

Abstract
Lung cancer is a leading cause of cancer death due to the high incidence of metastasis; therefore novel and effective treatments are urgently needed. A current strategy is cancer specific targeted gene therapy. While many identified cancer specific promoters are highly specific, they tend to have low activity compared to the ubiquitous CMV promoter, limiting their application. We developed a targeted gene therapy expression system for lung cancer that is highly specific with strong activity. Our expression vector uses the survivin promoter, highly expressed in many cancers but not normal adult tissues. We enhanced the survivin promoter activity comparable to the CMV promoter in lung cancer cell lines using an established platform technology, while the survivin promoter remained weak in normal cells. In mouse models, the transgene was specifically expressed in the lung tumor tissue, compared with the CMV promoter that was expressed in both normal and tumor tissues. Additionally, the therapeutic gene BikDD, a mutant form of pro-apoptotic Bik, induced cell killing in vitro, and inhibited cell growth and prolonged mouse survival in vivo. Importantly, there was virtually no toxicity when BikDD was expressed with our expression system. Thus, the current report provides a therapeutic efficacy and safe strategy worthy of development in clinical trials treating lung cancer.
Keywords: Targeted Gene Therapy, Lung Cancer, BiKDD, VISA, Survivin
Introduction
Lung cancer remains the leading cause of cancer deaths in the world (Jemal et al., 2008). New approaches to the treatment of lung cancer are urgently needed. Of the current methods, targeted gene therapy is a novel strategy for treatment of cancer, especially for treating unresectable, metastasized, or therapy refractory tumors (Gunther et al., 2005). To succeed in cancer gene therapy, the efficient delivery of therapeutic genes to a target site is a major challenge. Although viruses are very effective gene delivery systems, there are concerns for safety issue due to immunogenicity of viral proteins, risk of oncogenesis and inadvertent creation of infectious viral particles (Russ and Wagner, 2007). Rapid advances in transfection technologies employing nonviral vectors, together with their relatively low toxicity, suggest that nonviral vectors may have significant potential for clinical applications (Huang, 2005). Most gene therapy protocols use strong promoters such as the CMV promoter to drive a transgene to validate the biological activity of the gene of interest (Fitzsimons et al., 2002). However, these types of strong promoters are usually ubiquitously expressed without tumor-targeting activity, thus limiting their applications due to potential side effects caused by unwanted expression of therapeutic gene in normal tissue. This issue is especially critical when the therapeutic gene is delivered systemically such as by intravenous injection. One way to circumvent the non-specific delivery is to develop a strong cancer-specific expression vector. Most of the identified lung cancer-specific promoters exhibit a high level of lung cancer specificity but are still much weaker than the CMV promoter (Chen et al., 2004; Fukazawa et al., 2007; Oshikiri et al., 2006). To overcome this issue, we have established a specific promoter amplification system, called “VISA” (VP16-GAL4-WPRE integrated systemic amplifier) system (Xie et al., 2007) to significantly amplify the cancer-specific promoter activity to a comparable level to CMV in cancer cells while still retaining low promoter activity in normal tissues. This VISA system combined with pancreatic-cancer-specific promoter to target expression of proapoptotic gene exhibited significant antitumor effects on pancreatic cancer.
Survivin is a member of the inhibitors of apoptosis (IAP) protein family that plays an important role in both control of cell division and inhibition of apoptosis (Ambrosini et al., 1997). Survivin is strongly expressed in embryonic, fetal organs and common cancers, but has not been reported in differentiated normal tissues with exception of the thymus, basal colonic epithelium (Altieri, 2003a) and neural stem cells during angiogenesis (O'Connor et al., 2000). Owing to its upregulation in most human tumors and its involvement in cancer progression and treatment resistance, survivin is currently undergoing extensive investigation as a novel therapeutic target (Zaffaroni et al., 2005). Our previous results also demonstrated that the survivin promoter possessed high activity in lung cancer cells and low activity in other cell types, including immortalized normal cell lines and other cancer types (Chen et al., 2004), suggesting that survivin may be a good cancer specific promoter in cancer gene therapy. In addition, we have developed a Bik (Bcl2 interacting killer) mutant, BikDD in which threonine 33 and serine 35 were changed to aspartic acid to mimic the phosphorylation at these two residues, and found the mutant’s anti-cancer activity to be more potent than wild type Bik (Day et al., 2006; Li et al., 2003; Xie et al., 2007). Here we demonstrate that the therapeutic effect becomes more significant through combining cancer-specific promoter, survivin with the promoter amplification VISA system to enhance transgene, BikDD expression in lung cancer cells. In addition, this vector produced virtually no toxicity compared with the CMV promoter vector. Thus, this Survivin-VISA expression vector in combination with the therapeutic gene BikDD may be a promising candidate for clinical trials in the near future.
Results
Enhancement of survivin promoter activity by the VISA system
To assess whether the VISA system can selectively enhance transgene expression driven by the survivin promoter in lung cancer cells but not in normal cells, three promoters, survivin, survivin combined with VISA system (Survivin-VISA), and CMV were used to drive luciferase expression (Fig. 1A). A panel of non-small cell lung cancer cell lines and normal cell lines were used to detect the promoter activity. With the survivin promoter, there was a significant difference in luciferase expression between the lung cancer cell lines and normal cell lines (P = 0.015), but the expression level was much weaker than with the CMV promoter (Fig. 1B). However, the Survivin-VISA vector greatly enhanced the surviving promoter activity with an average fold of 31±18 and up to 65-fold compared to the surviving promoter alone and even stronger than or comparable to CMV activity in most of the lung cancer cell lines by dual luciferase assay (Fig. 1C). Similar results were also observed when the luciferase activity was normalized by protein level (data not shown). Most importantly, the VISA vector was able to selectively amplify the survivin promoter in lung cancer cells but not in various normal cells (P = 0.028). The amplified promoter activity of Survivin-VISA is stronger or equal to the activity of CMV in lung cancer cells but still remains much weaker in normal cells. Thus, the Survivin-VISA vector indeed selectively enhanced the transgene expression in lung cancer cells, but remained low expression in normal cells. p53 has been described to negatively regulate the survivin promoter (Hoffman et al., 2002). Hence, the p53 status could be one factor predetermining transgene expression via the survivin-VISA vector. To address this, we checked the p53 status in the lung cancer cell lines from the reported literature (Hay et al., 1996; Tsai et al., 2006), listed in the Fig. 1C. Indeed, the transgene expression from Survivin-VISA vector was lower than that from CMV promoter in three lung cancer cell lines, CRL5802, CRL5806, and A549 harboring wild type p53, while the level was higher or comparable to CMV promoter activity in other lung cancer cells with mutant p53. Therefore, the functional p53 status could be one factor predetermining transgene expression via Survivin-VISA. We also investigated other factors, such as mitosis regulator, Cdc2 (Nigg, 2001) and proliferation rate for their potential activity to affect Survivin-VISA driven transgene expression. The results suggest that they are not related to the expression efficacy of transgene from the survivin promoter with correlation coefficience of 0.52 and 0.07, respectively (Supplementary figure 1).
