

Introduction
Isolated growth hormone (GH) deficiency (IGHD) resulting in short stature has an estimated birth incidence of 1 out of 4000-10000 (Millar et al., 2003) and is usually sporadic, but monogenic forms are known. Involvement of a number of autosomal genes has been demonstrated, including the GH-1 (Wainrajch et al., 1996; Hess et al., 2007), GH releasing hormone receptor (Wainrajch et al., 1996), and HESX1 (Thomas et al., 2001) genes. Further molecular heterogeneity is suggested, as the inherited basis of many familial cases remains unclear (Dattani, 2005). X-linked combined pituitary hormone deficiency has been demonstrated previously to show linkage to two loci. One locus at Xq21.3q22 is associated with agammaglobulinemia (Conley et al., 1991), whereas another is associated with the learning disability and spina bifida and results from duplication of Xq26.1-q27.3 (Solomon et al., 2002). SOX3 lies in this duplicated segment, and is an SRY-related high mobility group box transcription factor expressed in the developing pituitary (Collignon et al., 1996). In a single large family, Laumonnier et al., (2002) identified an in-frame 33bp duplication, encoding 11 additional alanines, in the polyalanine tract in SOX3 as the cause for X-linked learning disability associated with GH deficiency. Woods et al., (2005) subsequently demonstrated that a seven alanine repeat duplication in SOX3 could cause congenital hypopituitarism.
Participants and methods
Participants
A large family with several males affected by IGHD was referred to genetics. Their clinical presentation is described below and the pedigree is illustrated in Figure 1a.
Fig. 1.
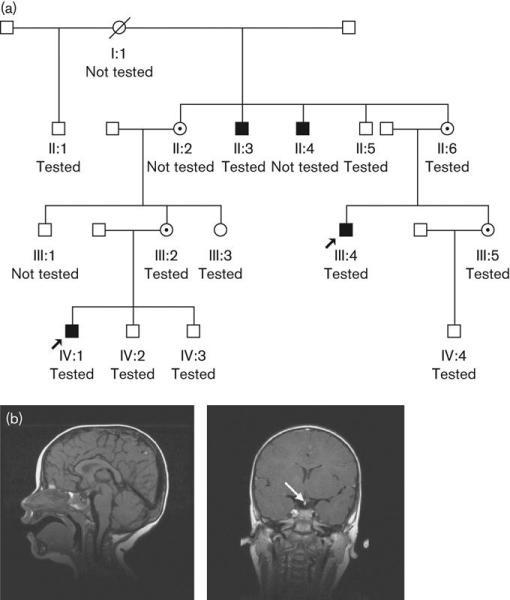
(a) Family pedigree. (b) Sagittal and coronal views of cranial magnetic resonance imaging scan showing ectopic posterior pituitary (arrowed).
Genetic testing
With informed consent, DNA was prepared from the two probands, indicated with arrows in Figure 1a, and 10 other family members. The entire open reading frame of SOX3 was sequenced in affected male III:4, with analysis on an ABI 3730 analyser (Applied Biosystems, Foster City, California, USA). A polymerase chain reaction based assay was then designed to allow sizing of the alleles by polyacrylamide gel electrophoresis in this affected male and all other family members. This assay was then used to assess whether the expansion was present in 50 control females of normal stature.
Results
Clinical presentation
Both II:2 and II:6 are sisters whose adult heights are around 155cm [standard deviation score (SDS) -1.2]. Of their three full brothers, one, II:5, has normal stature, and two short stature, II:3 (152cm, SDS -3.4) and II:4 (156cm, SDS -2.8). Their half brother, II:1, also has normal stature (180cm). All have normal intelligence. The affected brothers II:3 and II:4 had not had imaging investigations and were no longer under follow-up. No distinctive facial characteristics were apparent in either affected males or intervening females.
III:4 presented at 9 years with short stature (height SDS -2.8). Thyroid function was normal and bone age was 7.3 years. Peak GH levels during insulin tolerance and arginine stimulation tests were <2mU/l and 4.5mU/l, respectively. Growth velocity over 1.2 years was 3.9cm/year (<3rd centile). Human GH treatment was started at 9.2 years, and by 11 years his height SDS had increased to -1.7. He had delayed puberty, and was prescribed Sustanon at 14 years, which continued until 18 years. His final height was 165.2cm at 18.5 years (height SDS -1.4). Retesting at 18 years confirmed GH deficiency, with no evidence of adrenocorticotropic hormone or thyroid-stimulating hormone deficiency. Testicular volumes were lower than average, but early morning testosterone was at the lower end of the normal range. Magnetic resonance imaging demonstrated an ectopic posterior pituitary (EPP), (Fig. 1b). He remains on GH treatment as an adult, is intellectually normal and married. He attended college and runs his own computer company.
