportionately expanded c-kit+ CSCs in W/Wv hearts.
To establish the specificity of these changes, we examined the effect of c-kit dysfunction and PO on LV Sca-1+, MDR-1+, and Abcg-2+ CSC populations. Abcg-2 was mainly located in CD31+ ECs; we observed no Abcg-2+/CD31− interstitial cells in WT or W/Wv left ventricles. c-kit dysfunction or PO did not influence the number of Sca-1+/CD31− (2.6±0.36, 2.7±0.24, 2.3±0.5, and 2.2±0.26 Sca-1+/CD31− interstitial cells/mm2 in WT-sham, WT SAC, W/Wv sham and W/Wv SAC left ventricles, respectively; n=5 per group) or MDR-1+/CD31− CSCs (0.17±0.07, 0.11±0.06, 0.06±0.06, and 0.11±0.07 MDR-1+/CD31− interstitial cells/mm2 in WT sham, WT SAC, W/Wv sham, and W/Wv SAC left ventricles, respectively; n=5 per group). Together, these data indicate that c-kit+ CSCs are more abundant than Sca-1+ and MDR-1+ CSCs in WT and W/Wv left ventricles and suggest that there is no interplay between these seemingly distinct CSC populations.
PO Increases c-kit Expression in Cardiomyocytes
c-kit+ cardiomyocytes were found infrequently in the LV of WT and W/Wv mice subjected to 7 days of SAC but not at all in sham controls (data not shown). We sought to determine the relation between c-kit+ cardiomyocytes and CSCs in W/Wv SAC mice. Accordingly, we asked whether c-kit+ cardiomyocytes were more abundant adjacent to c-kit+ CSC clusters, as might be expected if their occurrence was dependent on c-kit+ CSCs. We tested this in W/Wv SAC mice. We found c-kit+ cardiomyocytes adjacent to clusters of c-kit+ CSCs in W/Wv mice (Figure 8c), but the frequency of these cells was related to the size of the cluster: ≈17-fold more c-kit+ cardiomyocytes were observed adjacent to large c-kit+ CSC clusters than adjacent to isolated (1 to 2 cells) c-kit+ CSCs (P<0.001) (Figure 8e). The close association of c-kit+ CSCs and c-kit+ cardiomyocytes is strongly suggestive of some involvement of CSCs in regulating c-kit in cardiomyocytes. The possibility that cardiomyocyte c-kit immunofluorescence in W/Wv left ventricles represents uptake of the soluble c-kit mutant W from CSCs can be ruled out because we used a c-kit antibody that is directed to an epitope not present in W. The close association of c-kit+ CSCs and c-kit+ cardiomyocytes could be attributable to the derivation of new cardiomyocytes, with retained c-kit expression, from c-kit+ CSCs. Another possibility is that c-kit+ CSC-derived cytokines and growth factors could perhaps have caused c-kit reexpression in adjacent cardiomyocytes.
Discussion
In mammalian cardiomyocytes, terminal differentiation is thought to occur in 2 discernable phases.12 The first phase involves the uncoupling of cytokinesis from karyokinesis during a wave of DNA synthesis that occurs soon after birth. In mice, this results in binucleation of cardiomyocytes.12 The adult newt heart, which is capable of myocardial regeneration after injury, is composed of mono- and binucleated cardiomyocytes, and both can proliferate.22 This suggests that the uncoupling of karyokinesis from cytokinesis in the early postnatal period does not in itself signify terminal differentiation. The second phase, which also occurs in early postnatal life, is characterized by the near total inability of cardiomyocytes to reenter the cell cycle, even when the myocardium is injured or subjected to hemodynamic stress.2,3,23 We show here that c-kit is expressed by cardiomyocytes for only a few days, beginning immediately after birth and coinciding with the onset of their terminal differentiation. In adult mice with the c-kit mutations W and Wv, which markedly suppress c-kit tyrosine kinase activity,8 cardiomyocytes are mainly binucleated. PO causes W/Wv cardiomyocytes to reenter the cell cycle and to divide. Studies with transgenic mice with cardiomyocyte-restricted overexpression of the c-kit dominant-negative mutant Wv indicate that cardiomyocyte c-kit dysfunction is the basis for PO-dependent cardiomyocyte cell cycle reentry. Taken together, these findings indicate that c-kit is required for cardiomyocyte terminal differentiation.