1.
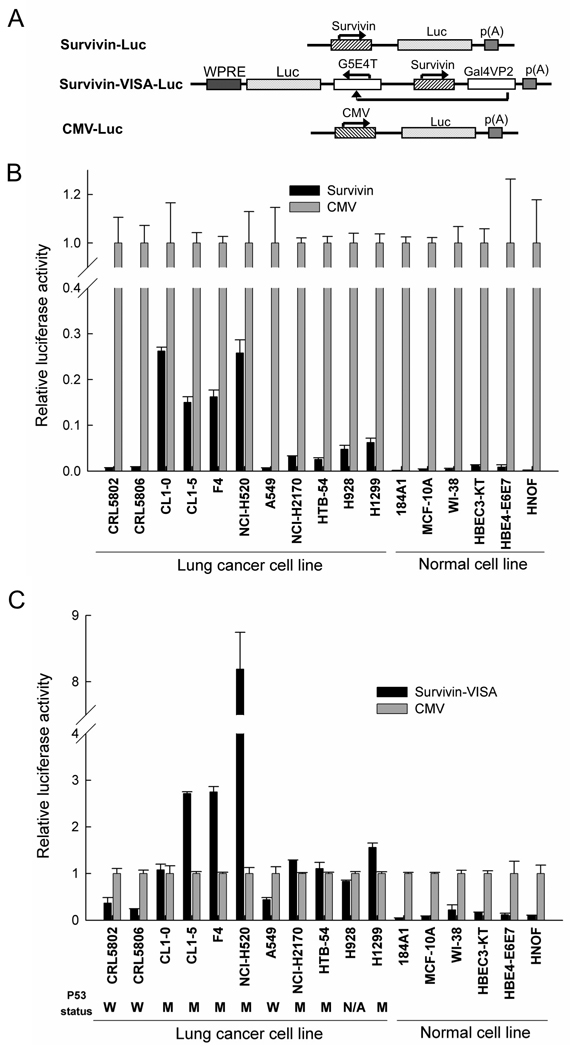
The VISA system can selectively enhance transgene expression driven by the Survivin promoter in lung cancer cell lines, but not in normal cells. (A) Schematic diagram of engineered promoter driving luciferase constructs. (B) The transcriptional activity of the Survivin, and CMV promoters was measured in lung cancer cell lines and normal cells by cotransfection with indicated plasmid DNA and pRL-TK in dual luciferase assay. The relative luciferase activity shown here represents the dual luciferase activity ratio (firefly versus renilla luciferase) in Survivin-VISA vector relative to that of the CMV promoter by setting CMV activity as 1. (C) The transcriptional activity of the Survivin-VISA, and CMV promoters was measured in lung cancer cell lines and normal cells as (B). The p53 status is listed under the indicated lung cancer cell lines. W indicated wild type; M indicated mutant; N/A indicates not available.
Cancer specific targeting of Survivin-VISA promoter in vivo
To examine whether the activity and specificity of Survivin-VISA is still retained in vivo, the CMV-Luc, and Survivin-VISA-Luc (SV-Luc) constructs (Fig. 1A) were i.v. delivered by liposome (Templeton et al., 1997) to tumor-free and tumor-bearing SCID mice in orthotopic lung cancer xenograft animal models. Bioluminescent imaging revealed a strong signal in the thoracic area of mice treated with CMV-Luc construct in tumor-free and tumor-bearing mice but a low signal in mice treated with SV-Luc construct (Fig. 2A upper). To monitor the source of signal more precisely, the mice were sacrificed immediately after in vivo imaging, and their organs were dissected for ex vivo imaging (Fig. 2A lower). In CMV-Luc injected groups, the quantified photon signal showed the high values in the lungs from tumor-free and tumor-bearing mice, while very low values in the lungs from SV-Luc injected tumor-free and tumor-bearing mice. Immunohistochemical analysis also showed strong luciferase expression in both of normal lung tissue and tumor tissues in the CMV-Luc injected group in contrast to very low luciferase expression in lung normal tissue but strong expression in tumor tissues in SV-Luc injected group (Fig. 2B). That confirmed the ex vivo imaging results that the CMV promoter strongly drove luciferase expression in both normal lung and lung tumor tissue while the SV expression vector selectively drove luciferase expression in the lung tumor tissue. The in vivo data demonstrated that the transgene expression in Survivin-VISA vector is more lung cancer specific than CMV vector and this vector may be suitable for gene therapy.
2.
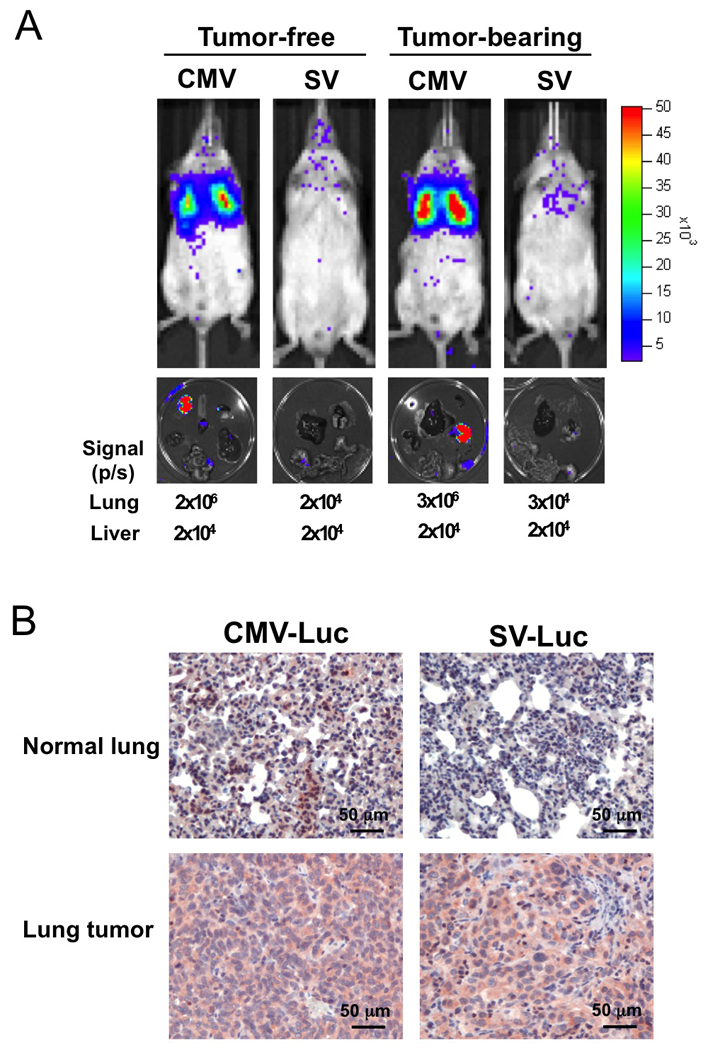
Survivin-VISA vector selectively expressed luciferase in the lung tumor tissue of SCID mice carrying orthotopic lung cancer xenograft, while CMV promoter strongly expressed luciferase in both of lung and lung tumor tissue of tumor-bearing mice. Fifty µg plasmid plus HLDC liposome were administrated into mice by tail vein injection, and the luciferase activity was detected with the noninvasive imaging system (IVIS imaging system, xenogen) after 48 hours. (A) The promoter specificity of CMV and Survivin-VISA (SV) was detected by driving luciferase expression in tumor-free and tumor-bearing mice (upper panel). The organs from mice were then dissected for ex vivo imaging (lower panel). The quantified signal from lungs and livers is also shown under the lower panel. (B) The lung organs from tumor-bearing mice were fixed and processed for immunohistochemical analysis of firefly luciferase expression by using anti-Luc antibody. The luciferase protein was stained in red and nucleus in blue.
