

Abstract
We show on Imaging Mass Spectrometry (IMS) data that the Random Forest classifier can be used for automated tissue classification and that it results in predictions with high sensitivities and positive predictive values, even when inter-sample variability is present in the data. We further demonstrate how Markov Random Fields and vector-valued median filtering can be applied to reduce noise effects to further improve the classification results in a post-hoc smoothing step. Our study gives clear evidence that digital staining by means of IMS constitutes a promising complement to chemical staining techniques.
Introduction
Currently, wet lab staining techniques are the method of choice for visualizing the spatial distribution of specific biomolecules or cell compartments. However, the high specificity of a wet lab stain is also its principal limitation: one slice of tissue can be treated with a small number of stains only (for exceptions, see [1]) and cannot be reanalyzed at will when different biomolecules take center stage. Imaging Mass Spectrometry (IMS) [2, 3], in contrast, is capable of monitoring a large number of molecules in the target range at the same time, in our case more than 4000 m/z-channels between 0 and 400 Da. IMS permits detailed analysis of spatial distributions of (bio-)molecules, including but not limited to proteins, peptides, lipids or metabolites [4, 5]. It has found wide application ranging from disease studies [6, 7, 8] to drug distribution and metabolism analysis [9, 10].
With this richness of information at hand, classification of tissue types (e.g. healthy vs. tumorous or different tumors) may become more reliable. Influential mass spectral peaks or groups of mass spectral peaks with their corresponding molecules may in turn be identified as biomarkers. The strength of the information richness is also a major burden - automated and reliable computational analysis of IMS data becomes indispensable.
Generally, current approaches for the analysis of IMS data fall in two categories. If no prior knowledge on the composition of a tissue sample is available, unsupervised methods like Principal Component Analysis (PCA) [11] or Probabilistic Latent Semantic Analysis (pLSA) [12] are employed to decompose and classify elements of a tissue sample. Recently, hierarchical clustering has been proposed [13] and the authors conclude from the good results obtained with their unsupervised method that “software based classification should be achievable”. In an increasing number of studies like ours, some prior knowledge on the spatial composition of a tissue sample is available and spatially resolved labels exist (i.e. training examples where the observed data points are annotated with labels from a given set, see Data section). In such a scenario, more powerful supervised methods like Support Vector Machines (SVM) [14], Random Forests [15] or other classifiers can be used to automatically distinguish between the classes of interest. These algorithms can constitute a valuable tool for pathologists or medical doctors that have to analyze large numbers of tissue samples. In that case, reliable classifiers can help minimize the risk to underdiagnose. In the field of Mass Spectrometry, different classifiers have been applied: k-nearest neighbors (knn) [16, 17], SVMs [18, 19, 20] and other approaches [21, 7]. Random Forests have also successfully been used [22, 23, 24, 25, 26, 27, 28], but to our knowledge not on IMS data and often only for binary classification tasks.
Random Forests have many favorable properties. Empirically, the algorithm is robust to overfitting, the method has high prediction accuracy, is capable of dealing with a large number of input variables, allows fast training (i.e. only a few seconds in the scenario described below), and performance is robust with respect to the exact choice of the two hyperparameters: number of trees, and size of the random feature subset evaluated at a node. Random Forests have successfully been applied to various kinds of spectral data, including remote sensing [29], astrophysics [30] and Magnetic Resonance Spectroscopic Imaging (MRSI) [31]. Previous studies have compared the performance of Random Forests to SVMs and other state-of-the-art methods in many fields of application and have concluded that they deliver comparable [32, 33, 34, 25] or even superior [24] performance. The fact that there is no clear benefit of using SVMs or Random Forests, but that Random Forests are “more” non-parametric, i.e. require less tuning, and that their training is fast, makes this algorithm a natural choice for our study. In the first part of the paper we demonstrate that Random Forests are a highly suitable automated approach for classifying IMS data. In spite of inter-tissue-sample variability being present in our data, the classifier results in predictions with high sensitivity and high positive predictive values.
