

Abstract
Human diabetes mellitus comprises two main clinical entities: type 1 and type 2 diabetes. While type 1 diabetes is autoimmune in origin, type 2 diabetes is due to a decreased sensitivity to insulin action (so called insulin resistance) associated to impaired beta cell function. However, it is becoming increasingly clear that there is a certain overlap between these two diseases. While some degree of insulin resistance is present in type 1 patients, markers of beta cell autoimmunity (either primary or secondary) can frequently be detected in type 2 subjects. In this scenario, anti-CD38 autoantibodies (aAbs) have been described in both type 1 and type 2 diabetic patients. Contrary to the other known islet aAbs, anti-CD38 are more prevalent in long-standing than in new onset type 1 diabetes, and more prevalent in type 2 than in type 1 disease. Moreover, anti-CD38 aAbs are endowed with unique stimulatory properties on Ca2+ mobilization and insulin secretion. These observations suggest that autoimmunity may be both cause and consequence of beta cell dysfunction, in either case imposing a further toll for the control of glucose homeostasis.
Keywords: Antigens, CD38; immunology; Autoantibodies; immunology; Diabetes Mellitus, Type 1; immunology; Diabetes Mellitus, Type 2; immunology; Humans; Insulin Resistance; Islets of Langerhans; immunology; Models, Immunological
Keywords: beta cell autoimmunity, insulin resistance, type 1 diabetes, type 2 diabetes
Diabetes mellitus is defined as a group of metabolic diseases characterized by hyperglycemia, resulting from defects in insulin secretion, insulin action, or both (1). In the big groove of diabetes classification, two archetypical clinical entities are the most relevant:
- type 1 diabetes, resulting from a T cell-mediated autoimmune destruction of pancreatic beta cells (2). It follows that the primary mechanism responsible for hyperglycemia is defective insulin secretion from loss of beta cell mass. While it commonly occurs in childhood and adolescence, type 1 diabetes can however appear at any age (1). Part of the difference in the age of onset is due to different rates of beta cell destruction. Indeed, an early age of onset is frequently associated with more aggressive beta cell autoimmunity.
- type 2 diabetes, resulting from a decreased tissue sensitivity to insulin action (so called insulin resistance). This is however usually associated with a relative (rather than absolute) insulin deficiency, given by the fact that beta cells try to compensate for the decreased insulin action by increasing insulin secretion, and ultimately fail to do so (3).
The view that the distinction between these two pathogenic mechanisms of beta cell autoimmunity and insulin resistance may not be as rigid is however gaining credit (4,5). We will summarize current evidence for this blurring of the type 1 – type 2 diabetes boundaries. With respect to beta cell autoimmunity, we have described new islet autoantibodies (aAbs) directed against the novel CD38 islet autoantigen, which where found to be present in both type 1 and type 2 diabetic patients. While the presence of these serum aAbs may be a sign of beta cell stress, it indicates that a component of beta cell autoimmunity, either as a primary cause or as a secondary consequence of islet destruction, is present in many diabetic patients irrespective of the primary pathogenic mechanism.
Insulin resistance in type 1 diabetes
Insulin resistance before and after onset of type 1 diabetes
The loss of first-phase insulin secretion (FPIR) constitutes a powerful predictor of development of diabetes among islet aAb+ relatives. However, there are individual differences in the age of onset of clinical disease, which likely reflect different rates of disease progression (6–8). These differences may at least in part be explained by different degrees of insulin sensitivity present in different subjects (9).
This hypothesis has recently been substantiated by L. Harrison and coworkers, who followed for 4 years the insulin secretion (measured as FPIR) and insulin sensitivity (as estimated by homeostasis model assessment of insulin resistance, HOMA-R) of islet aAb+ first-degree relatives of type 1 diabetes patients, comparing subjects who progressed with those who did not progress to diabetes. The progressors had significantly higher insulin resistance relative to their level of insulin secretion already at baseline, years before the onset of clinical symptoms (10).
The variable contribution of insulin sensitivity may be at play not only throughout the years between initiation of autoimmunity and metabolic decompensation. It could also be critical in the weeks preceding the first diagnosis of type 1 diabetes, where precipitating factors associated with insulin resistance are frequently present. In the face of a damaged beta cell and thus reduced insulin secretion, these changes are sufficient to precipitate hyperglycemia. Insulin resistance also plays an important role during the subsequent course of disease. In the first year of clinical disease, considerable evidence suggests that the occurrence of clinical remission referred to as “honeymoon phase” may result not only from improved secretion due to resolution of the acute islet inflammation, but also from a temporary resolution of the insulin-resistant state present at diagnosis (11–13). The fact that the contribution of improved insulin secretion to the honeymoon phenomenon is only partial was documented by a study in which beta cell function, as measured by stimulated C-peptide responses, did not change as patients entered clinical remission (11). Conversely, the dominant contribution of improved insulin sensitivity was shown by Yki-Jarvinen et al. (12,13), who performed direct measurements of insulin sensitivity by euglycemic–hyperinsulinemic clamp on type 1 patients 2 weeks, 3 months, and 1 year after diagnosis. These subjects demonstrated a decrease in insulin sensitivity at 2 weeks after diagnosis, as compared with healthy controls; however, by 3 months, the insulin sensitivity of the diabetic group was no longer different than controls. Importantly, this improvement in insulin sensitivity was concomitant with the honeymoon phase, and subjects in clinical remission showed higher glucose uptakes than those not in remission. Other studies documented that the reverse is also true, i.e. loss of clinical remission was associated with a deterioration of insulin sensitivity (14).
Pathogenesis of insulin resistance in type 1 diabetes
The factors at play in the insulin resistant state of type 1 diabetes are a matter of debate. The insulin resistance present at diagnosis could be a result of the acute metabolic decompensation, but its persistence beyond resolution of ketoacidosis suggests a more complex pathogenesis. Counter-regulatory hormones, or a direct effect of impaired glucose metabolism (so called “glucose toxicity”) (15) could play an additional role. Moreover, the importance of the genetic background of the patients in terms of insulin resistance genes is underlined by epidemiologic observations suggesting that the offspring of type 2 diabetic parents is more likely to develop not only type 2, but also type 1 diabetes (16–19).
An alternative interpretation on the pathogenesis of insulin resistance in type 1 diabetes is that an insulin-resistant state may be part of a more aggressive form of autoimmunity, characterized by a more marked inflammatory environment. Pro-inflammatory cytokines such as IL-1, IL-6 and TNF-alpha induce the hepatic production of acute phase reactants (20,21), as well as leptin production by the adipose tissue (22,23). These modifications favor the onset of an insulin-resistant state. Insulin resistance is also directly induced by TNF-alpha, which is a potent inhibitor of the tyrosine kinase activity of the insulin receptor. Despite the fact that C-reactive protein levels were repeatedly found not elevated before, at and after type 1 diabetes onset (24–26), several markers of inflammation have been found to be increased in more advanced disease and associated with micro- and macrovascular complications (27,28).
Is insulin resistance responsible for the changing epidemiology of type 1 diabetes?