III:2, who has normal stature, has a son, IV:1. His birth weight was 4.16kg (SDS +1.3) and length 56cm (SDS +2.9). His growth was normal until about 5-6 months, but by 12 months, length was 72.5cm (SDS -1.4). Investigations done because of the family history revealed no bone age delay, but a peak GH level during an arginine stimulation test was 16.2mU/l. Insulin-like growth factor-I was very low at 29μg/l (normal 88-474μg/l). A further GH stimulation test with glucagon showed poor GH reserve (peak 5.1mU/l), whereas peak GH during sleep was 8.7mU/l. Computed tomography scanning demonstrated pituitary hypoplasia, whereas magnetic resonance imaging showed a small anterior pituitary gland, EPP and absent distal pituitary stalk (Fig. 1b), classic appearances associated with both IGHD and hypopituitarism. GH replacement therapy was started at 22 months and his growth has normalised. At the age of 4.6 years, his height was 114.5cm (SDS +2) and weight 21kg (SDS +1.4). His developmental milestones were normal, and at the age of 6 years he was doing well in school.
Genetic testing
A 21bp in-frame insertion, resulting in the insertion of seven alanine residues within the normal polyalanine tract at amino acids 234-249, was demonstrated in III:4 (Fig. 2a). By polyacrylamide gel electrophoresis, this expanded allele was also confirmed in two other affected males, II:3 and IV:1 (Fig. 2b). Intervening females II:6 and III:2 were shown to be heterozygous for the mutation, confirming their obligate carrier status, and it was absent from II:1, II:5, IV:2 (shown), and from III:3, IV:3 and IV:4 (not shown). The mutation therefore cosegregates fully with the GH deficiency phenotype in the family, with a calculated logarithm of the odds ratio score of 2.68. The mutation was not present in 100 normal control female chromosomes. One further female carrier, III:5, was identified in this family. Her asymptomatic son IV:4 was subsequently able to be discharged from endocrinological follow-up, as he was found to have inherited her normal allele.
Fig. 2.
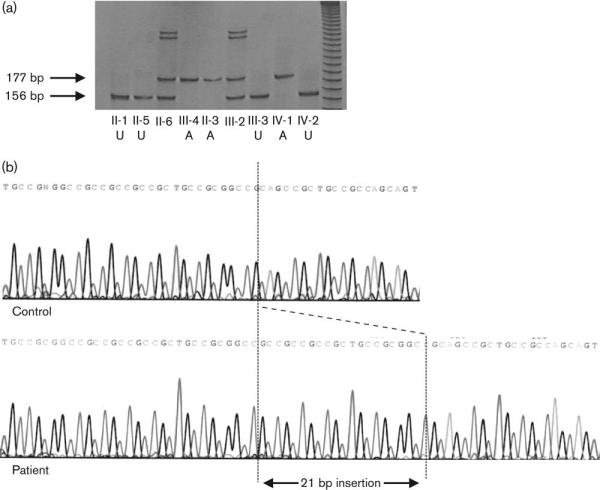
(a) Polyacrylamide gel electrophoresis demonstrating expanded allele in affected males: the 156bp polymerase chain reaction product represents wild type and the 177bp product the expanded allele. A, affected; U, unaffected.
Discussion
IGHD can be a difficult diagnosis to confirm on biochemical tests alone. Anterior pituitary hypoplasia, an absent stalk and EPP are useful findings to support the diagnosis of IGHD, and suggesting that the deficiency is likely to be permanent (Tillmann et al., 2000). The findings in the family described here demonstrate that mutation in SOX3 may be one cause of IGHD in association with such appearances.
The larger polyalanine expansion described by Laumonnier et al., (2002) (11 residues) is associated with not only GH deficiency but also learning disability. The seven residue expansion described here, and also by Woods et al., (2005) in association with hypopituitarism, is not. This demonstration of a range of phenotypes associated with the different sized expansions mirrors observations of similar mutations in transcription factor genes underlying a number of autosomal Mendelian disorders, including synpolydactyly (HOXD13 - MIM 186000) (Muragaki et al., 1996), hand-foot genital syndrome (HOXA13 - MIM 140000) (Utsch et al., 2002) and blepharophimosis, ptosis, epicanthus inversus syndrome (FOXL2 - MIM 110100) (Fokstuen et al., 2003).
The growth failure exhibited by affected males in this family was relatively modest compared with the classical GH deficiency. In addition, the contrast between this family’s presentation of isolated GH deficiency, and the combined pituitary hormone deficiency described by Woods et al., (2005) demonstrates the interfamilial phenotypic variability that may be seen, even with identically sized expansions. As IGHD is twice as common in males as females, a SOX3 mutation screen in selected affected males may be useful. The presence of pituitary structural abnormality, in particular EPP, may be a useful discriminator, as this was present in both affected individuals scanned in this family. Investigation of further males with IGHD, in particular those with an X-linked family history, could provide additional information regarding mutation prevalence. Early diagnosis of males in families like the one described here allows for optimal management, whereas normal testing results can obviate the need for follow up of previously at-risk males. Identification of carrier females has genetic counselling implications.
Acknowledgements
The authors are grateful to Professor S Shalet, The Christie Hospital, Manchester, and Dr S Salisbury, (now retired) from IWK Health Centre, Halifax, Nova Scotia, for their provision of updated clinical details, and Dr D Clarke for some sequencing data. Graeme C. Black is a Senior Research Fellow in Clinical Science funded by the Wellcome Trust. Genetic research within Central Manchester is supported by the NIHR through the Manchester Biomedical Research Centre. No competing interests are declared.