Owing to noise or instabilities in the data acquisition process, the classification maps obtained with Random Forests (or other classifiers) can have a “noisy” appearance. Single pixels that have been classified differently than their surrounding area can frequently be observed (see Results and Discussion). Typically, the “gold standard” label maps provided by human experts tend to be much more homogeneous (see Data section). In the second part of the paper, we therefore apply a post-hoc smoothing method that removes these “outliers” from the classification maps. To this end, we compare a Markov Random Field (MRF) [35, 36] approach to a vector-valued median filter [37] and show that post-hoc smoothing significantly improves the sensitivities and positive predictive values.
In our study, we analyze human breast cancer cells grown as tumor xenografts in mice. Within these tumor xenografts, five different regions (necrotic tissue, viable tumor, gelatine, tumor interface, glass/hole) can be identified (see Data section). The data comprises a total of 7 slices from two tumors (same cell line, no genetic variation). We describe and discuss in detail how the Random Forest algorithm combined with post-hoc smoothing techniques can be used for automated IMS data classification and show that it results in predictions with high sensitivity estimates and positive predictive values of about 90%.
Materials and Methods
Random Forest
The Random Forest classifier [15] is a decision tree based ensemble method. In contrast to single decision trees, a randomized tree ensemble is robust to overfitting [15]. In a typical training setup, a few hundred decision trees are constructed which in their entirety constitute the “forest”. The single trees are generated by the following algorithm:
Let S = {(x1,y1),…,(xN,yN)} be the set of available M-dimensional training samples, i.e. mass spectra xi with M channels and corresponding class labels yi, e.g. cancerous or healthy tissue 1. First, a bootstrap sample (in-bag sample) of size N is chosen by randomly sampling N times from S with replacement. This training set is used for building the tree, whereas the rest of the samples (out-of-bag sample) is used for estimating the out-of-bag error. Construction starts from the root that contains all training samples. At each node, a subset of size M̃; of the M features is chosen at random and that feature in the subset which allows for the best separation of the classes in the sample set in that node is determined. The node is subsequently split into two child nodes and the process is repeated until the tree is fully grown, i.e. all leaf nodes contain samples from one class only (see figure 1). Finally, the respective labels are assigned to the leaf nodes. Note that the trees are not pruned [38]. For each tree, the training error is estimated by predicting the classes of the out-of-bag samples and comparing the results with the true class memberships yi.
Figure 1.
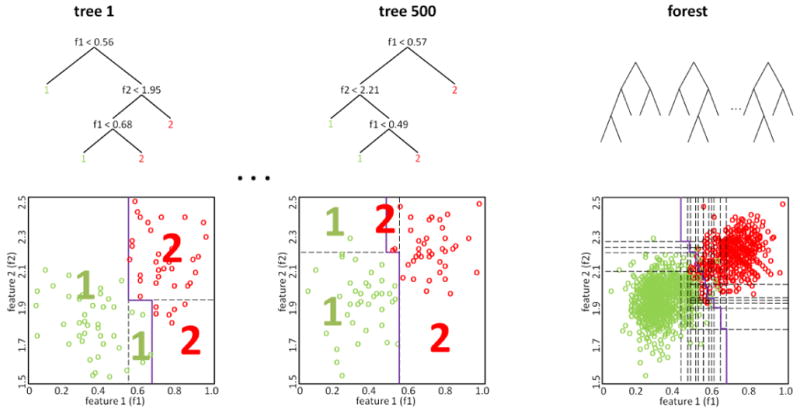
The Random Forest classifier is an ensemble of decision trees where the single trees are constructed from bootstrap samples. On the left and in the center, two trees of the forest are shown in detail: At each node, the feature which allows for the best class separation is chosen (with respect to the subset of features selected for that node). The corresponding partitioning of the feature space is shown below with the decision boundary plotted in purple. On the right, the decision boundary of the Random Forest is displayed. It is based on the majority votes of the individual trees.