Therapeutic gene BikDD driven by the Survivin-VISA promoter inhibits the lung cancer cells growth in vitro
To examine whether the Survivin-VISA vector can be used for gene therapy to drive a therapeutic gene to selectively kill lung cancer cells, we constructed CMV-BikDD and Survivin-VISA-BikDD (SV-BikDD) plasmids (Fig. 3A). The control vector alone was used as control group (Ctrl). The expression of BikDD driven by CMV or SV was detected by Western blot after 24h transient transfection in F4 lung cancer cells (Fig. 3B). We then examined whether SV-BikDD plasmid had selective killing activity in lung cancer cells. Eleven lung cancer cell lines and 6 normal cell lines were transiently cotransfected with Ctrl, CMV-BikDD, or SV-BikDD plus pRL-TK (the indicator for transfection efficiency control) and the renilla luciferase activity was detected (Fig. 3C). The relative cell viability was measured by setting Ctrl group in each cell lines to 100%. We found that CMV-BikDD exhibited a strong killing effect in both lung cancer cell lines and normal cells (P = 0.19). While SV-BikDD induced a significant cytotoxic effect in cancer cells, and a weak effect in normal cells (P = 0.006), indicating that the Survivin-VISA vector drove BikDD expression selectively in lung cancer cells in vitro.
3.
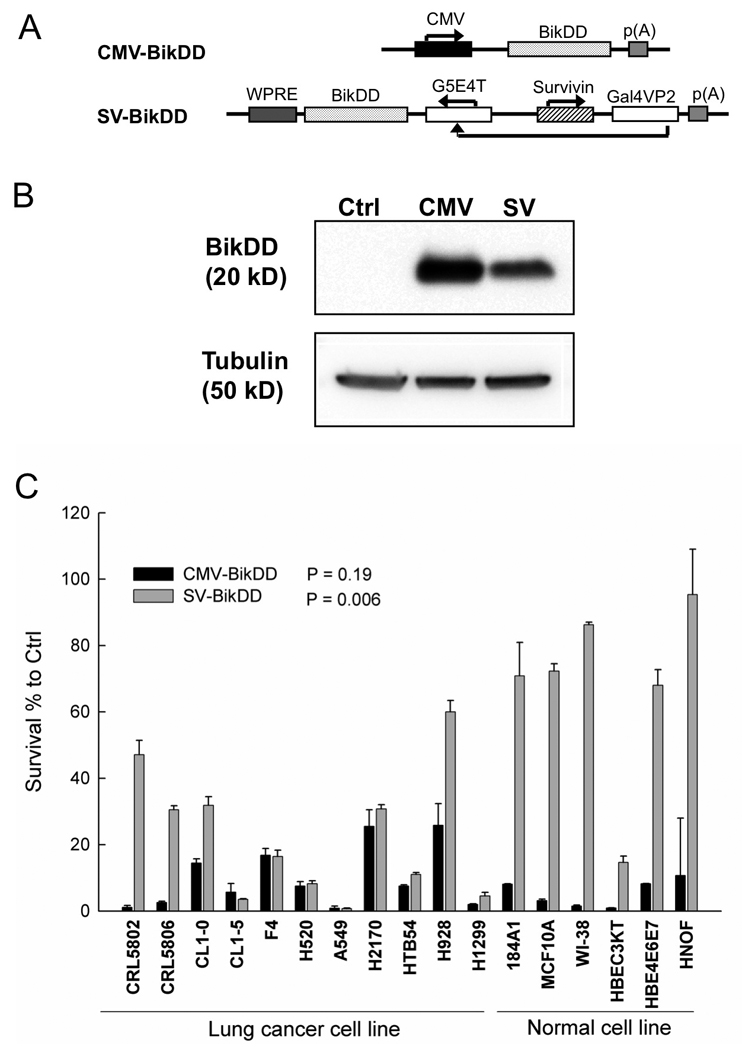
The therapeutic gene BikDD was expressed in lung cancer cells and inhibited lung cancer cells growth. (A) The schematic diagram of therapeutic constructs. (B) BikDD driven by CMV and SV was detected in F4 lung cancer cell lysate after 24h plasmids transient transfection by Western blot. (C) In vitro cell killing effect of BikDD. Indicated therapeutic plasmids and pRL-TK were co-transfected into cells by lipofetamine 2000. The renilla luciferase activity was detected after 48 hours. Relative cell viability was measured by setting Ctrl as 100% cell viability. The p values between lung cancer cell lines and normal cell lines with CMV-BikDD and SV-BikDD treatment were 0.19 and 0.006, respectively.