In line with this increased attention to insulin resistance in type 1 diabetes, there is also epidemiological evidence suggesting that the characteristics of the disease may be under change. The group of E. Gale in Bristol recently compared the frequency of HLA Class II haplotypes of type 1 diabetic patients diagnosed between 1985 and 2000 with that of patients diagnosed more than 50 years earlier. The proportion of high-risk susceptibility genotypes was much higher in the earlier cohort, suggesting that the weight of environmental factors in the more recent pathogenesis of type 1 diabetes is increasing (29). A previous Finnish study has also reported a higher concentration of high risk haplotypes in individuals who developed type 1 diabetes between 1939 and 1965 than in individuals who contracted the disease between 1990 and 2001. Moreover, this analysis showed a heightened proportion of individuals with the protective haplotype DR15-DQB2*0602 in the recently diagnosed population (30). The main suspected environmental factor is the change in lifestyle with regards to diet and physical exercise, leading to higher levels of insulin resistance. Such precipitating factors may unmask type 1 diabetes in individuals carrying low risk haplotypes, who were before not as prone to disease.
Beta cell autoimmunity in type 2 diabetes
The role of inflammation
The partial pathogenic overlap between autoimmune type 1 and insulin-resistant type 2 diabetes is not only limited to the component of insulin resistance recognized in type 1 diabetes. Conversely, an autoimmune, likely secondary, component is increasingly recognized in many forms of “so called” type 2 diabetes (31). Beta cell dysfunction has traditionally been thought to arise from the prolonged, increased secretory demand placed on the beta cells by insulin resistance (3) (Fig. 1), as well as by the deleterious effects of hyperglycemia (so called “glucose toxicity” or “glucose desensitization”) (15).
Fig. 1.
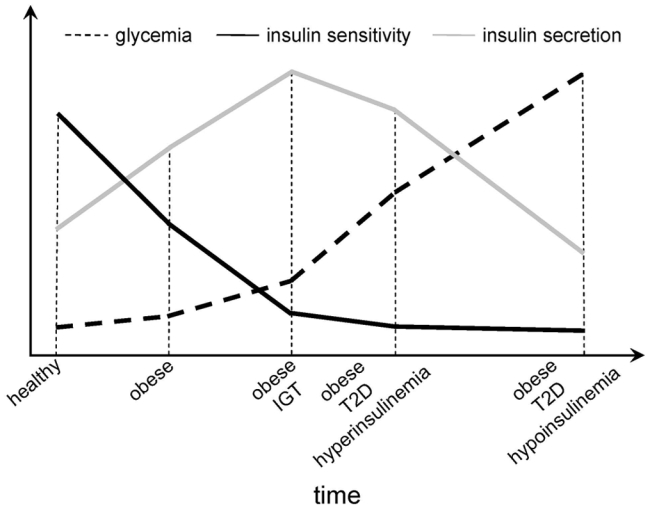
The natural evolution of insulin sensitivity and insulin secretion in different metabolic situations and with progression to type 2 diabetes (T2D).
However, the possibility that an autoimmune component of beta cell damage might be present has long been overlooked. In a provocative genetic study, L. Groop and collaborators confirmed previous observations (16–18) that type 1 and type 2 diabetes cluster in the same families. More importantly, a type 1 diabetes genetic background, in terms of DR3-DR4 HLA Class II risk haplotypes, predisposed type 2 relatives not only to an Ab+ status, but also to impaired insulin secretion, irrespective of Ab status (19).
A concept of type 2 diabetes as “disease of the innate immune system” (20) is also gaining momentum (32). This low-grade inflammation present in type 2 diabetes, which is also shared by atherosclerotic processes, is generally considered to be a non-specific consequence of metabolic stress. This type of ‘inflammatory’ response would not require the infiltration of ‘inflamed’ tissues by (auto)antigen-reactive immune cells. Although it is conceivable that inflammation is fueled by non-antigen-specific reactions, the possibility exists that a chronic inflammatory state may be driven by autoantigens. In support of this hypothesis, Pietropaolo et al. have reported that Ab+ type 2 diabetic patients aged more than 65 years have higher levels of fibrinogen and C-reactive protein as compared to Ab− patients and healthy controls of the same age (33). Theorethically, the autoantigenic drive for this chronic inflammatory state could be linked to several mechanisms. For example, autoantigens normally sequestered inside the cells and therefore not visible to the immune system could be released following beta cell apoptosis (34). Alternatively, the overwhelmed beta cells could start expressing immunogenic “stress” proteins not normally displayed, inducing an autoimmune response against these novel antigens (Ags).
Latent autoimmune diabetes of the adults (LADA)
A second turning point in our view of type 2 diabetes came from the repeated observation that ~10–15% of patients with clinical type 2 diabetes are positive for one or more islet aAbs. These patients can be categorized as having a slowly evolving form of type 1 diabetes, later christened LADA (Latent Autoimmune Diabetes of the Adults) (35,36). In the UK Prospective Diabetes Study (UKPDS), islet cell antibodies (ICA) and anti-GAD aAbs were measured at diagnosis in 3,672 white patients with type 2 diabetes, aged between 25 and 65 years. The proportion of patients with ICA and/or anti-GAD decreased with increasing age at diagnosis, from 21–34% of patients aged 25–34 to 4–7% of patients aged 55–65. Among patients younger than 35 at diagnosis, those with ICA or anti-GAD had lower body-mass index (BMI) and higher levels of HbA1c (7). Moreover, these patients typically had lower residual C-peptide levels and beta cell function (37). These immune and metabolic features are more reminiscent of type 1 diabetes and underlie a more aggressive clinical phenotype. Indeed, 94% of patients with ICA and 84% of those with anti-GAD required insulin therapy by 6 years, compared with 14% of those without the aAbs. These features were less marked but yet appreciable in older patients (7). Also in LADA patients, the interplay of insulin resistance and beta cell autoimmunity is likely critical to define whether these silent defects will become clinically evident.
Anti-CD38 autoantibodies
As beta cell autoimmunity is increasingly recognized in type 2 diabetes, the recent description of anti-CD38 aAbs in both type 1 and type 2 disease is of great interest. Several lines of evidence suggest the the humoral response against CD38 may follow rather than accompany beta cell destruction. Despite this later appearance, and contrary to the other known islet aAbs, anti-CD38 aAbs may also exert some pathogenetic effects.
The CD38 target antigen
Human CD38 (ADP-ribosyl cyclase/cADPR hydrolase) is a surface molecule widely expressed by several blood cells and tissues (Tab. 1). While its original description has been in peripheral blood B and T lymphocytes (38,39), CD38 is also expressed in virtually all blood cell populations (40–42), in lymphocytes subsets of selected districts (43–45), and in solid tissues, including muscle cells (46) and pancreatic islets (47).