In W/Wv mice, PO increased LV cardiomyocyte cell cycle reentry by up to 4% from a baseline of <0.02% (>200-fold increase), which was associated with the activation of a network of genes that directly regulate cell division (supplemental Table V). Despite the high proliferative capacity of adult W/Wv cardiomyocytes, there were few if any differences in the gene expression profiles of the unstressed adult W/Wv and WT cardiomyocytes. It is noteworthy that expression levels of genes that regulate S phase transition in cardiomyocytes, eg, A and D type cyclins and p27kip,1,14,24,25 were not altered in W/Wv cardiomyocytes from the unstressed adult heart. Thus, cardiomyocyte terminal differentiation may not simply involve suppressed cell cycle protein expression, as has been previously suggested.26 Further studies are now required to address how c-kit causes cardiomyocytes to terminally withdraw from the cell cycle.
Mechanisms controlling heart function serve to match heart size to functional demand.2,3 Fine tuning of myocardial mass to ventricular work occurs even in the embryo, but unlike the adult heart, where this adjustment is by cardiomyocyte hypertrophy, the fetal heart adapts through hyperplasia.27 Similarly, it has been observed that cardiomyocyte hyperplasia abrogates hypertrophic growth in injured hearts of adult transgenic mice expressing p193 and p53 mutants.28 The differences between W/Wv and WT mice in the onset of the increase in LV wall thickness in response to PO are likely attributable to the delayed onset of cardiomyocyte hypertrophy in W/Wv mice. We speculate that in W/Wv SAC LV the initial cardiomyocyte hyperplastic response decreases the severity, and delays the onset, of cardiomyocyte hypertrophy. The positive effect of the c-kit dysfunction on LV function in mice with PO, in the face of unaltered capillary density, is related to this hyperplastic growth because of the significant positive correlation between the number of proliferating cardiomyocytes and LV VCFr. Moreover, the increase in LV contractility in W/Wv SAC mice was coeval with the greatest improvement in mortality in W/Wv SAC mice relative to the WT SAC mice. These findings suggest that because WT cardiomyocytes are terminally differentiated, their response to PO is necessarily restricted to hypertrophic enlargement, which appears to be maladaptive even at the earliest stage of PO.
Our findings contrast with those of Fazel et al,13 who showed that after myocardial infarction, angiogenesis is impaired and survival decreases markedly in W/Wv mice. We found that after PO, capillary growth occurs concurrently with LV growth to maintain capillary density and is likely attributable to EC proliferation as evidenced by equivalent PO-dependent increases in LV BrdUrd+ capillary ECs in WT and W/Wv mice. After myocardial infarction, bone marrow–derived c-kit+ cells act to regulate angiogenesis.13 Survival of W/Wv mice after myocardial infarction may be reduced because c-kit dysfunction interferes with the mobilization of these cells to the heart.13 This does not appear to be the case in our studies: capillary growth after PO is not affected by c-kit dysfunction, and this could explain the differences in mortality in W/Wv mice subjected to myocardial infarction versus PO.
Stem cell factor and c-kit play a critical role in MC and melanocyte development and in the differentiation of spermatogonial stem cells.7,9,10 However, the function of c-kit in CSCs has not been clear; it has been used as a cell marker.20 In this study, we explored the in vivo function of c-kit in CSCs. Although several growth factors and cytokines can regulate CSC differentiation in vitro,29 our findings indicate that c-kit is critical for the in vivo differentiation of c-kit+ CSCs into cardiac progenitors. Recently, Dawn et al30 have shown that stem cell factor, in combination with granulocyte colony–stimulating factor, regenerates cardiac tissue and improves LV function in mice with myocardial infarction. Because stem/progenitor cells refresh adult cardiomyocytes in the postinfarct myocardium,5 our findings suggest that stem cell factor may improve LV function in the postinfarct heart in part by increasing the number of cardiac progenitors through increased differentiation.
We conclude that c-kit is an important developmental cue; not only for cardiomyocyte terminal differentiation but also for regulating the number of cardiac progenitors in the LV. Furthermore the proliferative capability of W/Wv cardiomyocytes improves cardiac adaptation to PO. Pharmacological strategies that block or bypass cell cycle checkpoints involved in preventing adult cardiomyocyte proliferation could thus have therapeutic benefits beyond those that simply result in the regression of LV hypertrophy.
Supplementary Material
Supplemental Table 1. PCR primers used for q-RT-PCR analyses.