Footnotes
List of main features
Short stature, proportionate.
Pituitary, general abnormalities.
Isolated growth hormone deficiency.
Inheritance: X-linked recessive.
References
- Collignon J, Sockanathan S, Hacker A, Cohen-Tannoudji M, Norris D, Rastan S, et al. A comparison of the properties of Sox-3 with Sry and two related genes, Sox-1 and Sox-2. Development. 1996;122:509–520. doi: 10.1242/dev.122.2.509. [DOI] [PubMed] [Google Scholar]
- Conley ME, Burks AW, Herrod HG, Puck JM. Molecular analysis of X-linked agammaglobulinemia with growth hormone deficiency. J Pediatr. 1991;119:392–397. doi: 10.1016/s0022-3476(05)82051-7. [DOI] [PubMed] [Google Scholar]
- Dattani MT. Growth hormone deficiency and combined pituitary hormone deficiency: does the genotype matter? Clin Endocrinol. 2005;63:121–130. doi: 10.1111/j.1365-2265.2005.02289.x. [DOI] [PubMed] [Google Scholar]
- Fokstuen S, Antonarakis SE, Blouin JL. FOXL2-mutations in blepharophimosis-ptosis-epicanthus inversus syndrome (BPES); challenges for genetic counseling in female patients. Am J Med Genet. 2003;117A:143–146. doi: 10.1002/ajmg.a.10024. [DOI] [PubMed] [Google Scholar]
- Hess O, Hujeirat Y, Wainrajch MP, Allon-Shalev S, Zadik Z, Lavi I, Tenenbaum-Rakover Y. Variable phenotypes in familial isolated growth hormone deficiency caused by a G6664A mutation in the GH-1 gene. J Clin Endocrinol Metab. 2007;92:4387–4393. doi: 10.1210/jc.2007-0684. [DOI] [PubMed] [Google Scholar]
- Laumonnier F, Ronce N, Hamel BC, Thomas P, Lespinasse J, Raynaud M, et al. Transcription factor SOX3 is involved in X-linked mental retardation with growth hormone deficiency. Am J Hum Genet. 2002;71:1450–1455. doi: 10.1086/344661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar DS, Lewis MD, Horan M, Newsway V, Easter TE, Gregory JW, et al. Novel mutations of the growth hormone 1 (GH1) gene disclosed by modulation of the clinical selection criteria for individuals with short stature. Hum Mutat. 2003;21:424–440. doi: 10.1002/humu.10168. [DOI] [PubMed] [Google Scholar]
- Muragaki Y, Mundlos S, Upton J, Olsen BR. Altered growth and branching patterns in synpolydactyly caused by mutations in HOXD13. Science. 1996;272:548–551. doi: 10.1126/science.272.5261.548. [DOI] [PubMed] [Google Scholar]
- Solomon NM, Nouri S, Warne GL, Lagerstrom-Fermer M, Forrest SM, Thomas PQ. Increased gene dosage at Xq26-q27 is associated with X-linked hypopituitarism. Genomics. 2002;79:553–559. doi: 10.1006/geno.2002.6741. [DOI] [PubMed] [Google Scholar]
- Thomas PQ, Dattani MT, Brickman JM, McNay D, Warne G, Zacharin M, et al. Heterozygous HESX1 mutations associated with isolated congenital pituitary hypoplasia and septo-optic dysplasia. Hum Mol Genet. 2001;10:39–45. doi: 10.1093/hmg/10.1.39. [DOI] [PubMed] [Google Scholar]
- Tillmann V, Tang VWM, Price DA, Hughes DG, Wright NB, Clayton PE. Magnetic resonance imaging of the hypothalamic-pituitary axis in the diagnosis of growth hormone deficiency. J Paediatr Endocrinol Metab. 2000;13:1577–1583. doi: 10.1515/jpem.2000.13.9.1577. [DOI] [PubMed] [Google Scholar]
- Utsch B, Becker K, Brock D, Lentze MJ, Bidlingmaier F, Ludwig M. A novel stable polyalanine poly(A). expansion in the HOXA13 gene associated with hand-foot-genital syndrome: proper function of poly(A)-harbouring transcription factors depends on a critical repeat length? Hum Genet. 2002;110:488–494. doi: 10.1007/s00439-002-0712-8. [DOI] [PubMed] [Google Scholar]
- Wainrajch MP, Gertner JM, Harbison MD, Chua SC, Jr, Leibel RL. Nonsense mutation in the human growth hormone-releasing hormone receptor causes growth failure analogous to the little (lit) mouse. Nat Genet. 1996;12:88–90. doi: 10.1038/ng0196-88. [DOI] [PubMed] [Google Scholar]
- Woods KS, Cundall M, Turton J, Rizotti K, Mehta A, Palmer R, et al. Over- and underdosage of SOX3 is associated with infundibular hypoplasia and hypopituitarism. Am J Hum Genet. 2005;76:833–849. doi: 10.1086/430134. [DOI] [PMC free article] [PubMed] [Google Scholar]