The structure of human CD38 is depicted in Fig. 2. It is a 45 kD cell surface receptor with a very short intracytoplasmic tail, a transmembrane domain and a long extracellular portion which is highly glycosylated and which also contains the catalytic site of its enzymatic activities. The isoform expressed in pancreatic islets shows a slightly higher molecular weight (Fig. 3), possibly due to an islet-specific glycosylation pattern (R. Mallone, unpublished observations). Another low molecular weight isoform generated by alternative splicing has been described (48). Moreover, high molecular weight forms are generated by post-translational modification (49), with generation of multimers by action of tissue transglutaminase (50). This variety of molecular isoforms may give reason of how tolerance towards an Ag so widely expressed may be lost. Moreover, post-translational modifications can dramatically alter Ag presentation, and the role of tissue transglutaminase in these phenomena is well exemplified by celiac disease (51).
Fig. 2.

Structure of human CD38.
Fig. 3.

Western blot analysis of CD38 on isolated human islet cells, as compared to CD38 expressed on thymocytes, the Jurkat T cell line and human endothelial cells (HEC, negative control).
CD38 is endowed with a double function of receptor and ectoenzyme (52–54). As a receptor, it is able to transduce signals following interaction with natural ligand(s), including the endothelial surface molecule CD31 (55). The interaction with these natural ligands can be mimicked in vitro by some anti-CD38 monoclonal antibodies (mAbs) endowed with agonistic properties, leading to Ca2+ mobilization, phosphorylation of several cytoplasmic substrates within different transduction pathways (40), secretion of inflammatory cytokines (56).
As an ectoenzyme, CD38 displays two coupled catalytic activity. The first one, ADP-ribosyl cyclase, converts nicotinamide adenine dinucleotide (NAD+) to cyclic ADP-ribose (cADPR), a potent Ca2+ mobilizer which acts independently of the inositol triphosphate pathway (57–59). The second catalytic activity, cADPR hydrolase, converts the cADPR previously generated in the inactive metabolite ADPR.
Model of CD38-mediated insulin secretion
A role for CD38 in the mechanisms of insulin secretion, based on its coupled ADP-ribosyl cyclase and cADPR hydrolase activities, has been proposed by Okamoto et al. (60–63). A schematic representation of this model of CD38-mediated insulin secretion as opposed to the classical model proposed by Ashcroft et al. is depicted in Fig. 4. In the classical model, the ATP produced in the beta cell by glucose metabolism induces the closure of membrane K+ channels, with subsequent membrane depolarization. This depolarization in turn activates voltage-operated membrane Ca2+ channels, with influx of Ca2+ inside the cells. This Ca2+ current would then activate further Ca2+ mobilization from intracellular stores and subsequent exocytosis of insulin granules (64).
Fig. 4.

The Okamoto model of CD38-mediated insulin secretion. On the left, the classical model proposed by Ashcroft is depicted. See text for details.
Key to the alternative Okamoto model of insulin secretion is that the ATP produced by glucose metabolism inhibits the cADPR hydrolase activity of CD38, with subsequent accumulation of the cADPR produced by the cyclase activity (65). cADPR binds to the ryanodine receptors expressed on intracellular Ca2+ stores, leading to an increase in the cytoplasmic Ca2+ concentrations and subsequent exocytosis of insulin granules. Strong evidence for this model comes from in vivo mouse studies. CD38-transgenic mice which selectively express high CD38 levels on beta cells have insulin responses twice as high as compared to wild type animals (61). Complementary support for the Okamoto model comes from CD38 knock-out mice, which do not show any cADPR production in the beta cells following glucose stimulation. Similarly, Ca2+ release and insulin secretion are reduced to less than half the levels measured in wild type animals, and these knock-out mice have an altered glucose tolerance. All these defects are corrected when the knock-out mice are crossed with CD38-transgenic mice (62).
Anti-CD38 autoantibodies in type 2 diabetes
Anti-CD38 aAbs have been first described in a Japanese cohort of type 2 diabetic patients. In this group, 13.8% of the patients, but only 1.3% of the healthy controls, tested positive for serum anti-CD38, as determined by a Western blot assay on recombinant CD38. A lower fraction (10.1%) of a small cohort of type 1 diabetic patients also tested positive for anti-CD38 (66). These results have subsequently been confirmed by an Italian study, documenting a prevalence of 9.7% among type 2 and 13.1% among type 1 diabetic patients, using a similar Western blot assay (67). Interestingly, and contrary to the Japanese report, anti-CD38+ sera were here found to be stimulatory, rather than inhibitory, on insulin secretion. This discrepancy may be explained by the fact that the agonistic activity of anti-CD38+ sera on insulin secretion was assayed on human islets in the Italian study, and on rat islets in the Japanese one. A potential bias was therefore introduced in the latter study by selecting only anti-CD38+ sera cross-reactive with rat CD38.
To study the prevalence of anti-CD38 aAbs in different diabetic cohorts, we devised an enzymatic immunoassay on immobilized recombinant soluble CD38 produced in the yeast P. pastoris (68). This assay displayed a very good performance (4.3% intra-assay and 11.6% inter-assay variability), and proved more sensitive and as specific as the Western blot assay, with the advantage of a format more amenable to large scale studies (69). The higher sensitivity of the enzymatic immunoassay over the Western blot system may be due to the fact that the recombinant CD38 used in the latter is denatured by the electrophoresis in sodium dodecyl sulfate. Consequently, only aAbs recognizing linear epitopes can bind, while conformational epitopes are missed and the overall prevalence of anti-CD38 aAbs potentially underestimated.
Using a receiver-operator characteristic (ROC) analysis, the 97.5 centile, corresponding to 6,927 arbitrary units (AU)/ml, was chosen as the anti-CD38 enzymatic immunoassay cutoff, allowing for the best sensitivity and specificity (47). A summary of the results is shown in Fig. 5.
Fig. 5.

Prevalence and titers of anti-CD38 autoantibodies in different diabetic cohorts and corresponding control healthy groups matched for age and gender.
We first studied a cohort of type 2 diabetic subjects, finding a prevalence of anti-CD38 aAbs of 17.5% versus 0.9% in matched healthy controls (p<0.0001), thus confirming the Japanese report. The majority of anti-CD38+ sera (57.1%) displayed agonistic properties, i.e., they proved capable of triggering Ca2+ release when incubated with lymphocytic cell lines. The serum fraction responsible for this agonistic effect was an Ig one, since its removal led to a consistent decrease in the Ca2+-mobilizing activity. Moreover, this Ig fraction was specific for CD38, as preincubation of anti-CD38+ sera with recombinant soluble CD38 triggered minimal Ca2+ currents (Fig. 6) (69). The above mentioned Italian report by Pupilli et al. also documented a stimulatory effect of anti-CD38 aAbs on insulin release in cultured human islets (67), which correlated with a similar agonistic activity on Ca2+ mobilization on both human islets and T lymphocytic lines (70). Strikingly, these in vitro functional properties were paralleled by a distinctive phenotype of anti-CD38+ type 2 patients in our study. Indeed, the presence of anti-CD38 aAbs was associated with significantly higher levels of fasting plasma C-peptide and insulin, as compared with anti-CD38− counterparts, as if the aAbs were capable of exerting the same agonistic effects in vivo. A trend towards a higher BMI and a lower frequency of insulin therapy was also observed (69).