Survivin-VISA-BikDD inhibits tumor growth and prolongs mouse survival time in orthotopic models of F4-Luc and H1299-Luc xenografts
To evaluate the antitumor effects of SV-BikDD in human lung cancer in vivo, we established an orthotopic animal model by intrathoracic injection with human lung cancer cells, F4-Luc in SCID mice. Mice were treated with systemic delivery of liposomal DNA complex and luciferase activity was measured to reflect the tumor size with IVIS imaging system without sacrificing them. The signal increased much more slowly in the CMV-BikDD and SV-BikDD treatment groups than the Ctrl group (p <0.01), indicating that both CMV-BikDD and SV-BikDD treatment inhibited tumor growth (Fig. 4A). The survival curve also demonstrated that CMV-BikDD and SV-BikDD significantly prolonged the survival time compared with the Ctrl group (P < 0.01) (Fig. 4B). Both of the CMV-BikDD and SV-BikDD treatment groups expressed BikDD mRNA in the lung tissue from mice bearing lung cancer as demonstrated using RT-PCR after treatment (Fig. 4C), which shows a higher BikDD expression driven by SV in lung tissue than CMV but no BikDD expression in the liver. Endogenous Bik was undetectable from all tissue RNA although the primer set could amplify the Bik fragment from plasmids (data not shown). We also used another orthotopic xenograft animal model with H1299-Luc lung cancer tumors to determine the therapeutic effect of the plasmids. Figure 5A showed that SV-BikDD treatment inhibited the tumor growth more strongly than CMV-BikDD and Ctrl groups (p < 0.05). Additionally, SV-BikDD treatment prolonged longer survival time than in the CMV and Ctrl groups (p < 0.05) (Fig. 5B). The mRNA level of BikDD driven by Survivin-VISA vector was also higher than that from CMV vector in lung but not in liver, similar to the results shown in Figure 4C (data not shown). We then asked whether SV-BikDD gene therapy was able to prolong the survival of mice suffering metastatic lung cancer. To examine this, SCID mice were intravenously injected with H1299-Luc lung cancer cells to mimic metastatic circulating cancer cells and then treated with systemic delivery of liposomal DNA complex. While both CMV-BikDD and SV-BikDD treatment prolonged the survival time compared to Ctrl group (Fig. 5C), the survival curve of SV-BikDD was more significant than CMV-BikDD with p values of 0.01 and 0.05, respectively. Thus, SV-BikDD suppressed tumor growth and prolonged survival time more effectively than did CMV-BikDD in multiple orthotopic models of human lung cancer in immunodeficient mice. To determine if SV-BikDD is also effective in immunocompentent mice, we used a syngeneic cancer model. Mouse lung cancer cell line, TC1 was used to establish a syngeneic cancer model in C57BL/6 mice. As shown in Supplementary Fig. 2, expression of BikDD driven by the Survivin-VISA vector inhibited tumor growth more effectively and significantly than that driven by CMV promoter compared with Ctrl treatment in mouse TC1 lung cancer syngeneic models. Thus, the potent therapeutic efficacy of SV-BikDD gene therapy can also be observed in immunocompetent mice.
4.
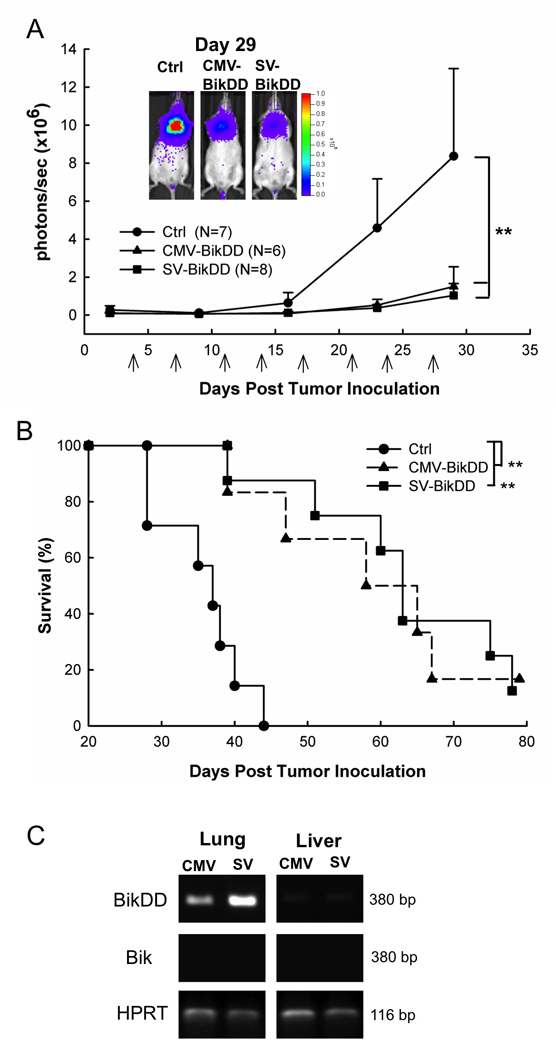
The therapeutic gene, BikDD driven by Survivin-VISA and CMV promoter inhibited tumor growth and prolonged survival time than Ctrl group in the F4-Luc lung cancer orthotopic xenograft model. (A) SCID mice with intrathoracic injection of F4-Luc lung cancer cells were intravenously injected with 25 µg of liposomal plasmid DNA. Arrows indicate the therapy time points. The photon signals were quantified with Xenogen’s living imaging system to reflect the tumor size. Error bars indicate SEM. (B) Kaplan-Meier survival analysis was performed. The mean survival time was 36±2, 59±5, and 64±5 days in Ctrl, CMV-BikDD, and SV-BikDD groups, respectively. (C) BikDD driven by CMV or Survivin-VISA promoter was detected in the lung tissue from the mouse bearing lung cancers by RT-PCR two days later after the last treatment. ** indicated p value < 0.01.
5.