Supplemental Table 2. Echocardiographic and Micromanometric Measurements
Supplemental Table 3. Gene expression in isolated cardiomyocytes from WT and W/Wv left ventricles after 7 days of a sham operation (sham). Purified total RNA from cardiomyocytes was analyzed with Illumina Mouse-6 Array chips as described in Methods. Average signals (S) and their standard deviations (StDev) from 6-7 independent hybridizations, in which the mean difference between WT-sham and WT-SAC mouse groups was significantly different (P-value less than 0.05), are shown. P-values were calculated from DiffScores (not shown)---an Illumina-devised statistical confidence measure that the gene's expression has changed with respect to a reference group (where DiffScores > 13 or < -13, > 20 or < -20, and > 30 or < -30 represent significance levels < 0.05, < 0.01, and < 0.001, respectively). Signal detection values were determined and normalized by using Bead Studio and the difference in average signal between WT-Sham and W/Wv-Sham left ventricular cardiomyocyte pools is given as relative expression (R.E.). Columns under D indicate absent (A) or present (P) expression.
Supplemental Table 4. Gene Expression in Isolated Cardiomyocytes from WT and W/Wv Left Ventricles after 7 days of SAC. Purified total RNA from cardiomyocytes was analyzed with Illumina Mouse-6 Array chips as described in Methods. Average signals (S) and their standard deviations (StDev) from 5-6 independent hybridizations, in which the mean difference between WT-sham and WT-SAC mouse groups was significantly different (P-value less than 0.05), are shown. P-values were calculated from DiffScores (not shown)---an Illumina-devised statistical confidence measure that the gene's expression has changed with respect to a reference group (where DiffScores > 13 or < -13, > 20 or < -20, and > 30 or < -30 represent significance levels < 0.05, < 0.01, and < 0.001, respectively). Signal detection values were determined and normalized by using Bead Studio and the difference in average signal between WT-SAC and W/Wv-SAC left ventricular cardiomyocyte pools is given as relative expression (R.E.). Columns under D indicate absent (A) or present (P) expression.
Supplemental Table 5. c-kit Dysfunction Regulates Cell-cycle Gene Expression in Cardiomyocytes from Hypertensive Mice
Acknowledgments
Sources of Funding: Supported by NIH grants R01HL79040 and P50HL077100 and Australian National Health and Medical Research Council Program grant 354400.
Footnotes
Disclosures: None.
References
- 1.Rubart M, Field LJ. Cardiac regeneration: repopulating the heart. Annu Rev Physiol. 2006;68:29–49. doi: 10.1146/annurev.physiol.68.040104.124530. [DOI] [PubMed] [Google Scholar]
- 2.MacLellan WR, Schneider MD. Genetic dissection of cardiac growth control pathways. Annu Rev Physiol. 2000;62:289–313. doi: 10.1146/annurev.physiol.62.1.289. [DOI] [PubMed] [Google Scholar]
- 3.Jessup M, Brozena S. Heart failure. N Engl J Med. 2003;348:2007–2018. doi: 10.1056/NEJMra021498. [DOI] [PubMed] [Google Scholar]
- 4.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal-Ginard B, Anversa P. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–776. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 5.Hsieh PC, Segers VF, Davis ME, MacGillivray C, Gannon J, Molkentin JD, Robbins J, Lee RT. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat Med. 2007;13:970–974. doi: 10.1038/nm1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Linke A, Müller P, Nurzynska D, Casarsa C, Torella D, Nascimbene A, Castaldo C, Cascapera S, Böhm M, Quaini F, Urbanek K, Leri A, Hintze TH, Kajstura J, Anversa P. Stem cells in the dog heart are self-renewing, clonogenic, and multipotent and regenerate infarcted myocardium, improving cardiac function. Proc Natl Acad Sci U S A. 2005;102:8966–8971. doi: 10.1073/pnas.0502678102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 8.Herbst R, Shearman MS, Obermeier A, Schlessinger J, Ullrich A. Differential effects of W mutations on p145c-kit tyrosine kinase activity and substrate interaction. J Biol Chem. 1992;267:13210–13216. [PubMed] [Google Scholar]