Fig. 6.

Agonistic activity of anti-CD38+ sera on Ca2+ release in Jurkat T cells. A, agonistic anti-CD38+ serum. B, the same serum after preincubation with protein G-sepharose to remove the Ig fraction. C, the same serum after preincubation with recombinant soluble CD38 to block the CD38-specific Ig fraction. Data are presented as density plots of the shift in the fluorescence of the Ca2+-sensitive dye Fluo-3 over a 8.5 min time course.
Anti-CD38 autoantibodies in LADA
The high prevalence of anti-CD38 aAbs in type 2 diabetes prompted us to investigate whether anti-CD38 could be a new marker of LADA. However, we did not find a higher prevalence of anti-CD38 aAb in a small cohort of LADA patients analyzed (14.9% versus 1.5% of matched healthy controls, p=0.005; Fig. 5) (47). This is also in line with the prevalent phenotype of anti-CD38+ type 2 patients, who displayed higher residual insulin secretion, higher BMI and less need for insulin (69), a phenotype somehow opposite to that of most LADA patients. Also among LADA patients, all the anti-CD38+ sera analyzed displayed agonistic properties on Ca2+ mobilization. Accordingly, anti-CD38 aAbs were associated with a trend towards higher fasting plasma C-peptide values. This trend was more effectively observed when only anti-CD38+ subjects displaying agonistic aAbs were considered (47).
Anti-CD38 autoantibodies in type 1 diabetes
A consistent prevalence of anti-CD38 aAbs was also found in type 1 diabetic patients with long-standing disease (11.7% versus 0.9%, p=0.0008; Fig. 5). Interestingly, this cohort was the only one where no association was found between a positive aAb status and more preserved beta cell function. This finding is not unexpected considering that the association between anti-CD38 and residual insulin secretion may reflect the stimulating effect of aAbs on beta cells. Such effect would not take place in this situation, since the target beta cells have long disappeared.
Similarly to other islet aAbs, higher titers and prevalence for anti-CD38 aAbs at diagnosis could be expected. We therefore analyzed a large cohort of new-onset type 1 diabetes patients (Fig. 5). Surprisingly, the prevalence of anti-CD38 aAbs was much lower in this group (3.8% versus 0.6% in healthy controls; p=0.04), contrary to all the other known markers of beta cell autoimmunity. Consistent with the higher residual beta cell mass still present at diagnosis as compared to long-standing type 1 diabetes, the association between anti-CD38 aAbs and more preserved insulin secretion was again evident in new-onset patients, and more so when the analysis was restricted to anti-CD38+ patients displaying agonistic aAbs (47).
This difference in prevalence between newly diagnosed and long-standing type 1 diabetes subjects suggests a late appearance of anti-CD38 aAbs. This hypothesis was recently confirmed in a retrospective longitudinal study, using serum samples from type 1 diabetes patients collected during the Childhood Diabetes in Finland (DiMe) study (71). For these patients, serum samples are available that were taken at diagnosis and at 2, 5 and 10 years after onset. Study of this longitudinal cohort (n=71) confirmed that the prevalence of anti-CD38 aAbs becomes higher with longer duration of disease. Prevalence at diagnosis was found to be 4.3% (versus 3.8% previously reported in the Italian cohort). This prevalence raised to 8.9% at 5 years and to 13.0% at 10 years. The prevalence among matched healthy controls was 0.7%. The difference between prevalence at diagnosis and at 5 and 10 years was statistically significant (p<0.05). Interestingly, the only two patients who were anti-CD38+ at diagnosis remained positive throughout the disease. For all other patients, anti-CD38 aAbs only appeared at 5 or 10 years after diagnosis (R. Mallone, M. Knip, H. Reijonen et al., unpublished data).
Hypotheses
1. A new autoimmune marker of beta cell stress?
To discuss the significance of anti-CD38 aAbs as markers of diabetes, their association with the other known islet aAbs should be considered. We performed this analysis on type 1 diabetic patients with new-onset and long-standing disease, as well as in the LADA cohort. The only marginal association with anti-CD38 was found in newly diagnosed type 1 diabetes for anti-GAD aAbs: all anti-CD38+ patients were also anti-GAD+ versus 63.9% among anti-CD38− patients (p=0.05). Moreover, anti-CD38 aAbs were not a subfraction of the islet cell antibody (ICA) immunofluorescence reactivity (47), as is the case for anti-GAD and anti-IA-2.
These results suggest that anti-CD38 aAbs are new, distinct markers of beta cell autoimmunity, with a later appearance in disease progression. This later appearance may be linked to different kinetics of Ag availability for the CD38 molecule. It is possible that CD38 may become differentially expressed, either in terms of density or of different isoforms, under conditions of beta cell stress. These stress conditions also lead to beta cell apoptosis, which could release sequestered pools of Ags, thus further triggering the autoimmune response. These mechanisms would also give reason for the higher prevalence of anti-CD38 aAbs in type 2 diabetic patients, who do not present other autoimmune features as is the case for LADA. Indeed, conditions of beta cell stress and apoptosis are invariably present in type 2 diabetes, where beta cells try to challenge the heavier toll imposed by insulin resistance. Subsequent mainteinance of anti-CD38 aAb titers in advanced disease could be triggered by extra-pancreatic expression of CD38.
An open issue is whether anti-CD38 aAbs are specific markers of diabetes or are also represented in other autoimmune conditions. Indeed, CD38 is expressed in a number of tissues which are targets of autoimmunity. In the human thyroid for example, CD38 is expressed in intraparenchymatous fibrous septa (46). Anti-CD38 aAbs have been found in 7.7% of patients with Graves’ disease and in 10.4% of patients with chronic thyroiditis (72). Similar frequencies (6–13%) are also found in Graves’ disease (73–75) and Hashimoto’s thyroiditis (73) for anti-GAD aAbs. Whether anti-CD38 aAbs have any functional effects on the thyroid remains to be established.
2. A pathogenic role for anti-CD38 autoantibodies?
It is widely accepted that aAbs do not play any pathogenic role in diabetes. Contrary to other autoimmune entities such as Graves’ disease or myasthenia gravis, beta cell autoimmunity is T-cell mediated. This concept is supported by several lines of evidence, but the most striking demonstration comes from an “experiment of nature”. A case report described a patient affected by X-linked agammaglobulinemia (XLA, also known as Bruton’s disease, a severe hereditary form of B cell deficiency) who developed type 1 diabetes despite complete absence of B cells and antibodies (76). The only pathogenic contribution of aAbs could consist in skewing Ag presentation towards certain T cell epitopes (77,78).