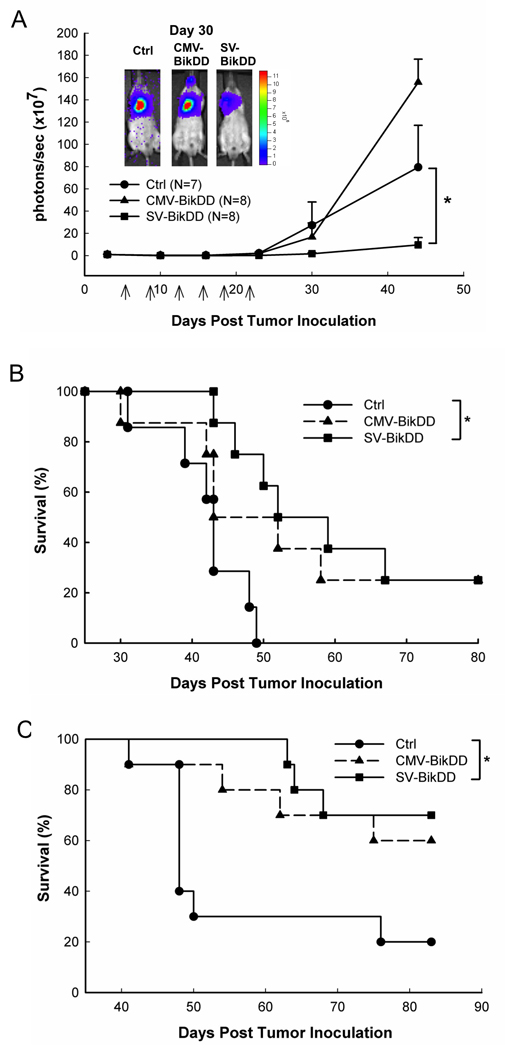
The therapeutic gene, BikDD driven by Survivin-VISA promoter inhibited tumor growth and prolonged survival time than that driven by CMV promoter in H1299-Luc lung cancer orthotopic xenograft models. (A) SCID mice with intrathoracic injection of H1299-Luc lung cancer cells were intravenously injected with 25 µg of liposomal plasmid DNA. Arrows indicate the therapy time points. The photon signals were quantified with Xenogen’s living imaging system to reflect the tumor size. Error bars indicate SEM. (B) Kaplan-Meier survival analysis was performed. The mean survival time is 42±2, 53±6, and 60±5 days in Ctrl, CMV-BikDD, and SV-BikDD groups, respectively. (C) SCID mice with intravenous injection of H1299-Luc lung cancer cells were treated with 25 µg of liposomal plasmid DNA as panel A. Each group has 10 mice. The survival curve was presented in Kaplan-Meier survival analysis. The mean survival time is 57±5, 73±5, and 78±3 days in Ctrl, CMV-BikDD, and SV-BikDD groups, respectively. * indicated p value < 0.05.
Survivin-VISA-BikDD has no acute toxicity compared to CMV-BikDD
A critical question raised in gene therapy is whether therapeutic plasmids for cancer treatment are toxic to normal cells/tissue. To evaluate whether Survivin-VISA-BikDD treatment is safer than CMV-BikDD, a single high dose of 50 µg plasmid DNA was injected through tail vein of Balb/c mice with normal immunity. All of the animals survived at this dosage and blood samples were collected to determine the serum levels of liver aspartate aminotransferase (AST), alanine aminotransferase (ALT) and blood urea nitrogen (BUN) (Fig. 6A). BUN levels were not elevated at any measurement time point (data not shown), indicating a lack of nephrotoxicity. Treatment with Ctrl and SV-BikDD did not increase the level of AST and ALT, however, serum level of AST and ALT in CMV-BikDD treatment group were readily detectable on Day 1 and decreased on Day 2, indicating CMV-BikDD caused severe acute liver toxicity. To assess apoptosis in vivo, mice were sacrificed on Day3 and the apoptosis was determined in the lung and liver using the terminal deoxynucleotidyl transferase [TdT]-mediated dUTP nick end labeling (TUNEL) assay (Fig. 6B). CMV-BikDD treatment led to remarkable amount of apoptosis in the normal lung and liver tissue compared with Ctrl and SV-BikDD treatment from the figure. After analysis, CMV-BikDD treatment significantly induced high percentage of apoptotic cells in normal lung and liver tissue compare to Ctrl treatment (p < 0.01), but SV-BikDD treatment did not. Taken together these results indicate that CMV-BikDD treatment can lead to normal lung and liver toxicity while SV-BikDD treatment may be safer for gene therapy.
6.
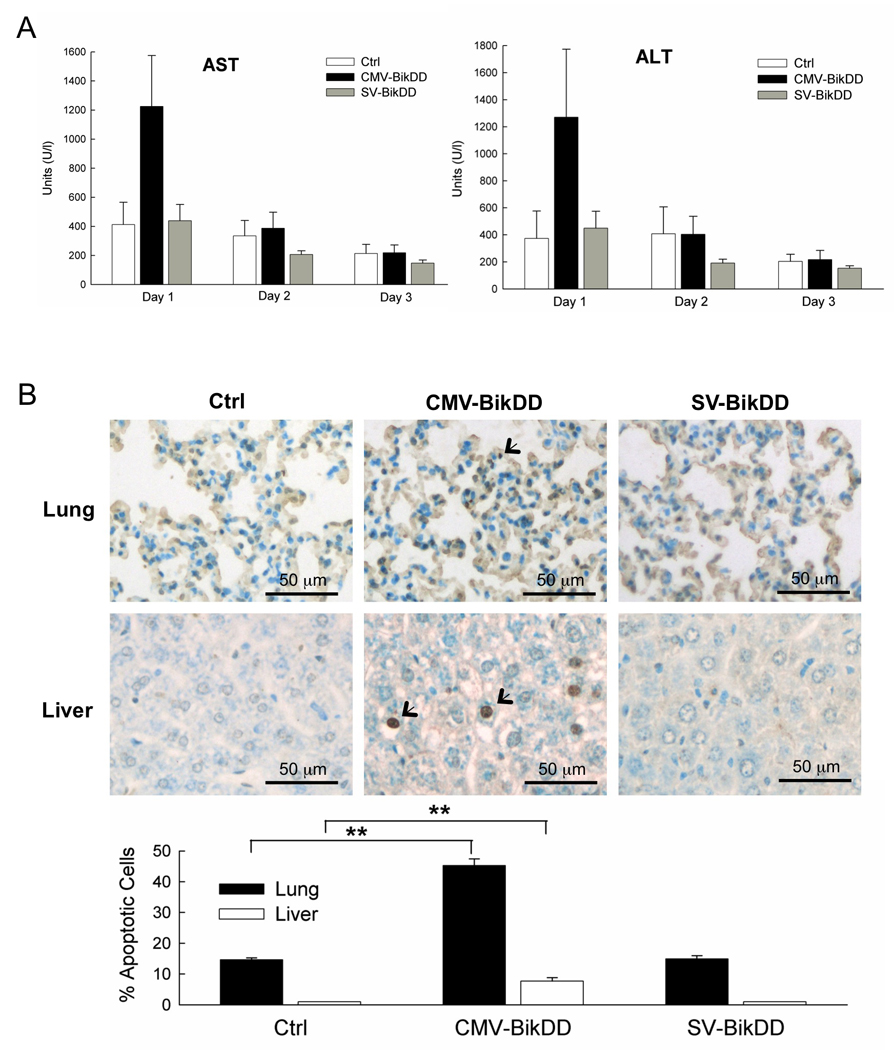
SV-BikDD treatment had no systemically acute toxicity compared with CMV-BikDD treatment in immunity complete mice. Male Balb/c mice were given single dose of 50 µg plasmid DNA in a liposomal complex via the tail vein injection (n = 5 mice/group). (A) Serum level of AST and ALT in mice was monitored after plasmid DNA injection. Error bars indicate SEM. (B) In vivo apoptosis of tissue specimens was detected by TUNEL assay and was quantified % of apoptotic cells from 4 fields. ** indicated p value < 0.01.