- 9.Galli SJ, Kitamura Y. Genetically mast-cell-deficient W/Wv and Sl/Sld mice. Their value for the analysis of the roles of mast cells in biologic responses in vivo. Am J Pathol. 1987;127:191–198. [PMC free article] [PubMed] [Google Scholar]
- 10.Ohta H, Yomogida K, Dohmae K, Nishimune Y. Regulation of proliferation and differentiation in spermatogonial stem cells: the role of c-kit and its ligand SCF. Development. 2000;127:2125–2131. doi: 10.1242/dev.127.10.2125. [DOI] [PubMed] [Google Scholar]
- 11.Nocka K, Tan JC, Chiu E, Chu TY, Ray P, Traktman P, Besmer P. Molecular bases of dominant negative and loss of function mutations at the murine c-kit/white spotting locus: W37, Wv, W41 and W. EMBO J. 1990;9:1805–1813. doi: 10.1002/j.1460-2075.1990.tb08305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soonpaa MH, Kim KK, Pjak L, Franklin M, Field LJ. Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol. 1996;271:H2183–H2189. doi: 10.1152/ajpheart.1996.271.5.H2183. [DOI] [PubMed] [Google Scholar]
- 13.Fazel S, Cimini M, Chen L, Li S, Angoulvant D, Fedak P, Verma S, Weisel RD, Keating A, Li RK. Cardioprotective c-kit+ cells are from the bone marrow and regulate the myocardial balance of angiogenic cytokines. J Clin Invest. 2006;116:1865–1877. doi: 10.1172/JCI27019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soonpaa MH, Koh GY, Pajak L, Jing S, Wang H, Franklin MT, Kim KK, Field LJ. Cyclin D1 overexpression promotes cardiomyocyte DNA synthesis and multinucleation in transgenic mice. J Clin Invest. 1997;99:2644–2654. doi: 10.1172/JCI119453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kühn B, del Monte F, Hajjar RJ, Chang YS, Lebeche D, Arab S, Keating MT. Periostin induces proliferation of differentiated cardiomyocytes and promotes cardiac repair. Nat Med. 2007;13:962–969. doi: 10.1038/nm1619. [DOI] [PubMed] [Google Scholar]
- 16.Du XJ, Fang L, Gao XM, Kiriazis H, Feng X, Hotchkin E, Finch AM, Chaulet H, Graham RM. Genetic enhancement of ventricular contractility protects against pressure-overload-induced cardiac dysfunction. J Mol Cell Cardiol. 2004;37:979–987. doi: 10.1016/j.yjmcc.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 17.Andersen CB, Ladefoged SD, Larsen S. Acute kidney graft rejection. A morphological and immunohistological study on “zero-hour” and follow-up biopsies with special emphasis on cellular infiltrates and adhesion molecules. APMIS. 1994;102:23–37. [PubMed] [Google Scholar]
- 18.Chen R, Ning G, Zhao ML, Fleming MG, Diaz LA, Werb Z, Liu Z. Mast cells play a key role in neutrophil recruitment in experimental bullous pemphigoid. J Clin Invest. 2001;108:1151–1158. doi: 10.1172/JCI11494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Limana F, Zacheo A, Mocini D, Mangoni A, Borsellino G, Diamantini A, De Mori R, Battistini L, Vigna E, Santini M, Loiaconi V, Pompilio G, Germani A, Capogrossi MC. Identification of myocardial and vascular precursor cells in human and mouse epicardium. Circ Res. 2007;101:1255–1265. doi: 10.1161/CIRCRESAHA.107.150755. [DOI] [PubMed] [Google Scholar]
- 20.Anversa P, Kajstura J, Leri A, Boli R. Life and death of cardiac stem cells: a paradigm shift in cardiac biology. Circulation. 2006;113:1451–1463. doi: 10.1161/CIRCULATIONAHA.105.595181. [DOI] [PubMed] [Google Scholar]
- 21.Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006;441:1068–1074. doi: 10.1038/nature04956. [DOI] [PubMed] [Google Scholar]
- 22.Matz DG, Oberpriller JO, Oberpriller JC. Comparison of mitosis in binucleated and mononucleated newt cardiac myocytes. Anat Rec. 1998;251:245–255. doi: 10.1002/(SICI)1097-0185(199806)251:2<245::AID-AR14>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 23.Soonpaa MH, Field LJ. Survey of studies examining mammalian cardiomyocyte DNA synthesis. Circ Res. 1998;83:15–26. doi: 10.1161/01.res.83.1.15. [DOI] [PubMed] [Google Scholar]