Anti-CD38 aAbs might constitute an exception to this rule (Fig. 7). Besides their distinctive pattern of expression in different diabetic groups, they are also characterized by unique functional properties, being capable of triggering Ca2+ release and insulin secretion in target CD38+ cells. We hypothesize that these stimulatory activities in vitro could be operating in vivo as well. Indeed, a higher residual insulin secretion was a common trait when circulating anti-CD38 aAbs were found, with the only exception of long-standing type 1 diabetes where, expectedly, no target beta cells are left for triggering. Whether this stimulating effect would be really beneficial in the long-term is questionable: triggering of insulin secretion could be initially favorable in the insulinopenic setting of early diabetes. However, this pathogenic mechanism could also be viewed as a “Graves’ disease of the beta cell”, and sustained stimulation could add a further challenge and possibly lead to faster exhaustion of the limited beta cell resources. Prolonged Ca2+ influx in the beta cell is also known to trigger several deleterious effects such as desensitisation (79) and apoptosis (80). Indeed, it has been shown on cultured human islet cells that incubation in the presence of Ca2+-mobilizing anti-CD38+ sera acutely stimulates insulin release, but subsequently leads to impairment of hormone secretion, which is accompanied by enhanced apoptosis (81).
Fig. 7.

Possible pathogenic effects of anti-CD38 autoantibodies. The parts depicted in grey are currently speculative only. See text for details.
CD38 is also expressed in a number of extrapancreatic tissues, and the pathogenetic role of anti-CD38 aAbs could thus not be limited to beta cells. Among the CD38+ tissues, peripheral blood mononuclear cells (PBMCs) deserve special consideration with regard to the prevalent agonistic properties of anti-CD38 aAbs. In PBMCs, binding of agonistic anti-CD38 mAbs induces release of proinflammatory cytokines, e.g., IL-6 (56), and similar effects could be triggered by anti-CD38 aAbs. Cytokine release could be an important contribution to the insulitis of type 1 diabetes, and would be in line with the models of type 2 diabetes as a disease of the innate immune system discussed above (20,32). Moreover, release of proinflammatory cytokines could also help promoting a state of insulin resistance. To substantiate this hypothesis, the in vitro potential of anti-CD38 aAbs to induce cytokine secretion should be addressed. Another issue is whether anti-CD38 autoantibodies can reach functionally relevant titers in the peripheral circulation or in confined microenvironments.
Another functional effect of CD38 aAbs could be to promote Ag processing and subsequent presentation of certain epitopes to T lymphocytes by several different mechanisms (82): 1) increased Ag uptake: aAbs can increase delivery of soluble Ags to Ag-presenting cells through Fc receptor-mediated binding; 2) induction of Ag downregulation: intracellular delivery and processing of the surface CD38 autoantigen, which is also expressed on Ag-presenting monocytes/macrophages and B cells, may be increased upon aAb binding and subsequent CD38 downregulation from the cell surface; 3) modification of Ag processing: high affinity Abs do not dissociate from Ag at the mildly acidic pH of the Ag-processing compartment and, therefore, the substrate for proteases becomes the Ag-Ab complex rather than the Ag alone. By these means, Ab binding can suppress the generation of some epitopes, while boosting the generation of others (83). However, the quest for CD38-specific T cells has been so far rewarded with little success, at least for the DR0401-restricted CD4+ responses investigated (R. Mallone, H. Reijonen, C.J. Greenbaum, G.T. Nepom, unpublished observations).
Conclusion
With efforts to ameliorate the etiological classification of diabetes, a partial overlap between type 1 and type 2 diabetes is increasingly recognized. The relative weight of beta cell autoimmunity versus insulin resistance determines the final outcome in terms of type 1 or type 2 diabetes, as well as in terms of age of onset (Fig. 8). The recognition of anti-CD38 aAbs as late markers of autoimmune- as well as metabolic-mediated beta cell destruction adds a further piece to this evolving puzzle. Development of sensitive radioimmunoassays for the detection of anti-CD38 aAbs and characterization of CD38-specific CD4+ and/or CD8+ T cell responses that should accompany aAb production are needed to advance our current knowledge.
Fig. 8.

Relative contributions of insulin resistance and beta cell autoimmunity to different forms of diabetes, and the corresponding prevalence of anti-CD38 autoantibodies.
Table 1.
Tissue distribution of human CD38.
Peripheral Blood
|
Lymphocyte subsets of selected districts
|
Solid tissues
|
Contributor Information
Roberto Mallone, Diabète de Type 1 : mécanismes et traitements immunologiques INSERM : U580, Université Paris Descartes - Paris V, FR.
Paolo Cavallo Perin, Department of Internal Medicine University of of Turin Medical School, IT.
References
- 1.American Diabetes Association Diagnosis and classification of diabetes mellitus. Diabetes Care. 2004;27 (Suppl 1):S5–S10. doi: 10.2337/diacare.27.2007.s5. [DOI] [PubMed] [Google Scholar]
- 2.Atkinson MA, Maclaren NK. The pathogenesis of insulin-dependent diabetes mellitus. N Engl J Med. 1994;331:1428–1436. doi: 10.1056/NEJM199411243312107. [DOI] [PubMed] [Google Scholar]
- 3.DeFronzo RA. Lilly lecture 1987. The triumvirate: beta-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes. 1988;37:667–687. doi: 10.2337/diab.37.6.667. [DOI] [PubMed] [Google Scholar]
- 4.Rosenbloom AL. Obesity, Insulin Resistance, beta-Cell Autoimmunity, and the Changing Clinical Epidemiology of Childhood Diabetes. Diabetes Care. 2003;26:2954–2956. doi: 10.2337/diacare.26.10.2954. [DOI] [PubMed] [Google Scholar]
- 5.Wilkin TJ. The accelerator hypothesis: weight gain as the missing link between Type I and Type II diabetes. Diabetologia. 2001;44:914–922. doi: 10.1007/s001250100548. [DOI] [PubMed] [Google Scholar]