Discussion
Evidence from studies of in vitro human cells and in vivo xenograft animal models with human cancer cells have revealed that inhibiting the function of survivin in human cancer cells can suppress tumor growth with little toxicity to normal cells/organs (Altieri, 2003b; Li and Ling, 2006). Our study here also demonstrates that survivin promoter is specifically activated in lung cancer cell lines (Fig. 1B), indicating survivin is a cancer specific promoter although the activity is weaker than CMV promoter and limits its application in gene therapy. Our developed VISA system resolves this problem by enhancing the surviving promoter activity in vitro in lung cell lines and in vivo in lung tumor tissue of mice but remaining low in normal cells (Fig. 1C and 2B). In fact, the VISA system can enhance survivin promoter activity in both lung cancer cells (0.09 to 1.89-fold) and normal cells (0.005 to 0.124-fold) to about 20 to 25-fold with CMV activity set to 1. However, since survivin is a cancer specific promoter, the amplification from VISA made the activity level of Survivin-VISA vector to become comparable or higher to CMV promoter in lung cancer cells, but in normal cells, Survivin-VISA was still much weaker than that of the CMV promoter (Fig. 1C). Thus, it is possible for the VISA system to serve as a powerful tool for gene therapy to overcome the typical promoter weakness of cancer specific promoters as compared with CMV that limits their applications. Combining the cancer specific promoter with the VISA system might enable novel strategies to treat different cancers without toxicity to normal human tissues.
The Survivin-VISA vector was specifically activated in lung cancer cells but low in normal cells (Fig. 1C) and was able to selectively kill cancer cells but not normal cells with the exception of HBEC3KT cells (Fig. 3C). The HBEC3KT cells were established by serially transfecting with retroviral constructs containing cyclin-dependent kinase (CDK) 4 and human telomerase reverse transcriptase (hTERT) in human bronchial epithelial cell (HBEC) lines, resulting in continuously growing cultures (Ramirez et al., 2004). CDK4 knockdown or inhibition has been reported to induce down regulation of survivin protein and survivin mRNA in pancreatic cancer cells (Retzer-Lidl et al., 2007). That indicated CDK4 expression is correlated with survivin expression and may activate surviving promoter activity. That may explain why the Survivin-VISA vector activated transgene expression in HBEC3KT and had a stronger killing effect than in the other normal cell lines.
While both SV-BikDD and CMV-BikDD were able to prolong survival in the F4-Luc mouse model (Fig. 4B), there was a significant therapeutic effect with SV-BikDD treatment but only a low therapeutic effect with CMV-BikDD treatment in H1299-Luc animal model (Fig. 5A and B). Based on the promoter activity normalized with total protein, the CMV promoter activity in F4 cells was higher than that in H1299 cells, with values of 1.41 × 109 and 1.05 × 109 RLU/mg, respectively. That may cause the better therapeutic effect of CMV-BikDD treatment in F4-Luc mouse model than in H1299-Luc mouse model. However, SV-BikDD treatments inhibited tumor growth and prolonged survival time in both orthotopic lung cancer animal models (Fig. 4 and 5). Furthermore, our data showed that mice treated with SV-BikDD survived longer than those treated with CMV-BikDD in the animal model of circulating metastatic cells (Fig. 5C). This novel strategy offers an opportunity to target circulating metastatic cancer cells or dormant tumor cells. Thus, SV-BikDD may be more advantageous for lung cancer than CMV-BikDD since metastasis is the major cause of death in lung cancer patients.
Material and Method
Cell Lines
The following cell lines were obtained from the American Type Culture Collection (Manassas, VA): human lung cancer cell lines (NCI-H520, A549, NCI-H2170, HTB54, H1299), immortalized normal mammary epithelial (184A1, MCF-10A), normal lung fibroblasts (WI-38), immortalized normal lung epithelial (HBE4-E6/E7). Immortalized normal lung epithelial cells (HBEC-3KT) were kindly provided by Dr. John D. Minna (Ramirez et al., 2004). Other human lung cancer cell lines, CRL-5802, CRL-5806, CL1-0, CL1-5, F4, H928, and H2981 were generated from National Taiwan University Hospital. Before use, all cell lines were tested and found to be free of Mycoplasma infection.
Construction of Plasmids
To construct the Survivin-VISA-Luc plasmid, the human survivin gene promoter (from our previous construct, pSRVN-Luc) (Chen et al., 2004) was amplified, digested with SpeI/EcoRV and inserted into pGL3-C-VISA-Luc (Xie et al., 2007). To construct the therapeutic plasmids, we replaced the luciferase gene with BikDD, which was digested with BglII/NheI and inserted. Then the pGL3 backbone (ampicillin resistant gene) except the Survivin-VISA-BikDD fragment was changed to pUK backbone (kanamycin resistant gene) through NotI and SalI ligation for antibiotic replacement.
Evaluation of Promoter Activity and Cytotoxicity
To normalize transfection efficiency, we used the Dual-Luciferase reporter assay system according to the manufacturer’s instructions (Promega, Madison, WI). Briefly, the detected promoter reporter plasmids: pGL3-CMV-Luc or pGL3-Survivin-VISA-Luc was co-transfected with the internal plasmid, pRL-TK. Cells were grown to about 80% confluent in 24-well plates and then, transiently transfected with 0.8 µg of tested plasmid DNA with 20 ng of pRL-TK by using lipofectamine 2000 (Invitrogen, Carlsbad, CA). The luciferase activity from cell lysate was determined after 48 hours in FB12 Luminometer (Berthold Technologies, Bad Wildbad, Germany). The dual luciferase ratio was presented as the luciferase activity of the tested plasmids divided by the luciferase activity of pRL-TK.
In cytotoxicity, pRL-TK was also used as a reporter gene in all experiments. The luciferase activity of control group (pUK21) was set as 100% in each cell line to detect the killing effect of therapeutic plasmids, pUK-CMV-BikDD and pUK-Survivin-VISA-BikDD.
Stable Cell Lines Expressing Firefly Luciferase
F4 and H1299 cells were transfected with pcDNA-Luc-Neo and selected with G418 (1 mg/ml). Ten individual clones with a high level luciferase activity were mixed as F4-Luc or H1299-Luc stable cell lines.