- 24.Cheng RK, Asai T, Tang H, Dashoush NH, Kara RJ, Costa KD, Naka Y, Wu EX, Wolgemuth DJ, Chaudhry HW. Cyclin A2 induces cardiac regeneration after myocardial infarction and prevents heart failure. Circ Res. 2007;100:1741–1748. doi: 10.1161/CIRCRESAHA.107.153544. [DOI] [PubMed] [Google Scholar]
- 25.Poolman RA, Li JM, Durand B, Brooks G. Altered expression of cell cycle proteins and prolonged duration of cardiac myocyte hyperplasia in p27KIP1 knockout mice. Circ Res. 1999;85:117–127. doi: 10.1161/01.res.85.2.117. [DOI] [PubMed] [Google Scholar]
- 26.Pasumarthi KB, Field LJ. Cardiomyocyte cell cycle regulation. Circ Res. 2002;90:1044–1054. doi: 10.1161/01.res.0000020201.44772.67. [DOI] [PubMed] [Google Scholar]
- 27.Clark EB, Hu N, Frommelt P, Vandekieft GK, Dummett JL, Tomanek RJ. Effect of increased pressure on ventricular growth in stage 21 chick embryos. Am J Physiol. 1989;257:H55–H61. doi: 10.1152/ajpheart.1989.257.1.H55. [DOI] [PubMed] [Google Scholar]
- 28.Nakajima H, Nakajima HO, Tsai SC, Field LJ. Expression of mutant p193 and p53 permits cardiomyocyte cell cycle reentry after myocardial infarction in transgenic mice. Circ Res. 2004;94:1606–1614. doi: 10.1161/01.RES.0000132279.99249.f4. [DOI] [PubMed] [Google Scholar]
- 29.Anversa P. Aging and longevity: the IGF-1 enigma. Circ Res. 2005;97:411–414. doi: 10.1161/01.RES.0000182212.09147.56. [DOI] [PubMed] [Google Scholar]
- 30.Dawn B, Guo Y, Rezazadeh A, Huang Y, Stein AB, Hunt G, Tiwari S, Varma J, Gu Y, Prabhu SD, Kajstura J, Anversa P, Ildstad ST, Bolli R. Postinfarct cytokine therapy regenerates cardiac tissue and improves left ventricular function. Circ Res. 2006;98:1098–1105. doi: 10.1161/01.RES.0000218454.76784.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. PCR primers used for q-RT-PCR analyses.
Supplemental Table 2. Echocardiographic and Micromanometric Measurements
Supplemental Table 3. Gene expression in isolated cardiomyocytes from WT and W/Wv left ventricles after 7 days of a sham operation (sham). Purified total RNA from cardiomyocytes was analyzed with Illumina Mouse-6 Array chips as described in Methods. Average signals (S) and their standard deviations (StDev) from 6-7 independent hybridizations, in which the mean difference between WT-sham and WT-SAC mouse groups was significantly different (P-value less than 0.05), are shown. P-values were calculated from DiffScores (not shown)---an Illumina-devised statistical confidence measure that the gene's expression has changed with respect to a reference group (where DiffScores > 13 or < -13, > 20 or < -20, and > 30 or < -30 represent significance levels < 0.05, < 0.01, and < 0.001, respectively). Signal detection values were determined and normalized by using Bead Studio and the difference in average signal between WT-Sham and W/Wv-Sham left ventricular cardiomyocyte pools is given as relative expression (R.E.). Columns under D indicate absent (A) or present (P) expression.
Supplemental Table 4. Gene Expression in Isolated Cardiomyocytes from WT and W/Wv Left Ventricles after 7 days of SAC. Purified total RNA from cardiomyocytes was analyzed with Illumina Mouse-6 Array chips as described in Methods. Average signals (S) and their standard deviations (StDev) from 5-6 independent hybridizations, in which the mean difference between WT-sham and WT-SAC mouse groups was significantly different (P-value less than 0.05), are shown. P-values were calculated from DiffScores (not shown)---an Illumina-devised statistical confidence measure that the gene's expression has changed with respect to a reference group (where DiffScores > 13 or < -13, > 20 or < -20, and > 30 or < -30 represent significance levels < 0.05, < 0.01, and < 0.001, respectively). Signal detection values were determined and normalized by using Bead Studio and the difference in average signal between WT-SAC and W/Wv-SAC left ventricular cardiomyocyte pools is given as relative expression (R.E.). Columns under D indicate absent (A) or present (P) expression.
Supplemental Table 5. c-kit Dysfunction Regulates Cell-cycle Gene Expression in Cardiomyocytes from Hypertensive Mice