- 6.Effects of age, duration and treatment of insulin-dependent diabetes mellitus on residual beta-cell function: observations during eligibility testing for the Diabetes Control and Complications Trial (DCCT) The DCCT Research Group. J Clin Endocrinol Metab. 1987;65:30–36. doi: 10.1210/jcem-65-1-30. [DOI] [PubMed] [Google Scholar]
- 7.Turner R, Stratton I, Horton V, et al. UKPDS 25: autoantibodies to islet-cell cytoplasm and glutamic acid decarboxylase for prediction of insulin requirement in type 2 diabetes. UK Prospective Diabetes Study Group. Lancet. 1997;350:1288–1293. doi: 10.1016/s0140-6736(97)03062-6. [DOI] [PubMed] [Google Scholar]
- 8.Leslie RD, Delli CM. Age-dependent influences on the origins of autoimmune diabetes: evidence and implications. Diabetes. 2004;53:3033–3040. doi: 10.2337/diabetes.53.12.3033. [DOI] [PubMed] [Google Scholar]
- 9.Greenbaum CJ. Insulin resistance in type 1 diabetes. Diabetes Metab Res Rev. 2002;18:192–200. doi: 10.1002/dmrr.291. [DOI] [PubMed] [Google Scholar]
- 10.Fourlanos S, Narendran P, Byrnes GB, et al. Insulin resistance is a risk factor for progression to type 1 diabetes. Diabetologia. 2004;47:1661–1667. doi: 10.1007/s00125-004-1507-3. [DOI] [PubMed] [Google Scholar]
- 11.Schober E, Schernthaner G, Frisch H, et al. Beta-cell function recovery is not the only factor responsible for remission in type I diabetics: evaluation of C-peptide secretion in diabetic children after first metabolic recompensation and at partial remission phase. J Endocrinol Invest. 1984;7:507–512. doi: 10.1007/BF03348458. [DOI] [PubMed] [Google Scholar]
- 12.Yki-Jarvinen H, Koivisto VA. Natural course of insulin resistance in type I diabetes. N Engl J Med. 1986;315:224–230. doi: 10.1056/NEJM198607243150404. [DOI] [PubMed] [Google Scholar]
- 13.Yki-Jarvinen H, Koivisto VA. Insulin sensitivity in newly diagnosed type 1 diabetics after ketoacidosis and after three months of insulin therapy. J Clin Endocrinol Metab. 1984;59:371–378. doi: 10.1210/jcem-59-3-371. [DOI] [PubMed] [Google Scholar]
- 14.Hramiak IM, Dupre J, Finegood DT. Determinants of clinical remission in recent-onset IDDM. Diabetes Care. 1993;16:125–132. doi: 10.2337/diacare.16.1.125. [DOI] [PubMed] [Google Scholar]
- 15.Yki-Jarvinen H. Glucose toxicity. Endocr Rev. 1992;13:415–431. doi: 10.1210/edrv-13-3-415. [DOI] [PubMed] [Google Scholar]
- 16.Gottlieb MS. Diabetes in offspring and siblings of juvenile- and maturity-onset-type diabetics. J Chronic Dis. 1980;33:331–339. doi: 10.1016/0021-9681(80)90042-9. [DOI] [PubMed] [Google Scholar]
- 17.Dahlquist G, Blom L, Tuvemo T, et al. The Swedish childhood diabetes study--results from a nine year case register and a one year case-referent study indicating that type 1 (insulin-dependent) diabetes mellitus is associated with both type 2 (non-insulin-dependent) diabetes mellitus and autoimmune disorders. Diabetologia. 1989;32:2–6. doi: 10.1007/BF00265396. [DOI] [PubMed] [Google Scholar]
- 18.Rich SS, Panter SS, Goetz FC, et al. Shared genetic susceptibility of type 1 (insulin-dependent) and type 2 (non-insulin-dependent) diabetes mellitus: contributions of HLA and haptoglobin. Diabetologia. 1991;34:350–355. doi: 10.1007/BF00405008. [DOI] [PubMed] [Google Scholar]
- 19.Li H, Lindholm E, Almgren P, et al. Possible human leukocyte antigen-mediated genetic interaction between type 1 and type 2 Diabetes. J Clin Endocrinol Metab. 2001;86:574–582. doi: 10.1210/jcem.86.2.7170. [DOI] [PubMed] [Google Scholar]
- 20.Pickup JC, Mattock MB, Chusney GD, et al. NIDDM as a disease of the innate immune system: association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia. 1997;40:1286–1292. doi: 10.1007/s001250050822. [DOI] [PubMed] [Google Scholar]
- 21.Feingold KR, Grunfeld C. Role of cytokines in inducing hyperlipidemia. Diabetes. 1992;41(Suppl 2):97–101. doi: 10.2337/diab.41.2.s97. [DOI] [PubMed] [Google Scholar]
- 22.Grunfeld C, Zhao C, Fuller J, et al. Endotoxin and cytokines induce expression of leptin, the ob gene product, in hamsters. J Clin Invest. 1996;97:2152–2157. doi: 10.1172/JCI118653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bornstein SR. Is leptin a stress related peptide? Nat Med. 1997;3:937. doi: 10.1038/nm0997-937. [DOI] [PubMed] [Google Scholar]
- 24.Bonifacio E, Mollenhauer U, Buuck D, et al. C-reactive protein concentration is not related to islet autoimmunity status in offspring of parents with type 1 diabetes. Clin Immunol. 2005;115:173–177. doi: 10.1016/j.clim.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 25.Basu S, Larsson A, Vessby J, et al. Type 1 diabetes is associated with increased cyclooxygenase- and cytokine-mediated inflammation. Diabetes Care. 2005;28:1371–1375. doi: 10.2337/diacare.28.6.1371. [DOI] [PubMed] [Google Scholar]
- 26.Waris ME, Koskinen JO, Simell O, et al. Onset of beta-cell autoimmunity is not associated with elevated concentration of C-reactive protein in children at genetic risk for Type 1 diabetes. Diabet Med. 2005;22:1123–1124. doi: 10.1111/j.1464-5491.2005.01593.x. [DOI] [PubMed] [Google Scholar]
- 27.Yngen M, Ostenson CG, Hu H, et al. Enhanced P-selectin expression and increased soluble CD40 Ligand in patients with Type 1 diabetes mellitus and microangiopathy: evidence for platelet hyperactivity and chronic inflammation. Diabetologia. 2004;47:537–540. doi: 10.1007/s00125-004-1352-4. [DOI] [PubMed] [Google Scholar]
- 28.Schram MT, Chaturvedi N, Schalkwijk CG, et al. Markers of inflammation are cross-sectionally associated with microvascular complications and cardiovascular disease in type 1 diabetes--the EURODIAB Prospective Complications Study. Diabetologia. 2005;48:370–378. doi: 10.1007/s00125-004-1628-8. [DOI] [PubMed] [Google Scholar]
- 29.Gillespie KM, Bain SC, Barnett AH, et al. The rising incidence of childhood type 1 diabetes and reduced contribution of high-risk HLA haplotypes. Lancet. 2004;364:1699–1700. doi: 10.1016/S0140-6736(04)17357-1. [DOI] [PubMed] [Google Scholar]
- 30.Hermann R, Knip M, Veijola R, et al. Temporal changes in the frequencies of HLA genotypes in patients with Type 1 diabetes--indication of an increased environmental pressure? Diabetologia. 2003;46:420–425. doi: 10.1007/s00125-003-1045-4. [DOI] [PubMed] [Google Scholar]