Animal Models of Lung Cancer and Treatments in vivo
CB17-SCID mice (BioLASCO, Taiwan), at 6–8 weeks of age, were used for human xenograft tumor animal model. The orthotopic lung cancer models of F4-Luc and H1299-Luc were established by inoculating with a suspension of cells through intrathoracic or intravenous injection (McLemore et al., 1988). After inoculation of cancer cells, the mice were imaged with the IVIS system and then randomly assigned in three groups. Therapeutic plasmids were purified with an Endo-free Mega Prep Kit (Qiagen, Valencia, CA) with manufacturer’s instructions. Plasmid DNA was dissolved in endotoxin-free TE buffer. The amount of endotoxin in plasmids was determined by a chromogenic Limulus amoebocyte clotting assay (QCL-1000 kit, BioWhittaker, Walkersville, MD) and in less 10 endotoxin units/mg of DNA. HLDC (Hung lab modified DOTAP:Cholesterol) was produced in our laboratory according to Dr. Nancy Tempeton’s protocol (Templeton et al., 1997). The mouse received intravenous injection of 100 µl of DNA:HLDC complex containing 25 µg of DNA each time, twice a week and total 6 – 8 therapy times.
IVIS system and Quantification
Mice tumor size was imaged by IVIS Imaging system (Xenogen, Alameda, CA). Mice were anesthetized with a mixture of oxygen and isoflurane and intraperitoneally injected with 100 ul of D-luciferin (Xenogen; 30 mg/ml in PBS). Ten minutes later, mice underwent imaging with the IVIS imaging system and analyzed with Living Imaging software.
Reverse Transcription PCR of BikDD
Total RNA was extracted from the lung and liver organs two days after last treatment using the TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. Extracted RNA was reverse transcribed using oligo(dT) primer and Superscript III reverse transcriptase. The primers are used for PCR : BikDD-For: 5’-ATGGAGGTTCTTGGCATGGATGACGA-3’; BikDD-Rev: 5’-AGGTGCAGGCCCCCGCTGAGCAGCGG-3’; B i k-For: 5’-ATGGAGGTTCTTGGCATGACTGACTC-3’; Bik-Rev is the same to BikDD-Rev; HPRT-For: 5 ’-GGAGCGGTAGCACCTCCT-3’; HPRT-Rev: 5’-CTGGTTCATCATCGCTAATCAC-3’. One µl aliquot of cDNA product was used for PCR amplification in a 50 µl reaction containing10 pmoles of forward and reverse primers, 0.2 mM dNTPs, 1X Taq PCR Reaction Buffer with 1.5 mM MgCl2, and 2 U of Taq DNA polymerase (Roche, Indianapolis, IN) using 35 cycles at 94°C for 15 sec, 58°C for 30 sec, and 68°C for 1 min, and a final 5-min extension at 68°C. Plasmids containing BikDD or Bik were used as positive control.
Immunohistochemical Stain
The lung tumor and other tissues dissected from mice were fixed in 10% neutal buffered formalin and embedded in paraffin wax. Five-micrometer sections were cut and stained with H&E. Immunostaining for firefly luciferase was performed as our previous protocol (Xie et al., 2007) by using the goat anti-firefly luciferase antibody (Abcam), horseradish peroxidase-conjugated avidin biotin complex (ABC) from the Vectastain Elite ABC Kit (Vector Laboratories, Inc., Burlingame, CA) and AEC chromogen (Vector Laboratories). The sections were counterstained with hematoxylin, and mounted.
In vivo Apoptosis Assays
To identify tissue undergoing apoptosis after BikDD expression, an in situ TUNEL assay (BioVision, Mountain View, CA) was performed according to the manufacturer’s instructions. The percentage of apoptotic cells were analyzed by random selecting 4 fields.
Acute Toxicity Analysis
Balb/c mice were used for acute toxic effects induced by high dose systemic administration of DNA (50 µg):HLDC complex. Mice were anethetized and blood collected by retro-orbital bleeding using a heparinized microcapillary tube. Levels of serum AST, ALT, and BUN were measured by using assay kits (Roche).
Statistical Analysis
Student’s t test was used to compare the different expression or cytotoxicity effect in cancer and normal group. All statistical tests were two sided. Survival curves were obtained by the Kaplan-Meier method. The difference of survival time between two groups was analyzed with the log-rank test. The significance level was set at p < 0.05.
Supplementary Material
Mitosis regulator, Cdc2 and proliferation rate were not related to the efficacy of transgene expression from the survivin promoter. (A) Western analysis of phospho-Cdc2 (Thr161) (from Cell signaling) and Cdc2 (from Santa Cruz) in human lung cancer cell lines. A tubulin control is included to verify equal protein loading in the lanes (left panel). The correlation of relative survivin promoter activity and relative p-Cdc2 protein expression was analyzed and shown in the right panel. (B) The proliferation rate of human lung cancer cell lines was detected by using thiazolyl blue tetrazolium bromide (MTT) assay. The relative fold of proliferation rate was calculated by setting the value in H928 as 1 (left panel). The correlation of relative surviving promoter activity and relative fold of proliferation rate was analyzed and shown in the right panel.
{kind=link}
Expression of BikDD driven by Survivin-VISA vector inhibits tumor growth more strongly than that driven by CMV promoter in mouse TC1 lung cancer syngeneic models. Mouse lung cancer cells, TC1 with indicated plasmids treatment were inoculated subcutaneously into 6 week old C57BL/6 mice. Tumor volume was monitored after inoculation of tumor cells. ** indicated p value < 0.01.
{kind=link}
Acknowledgments
This work was supported by Grants from NRPGM in DOH97-TD-G-111-041 and MDACC SPORE grants in pancreatic (P20 CA101936) and breast (1P50 CA116199) cancer (to M-C Hung) and DOH97-TD-111-TM003 (to L-Y. Li). YP Sher was also supported by a postdoctoral fellowship award from the National Health Research Institutes, Taiwan (PD9602).