- 31.Kahn SE. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia. 2003;46:3–19. doi: 10.1007/s00125-002-1009-0. [DOI] [PubMed] [Google Scholar]
- 32.Kolb H, Mandrup-Poulsen T. An immune origin of type 2 diabetes? Diabetologia. 2005;48:1038–1050. doi: 10.1007/s00125-005-1764-9. [DOI] [PubMed] [Google Scholar]
- 33.Pietropaolo M, Barinas-Mitchell E, Pietropaolo SL, et al. Evidence of islet cell autoimmunity in elderly patients with type 2 diabetes. Diabetes. 2000;49:32–38. doi: 10.2337/diabetes.49.1.32. [DOI] [PubMed] [Google Scholar]
- 34.Pittoni V, Valesini G. The clearance of apoptotic cells: implications for autoimmunity. Autoimmun Rev. 2002;1:154–161. doi: 10.1016/s1568-9972(02)00032-0. [DOI] [PubMed] [Google Scholar]
- 35.Zimmet PZ, Tuomi T, Mackay IR, et al. Latent autoimmune diabetes mellitus in adults (LADA): the role of antibodies to glutamic acid decarboxylase in diagnosis and prediction of insulin dependency. Diabet Med. 1994;11:299–303. doi: 10.1111/j.1464-5491.1994.tb00275.x. [DOI] [PubMed] [Google Scholar]
- 36.Zimmet P, Turner R, McCarty D, et al. Crucial points at diagnosis. Type 2 diabetes or slow type 1 diabetes. Diabetes Care. 1999;22 (Suppl 2):B59–B64. [PubMed] [Google Scholar]
- 37.Carlsson A, Sundkvist G, Groop L, et al. Insulin and glucagon secretion in patients with slowly progressing autoimmune diabetes (LADA) J Clin Endocrinol Metab. 2000;85:76–80. doi: 10.1210/jcem.85.1.6228. [DOI] [PubMed] [Google Scholar]
- 38.Terhorst C, van AA, LeClair K, et al. Biochemical studies of the human thymocyte cell-surface antigens T6, T9 and T10. Cell. 1981;23:771–780. doi: 10.1016/0092-8674(81)90441-4. [DOI] [PubMed] [Google Scholar]
- 39.Jackson DG, Bell JI. Isolation of a cDNA encoding the human CD38 (T10) molecule, a cell surface glycoprotein with an unusual discontinuous pattern of expression during lymphocyte differentiation. J Immunol. 1990;144:2811–2815. [PubMed] [Google Scholar]
- 40.Mallone R, Funaro A, Zubiaur M, et al. Signaling through CD38 induces NK cell activation. Int Immunol. 2001;13:397–409. doi: 10.1093/intimm/13.4.397. [DOI] [PubMed] [Google Scholar]
- 41.Zocchi E, Franco L, Guida L, et al. A single protein immunologically identified as CD38 displays NAD+ glycohydrolase, ADP-ribosyl cyclase and cyclic ADP-ribose hydrolase activities at the outer surface of human erythrocytes. Biochem Biophys Res Commun. 1993;196:1459–1465. doi: 10.1006/bbrc.1993.2416. [DOI] [PubMed] [Google Scholar]
- 42.Ramaschi G, Torti M, Festetics ET, et al. Expression of cyclic ADP-ribose-synthetizing CD38 molecule on human platelet membrane. Blood. 1996;87:2308–2313. [PubMed] [Google Scholar]
- 43.Zupo S, Rugari E, Dono M, et al. CD38 signaling by agonistic monoclonal antibody prevents apoptosis of human germinal center B cells. Eur J Immunol. 1994;24:1218–1222. doi: 10.1002/eji.1830240532. [DOI] [PubMed] [Google Scholar]
- 44.Kumagai M, Coustan-Smith E, Murray DJ, et al. Ligation of CD38 suppresses human B lymphopoiesis. J Exp Med. 1995;181:1101–1110. doi: 10.1084/jem.181.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deaglio S, Mallone R, Baj G, et al. Human CD38 and its ligand CD31 define a unique lamina propria T lymphocyte signaling pathway. FASEB J. 2001;15:580–582. doi: 10.1096/fj.00-0522fje. [DOI] [PubMed] [Google Scholar]
- 46.Fernandez JE, Deaglio S, Donati D, et al. Analysis of the distribution of human CD38 and of its ligand CD31 in normal tissues. J Biol Regul Homeost Agents. 1998;12:81–91. [PubMed] [Google Scholar]
- 47.Mallone R, Ortolan E, Pinach S, et al. Anti-CD38 autoantibodies: characterisation in new-onset type I diabetes and latent autoimmune diabetes of the adult (LADA) and comparison with other islet autoantibodies. Diabetologia. 2002;45:1667–1677. doi: 10.1007/s00125-002-0940-4. [DOI] [PubMed] [Google Scholar]
- 48.Nata K, Takamura T, Karasawa T, et al. Human gene encoding CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase): organization, nucleotide sequence and alternative splicing. Gene. 1997;186:285–292. doi: 10.1016/s0378-1119(96)00723-8. [DOI] [PubMed] [Google Scholar]
- 49.Mallone R, Ferrua S, Morra M, et al. Characterization of a CD38-like 78-kilodalton soluble protein released from B cell lines derived from patients with X-linked agammaglobulinemia. J Clin Invest. 1998;101:2821–2830. doi: 10.1172/JCI1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Umar S, Malavasi F, Mehta K. Post-translational modification of CD38 protein into a high molecular weight form alters its catalytic properties. J Biol Chem. 1996;271:15922–15927. doi: 10.1074/jbc.271.27.15922. [DOI] [PubMed] [Google Scholar]
- 51.Anderton SM. Post-translational modifications of self antigens: implications for autoimmunity. Curr Opin Immunol. 2004;16:753–758. doi: 10.1016/j.coi.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 52.Malavasi F, Funaro A, Roggero S, et al. Human CD38: a glycoprotein in search of a function. Immunol Today. 1994;15:95–97. doi: 10.1016/0167-5699(94)90148-1. [DOI] [PubMed] [Google Scholar]
- 53.Ferrero E, Malavasi F. The metamorphosis of a molecule: from soluble enzyme to the leukocyte receptor CD38. J Leukoc Biol. 1999;65:151–161. doi: 10.1002/jlb.65.2.151. [DOI] [PubMed] [Google Scholar]
- 54.Deaglio S, Mallone R, Baj G, et al. CD38/CD31, a receptor/ligand system ruling adhesion and signaling in human leukocytes. Chem Immunol. 2000;75:99–120. [PubMed] [Google Scholar]
- 55.Deaglio S, Morra M, Mallone R, et al. Human CD38 (ADP-ribosyl cyclase) is a counter-receptor of CD31, an Ig superfamily member. J Immunol. 1998;160:395–402. [PubMed] [Google Scholar]
- 56.Ausiello CM, la SA, Ramoni C, et al. Secretion of IFN-gamma, IL-6, granulocyte-macrophage colony-stimulating factor and IL-10 cytokines after activation of human purified T lymphocytes upon CD38 ligation. Cell Immunol. 1996;173:192–197. doi: 10.1006/cimm.1996.0267. [DOI] [PubMed] [Google Scholar]
- 57.Howard M, Grimaldi JC, Bazan JF, et al. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science. 1993;262:1056–1059. doi: 10.1126/science.8235624. [DOI] [PubMed] [Google Scholar]
- 58.Berridge MJ. Cell signalling. A tale of two messengers. Nature. 1993;365:388–389. doi: 10.1038/365388a0. [DOI] [PubMed] [Google Scholar]
- 59.Galione A. Cyclic ADP-ribose: a new way to control calcium. Science. 1993;259:325–326. doi: 10.1126/science.8380506. [DOI] [PubMed] [Google Scholar]
- 60.Takasawa S, Nata K, Yonekura H, et al. Cyclic ADP-ribose in insulin secretion from pancreatic beta cells. Science. 1993;259:370–373. doi: 10.1126/science.8420005. [DOI] [PubMed] [Google Scholar]
- 61.Kato I, Takasawa S, Akabane A, et al. Regulatory role of CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase) in insulin secretion by glucose in pancreatic beta cells. Enhanced insulin secretion in CD38-expressing transgenic mice. J Biol Chem. 1995;270:30045–30050. doi: 10.1074/jbc.270.50.30045. [DOI] [PubMed] [Google Scholar]
- 62.Kato I, Yamamoto Y, Fujimura M, et al. CD38 disruption impairs glucose-induced increases in cyclic ADP-ribose, [Ca2+]i, and insulin secretion. J Biol Chem. 1999;274:1869–1872. doi: 10.1074/jbc.274.4.1869. [DOI] [PubMed] [Google Scholar]
- 63.Okamoto H, Takasawa S, Nata K. The CD38-cyclic ADP-ribose signalling system in insulin secretion: molecular basis and clinical implications. Diabetologia. 1997;40:1485–1491. doi: 10.1007/s001250050854. [DOI] [PubMed] [Google Scholar]
- 64.Ashcroft FM, Harrison DE, Ashcroft SJ. Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature. 1984;312:446–448. doi: 10.1038/312446a0. [DOI] [PubMed] [Google Scholar]
- 65.Takasawa S, Tohgo A, Noguchi N, et al. Synthesis and hydrolysis of cyclic ADP-ribose by human leukocyte antigen CD38 and inhibition of the hydrolysis by ATP. J Biol Chem. 1993;268:26052–26054. [PubMed] [Google Scholar]
- 66.Ikehata F, Satoh J, Nata K, et al. Autoantibodies against CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase) that impair glucose-induced insulin secretion in noninsulin- dependent diabetes patients. J Clin Invest. 1998;102:395–401. doi: 10.1172/JCI1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pupilli C, Giannini S, Marchetti P, et al. Autoantibodies to CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase) in Caucasian patients with diabetes: effects on insulin release from human islets. Diabetes. 1999;48:2309–2315. doi: 10.2337/diabetes.48.12.2309. [DOI] [PubMed] [Google Scholar]
- 68.Munshi CB, Fryxell KB, Lee HC, et al. Large-scale production of human CD38 in yeast by fermentation. Methods Enzymol. 1997;280:318–330. doi: 10.1016/s0076-6879(97)80123-1. [DOI] [PubMed] [Google Scholar]
- 69.Mallone R, Ortolan E, Baj G, et al. Autoantibody response to CD38 in Caucasian patients with type 1 and type 2 diabetes: immunological and genetic characterization. Diabetes. 2001;50:752–762. doi: 10.2337/diabetes.50.4.752. [DOI] [PubMed] [Google Scholar]
- 70.Antonelli A, Baj G, Marchetti P, et al. Human anti-CD38 autoantibodies raise intracellular calcium and stimulate insulin release in human pancreatic islets. Diabetes. 2001;50:985–991. doi: 10.2337/diabetes.50.5.985. [DOI] [PubMed] [Google Scholar]
- 71.Savola K, Sabbah E, Kulmala P, et al. Autoantibodies associated with Type I diabetes mellitus persist after diagnosis in children. Diabetologia. 1998;41:1293–1297. doi: 10.1007/s001250051067. [DOI] [PubMed] [Google Scholar]
- 72.Antonelli A, Fallahi P, Nesti C, et al. Anti-CD38 autoimmunity in patients with chronic autoimmune thyroiditis or Graves’ disease. Clin Exp Immunol. 2001;126:426–431. doi: 10.1046/j.1365-2249.2001.01683.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kawasaki E, Abiru N, Yano M, et al. Autoantibodies to glutamic acid decarboxylase in patients with autoimmune thyroid disease: relation to competitive insulin autoantibodies. J Autoimmun. 1995;8:633–643. doi: 10.1006/jaut.1995.0047. [DOI] [PubMed] [Google Scholar]
- 74.Hallengren B, Falorni A, Landin-Olsson M, et al. Islet cell and glutamic acid decarboxylase antibodies in hyperthyroid patients: at diagnosis and following treatment. J Intern Med. 1996;239:63–68. doi: 10.1046/j.1365-2796.1996.417758000.x. [DOI] [PubMed] [Google Scholar]
- 75.Maugendre D, Verite F, Guilhem I, et al. Anti-pancreatic autoimmunity and Graves’ disease: study of a cohort of 600 Caucasian patients. Eur J Endocrinol. 1997;137:503–510. doi: 10.1530/eje.0.1370503. [DOI] [PubMed] [Google Scholar]
- 76.Martin S, Wolf-Eichbaum D, Duinkerken G, et al. Development of type 1 diabetes despite severe hereditary B-lymphocyte deficiency. N Engl J Med. 2001;345:1036–1040. doi: 10.1056/NEJMoa010465. [DOI] [PubMed] [Google Scholar]
- 77.Reijonen H, Daniels TL, Lernmark A, et al. GAD65-specific autoantibodies enhance the presentation of an immunodominant T-cell epitope from GAD65. Diabetes. 2000;49:1621–1626. doi: 10.2337/diabetes.49.10.1621. [DOI] [PubMed] [Google Scholar]
- 78.Jaume JC, Parry SL, Madec AM, et al. Suppressive effect of glutamic acid decarboxylase 65-specific autoimmune B lymphocytes on processing of T cell determinants located within the antibody epitope. J Immunol. 2002;169:665–672. doi: 10.4049/jimmunol.169.2.665. [DOI] [PubMed] [Google Scholar]
- 79.Grill V, Bjorklund A. Overstimulation and beta-cell function. Diabetes. 2001;50(Suppl 1):S122–S124. doi: 10.2337/diabetes.50.2007.s122. [DOI] [PubMed] [Google Scholar]
- 80.Chandra J, Zhivotovsky B, Zaitsev S, et al. Role of apoptosis in pancreatic beta-cell death in diabetes. Diabetes. 2001;50 (Suppl 1):S44–S47. doi: 10.2337/diabetes.50.2007.s44. [DOI] [PubMed] [Google Scholar]
- 81.Marchetti P, Antonelli A, Lupi R, et al. Prolonged in vitro exposure to autoantibodies against CD38 impairs the function and survival of human pancreatic islets. Diabetes. 2002;51 (Suppl 3):S474–S477. doi: 10.2337/diabetes.51.2007.s474. [DOI] [PubMed] [Google Scholar]
- 82.Lanzavecchia A. How can cryptic epitopes trigger autoimmunity? J Exp Med. 1995;181:1945–1948. doi: 10.1084/jem.181.6.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Simitsek PD, Campbell DG, Lanzavecchia A, et al. Modulation of antigen processing by bound antibodies can boost or suppress class II major histocompatibility complex presentation of different T cell determinants. J Exp Med. 1995;181:1957–1963. doi: 10.1084/jem.181.6.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]