References
- Altieri DC. Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene. 2003a;22:8581–8589. doi: 10.1038/sj.onc.1207113. [DOI] [PubMed] [Google Scholar]
- Altieri DC. Validating survivin as a cancer therapeutic target. Nat Rev Cancer. 2003b;3:46–54. doi: 10.1038/nrc968. [DOI] [PubMed] [Google Scholar]
- Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917–921. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- Chen JS, Liu JC, Shen L, Rau KM, Kuo HP, Li YM, et al. Cancer-specific activation of the survivin promoter and its potential use in gene therapy. Cancer Gene Ther. 2004;11:740–747. doi: 10.1038/sj.cgt.7700752. [DOI] [PubMed] [Google Scholar]
- Day CP, Rau KM, Qiu L, Liu CW, Kuo HP, Xie X, et al. Mutant Bik expression mediated by the enhanced minimal topoisomerase IIalpha promoter selectively suppressed breast tumors in an animal model. Cancer Gene Ther. 2006;13:706–719. doi: 10.1038/sj.cgt.7700945. [DOI] [PubMed] [Google Scholar]
- Hay RJ, Reid YA, McClintock PR, Chen TR, Macy ML. Cell line banks and their role in cancer research. J Cell Biochem Suppl. 1996;24:107–130. doi: 10.1002/jcb.240630507. [DOI] [PubMed] [Google Scholar]
- Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem. 2002;277:3247–3257. doi: 10.1074/jbc.M106643200. [DOI] [PubMed] [Google Scholar]
- Fitzsimons HL, Bland RJ, During MJ. Promoters and regulatory elements that improve adeno-associated virus transgene expression in the brain. Methods. 2002;28:227–236. doi: 10.1016/s1046-2023(02)00227-x. [DOI] [PubMed] [Google Scholar]
- Fukazawa T, Maeda Y, Durbin ML, Nakai T, Matsuoka J, Tanaka H, et al. Pulmonary adenocarcinoma-targeted gene therapy by a cancer- and tissue-specific promoter system. Mol Cancer Ther. 2007;6:244–252. doi: 10.1158/1535-7163.MCT-06-0408. [DOI] [PubMed] [Google Scholar]
- Gunther M, Wagner E, Ogris M. Specific targets in tumor tissue for the delivery of therapeutic genes. Curr Med Chem Anticancer Agents. 2005;5:157–171. doi: 10.2174/1568011053174855. [DOI] [PubMed] [Google Scholar]
- Huang L, Hung M-C, Wagner E. Non-Viral Vectors for Gene Therapy. 2nd Edition Part I & Part II. edn. Elsevier Academic Press; 2005. [Google Scholar]
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- Li F, Ling X. Survivin study: an update of "what is the next wave"? J Cell Physiol. 2006;208:476–486. doi: 10.1002/jcp.20634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YM, Wen Y, Zhou BP, Kuo HP, Ding Q, Hung MC. Enhancement of Bik antitumor effect by Bik mutants. Cancer Res. 2003;63:7630–7633. [PubMed] [Google Scholar]
- McLemore TL, Eggleston JC, Shoemaker RH, Abbott BJ, Bohlman ME, Liu MC, et al. Comparison of intrapulmonary, percutaneous intrathoracic, and subcutaneous models for the propagation of human pulmonary and nonpulmonary cancer cell lines in athymic nude mice. Cancer Res. 1988;48:2880–2886. [PubMed] [Google Scholar]
- Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol. 2001;2:21–32. doi: 10.1038/35048096. [DOI] [PubMed] [Google Scholar]
- O'Connor DS, Schechner JS, Adida C, Mesri M, Rothermel AL, Li F, et al. Control of apoptosis during angiogenesis by survivin expression in endothelial cells. Am J Pathol. 2000;156:393–398. doi: 10.1016/S0002-9440(10)64742-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshikiri T, Miyamoto M, Hiraoka K, Shichinohe T, Kawarada Y, Kato K, et al. Transcriptional targeting of adenovirus vectors with the squamous cell carcinoma-specific antigen-2 promoter for selective apoptosis induction in lung cancer. Cancer Gene Ther. 2006;13:856–863. doi: 10.1038/sj.cgt.7700953. [DOI] [PubMed] [Google Scholar]
- Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J, et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 2004;64:9027–9034. doi: 10.1158/0008-5472.CAN-04-3703. [DOI] [PubMed] [Google Scholar]
- Retzer-Lidl M, Schmid RM, Schneider G. Inhibition of CDK4 impairs proliferation of pancreatic cancer cells and sensitizes towards TRAIL-induced apoptosis via downregulation of survivin. Int J Cancer. 2007;121:66–75. doi: 10.1002/ijc.22619. [DOI] [PubMed] [Google Scholar]
- Russ V, Wagner E. Cell and tissue targeting of nucleic acids for cancer gene therapy. Pharm Res. 2007;24:1047–1057. doi: 10.1007/s11095-006-9233-9. [DOI] [PubMed] [Google Scholar]
- Templeton NS, Lasic DD, Frederik PM, Strey HH, Roberts DD, Pavlakis GN. Improved DNA: liposome complexes for increased systemic delivery and gene expression. Nat Biotechnol. 1997;15:647–652. doi: 10.1038/nbt0797-647. [DOI] [PubMed] [Google Scholar]
- Tsai MF, Wang CC, Chang GC, Chen CY, Chen HY, Cheng CL, et al. A new tumor suppressor DnaJ-like heat shock protein, HLJ1, and survival of patients with non-small-cell lung carcinoma. J Natl Cancer Inst. 2006;98:825–838. doi: 10.1093/jnci/djj229. [DOI] [PubMed] [Google Scholar]
- Xie X, Xia W, Li Z, Kuo HP, Liu Y, Ding Q, et al. Targeted expression of BikDD eradicates pancreatic tumors in noninvasive imaging models. Cancer Cell. 2007;12:52–65. doi: 10.1016/j.ccr.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Zaffaroni N, Pennati M, Daidone MG. Survivin as a target for new anticancer interventions. J Cell Mol Med. 2005;9:360–372. doi: 10.1111/j.1582-4934.2005.tb00361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mitosis regulator, Cdc2 and proliferation rate were not related to the efficacy of transgene expression from the survivin promoter. (A) Western analysis of phospho-Cdc2 (Thr161) (from Cell signaling) and Cdc2 (from Santa Cruz) in human lung cancer cell lines. A tubulin control is included to verify equal protein loading in the lanes (left panel). The correlation of relative survivin promoter activity and relative p-Cdc2 protein expression was analyzed and shown in the right panel. (B) The proliferation rate of human lung cancer cell lines was detected by using thiazolyl blue tetrazolium bromide (MTT) assay. The relative fold of proliferation rate was calculated by setting the value in H928 as 1 (left panel). The correlation of relative surviving promoter activity and relative fold of proliferation rate was analyzed and shown in the right panel.
Expression of BikDD driven by Survivin-VISA vector inhibits tumor growth more strongly than that driven by CMV promoter in mouse TC1 lung cancer syngeneic models. Mouse lung cancer cells, TC1 with indicated plasmids treatment were inoculated subcutaneously into 6 week old C57BL/6 mice. Tumor volume was monitored after inoculation of tumor cells. ** indicated p value < 0.01.