The importance of the different features (in our case mass channels) for the classification can be estimated with the permutation accuracy criterion [15]. In short, for each tree the prediction accuracy on the out-of-bag sample is calculated in a first step. Then this process is repeated M̃ times where in each run the values of the l-th feature are randomly permuted (l = 1, …,M̃). For each feature the difference between the two accuracies (non-permuted and permuted) is averaged over all trees. This mean decrease in accuracy is used as feature importance measure. In addition to the overall feature importance, a class-wise version can be calculated. Intuitively, a feature is considered unimportant if permuting its values does not (or only marginally) affect prediction accuracy.
After training of the forest, a sample (in our case spectrum) is classified by putting it down on each of the trees in the ensemble. Each tree constitutes a crisp classifier and returns the label corresponding to the leaf node in which the sample ends up. The Random Forest averages over all trees and the classification result is represented by a probability vector that reflects how many trees have voted for one specific class. This output can be interpreted as posterior probability that an object belongs to a particular class, given the features of that object. In short, the output of a Random Forest can be seen as an estimate for class probabilities. A crisp classification can be obtained by taking the mode of this distribution.
Smoothing
In order to remove salt and pepper noise structures from the classification maps (i.e. spatial maps, or images, of the predicted class probabilities), we apply a post-hoc smoothing by means of Markov Random Fields (MRF) and a vector-valued median algorithm.
Markov Random Fields
In the MRF, each pixel of the classification map is represented by a node with 4-connectivity (see figure 2). In brief, the MRF employed is defined as follows (for an extensive introduction, see [39]). Each node takes a value in the set of labels (here L = {necrotic tissue, viable tumor, gelatine, tumor inter: see Data section). Motivated by a local homogeneity assumption and the available label maps, a regularized solution is sought which is a good compromise of two factors: the single site potentials (SSP) that encourage the agreement of each label with the local classification result (data term) and the pair potentials (PP) that call for the consistency of each label with the labels of the surrounding pixels, and therefore encourage smoothness of the label map. According to this model, the optimum compromise or map of labels for all pixels, Z, is found as the maximizer of the log probability
Figure 2.
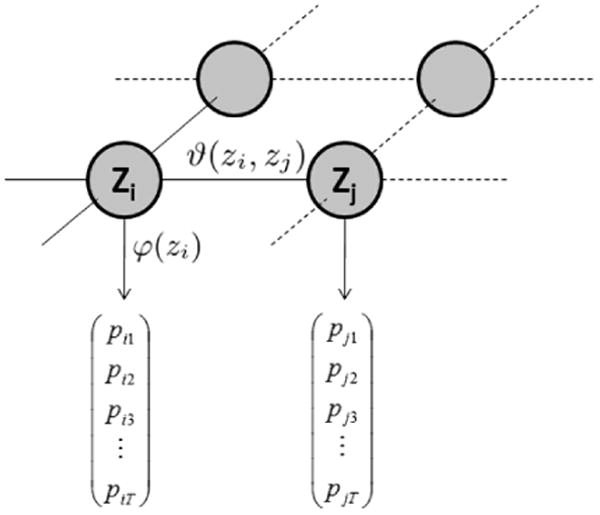
Markov Random Field (MRF) model. Each node represents a pixel in the classification map, the optimal label assignment depends on both single site potential (φ) and pair potential (ϑ). The former is defined by the Random Forest output that assigns probabilities to the T class labels whereas Potts potentials are used for the latter.
| (1) |
Here, neigh(i) identifies the set of neighboring nodes for node i and zi represents the label assigned to node i. The logarithm of φ(zi) is the single site potential for node i and the logarithm of ϑ (zi, zj) is the pair potential function for the neighboring nodes i and j weighted by the scalar λ. We use the Random Forest output as single site potential φ(zi), i.e. φ(zi) equals the probability that the sample corresponding to node i belongs to class zi. We further use Potts potentials [36] for ϑ(zi, zj), i.e. we assign a fit of 1 if i and j share the same label and 0 otherwise. To avoid numerical problems caused by zero entries in the potentials, we modify both, the SSPs and PPs by component-wise addition of a small constant ε > 0 and subsequent re-normalization.
An approximation maximizer of equation 1 can be found efficiently with Loopy Belief Propagation (BP) [40]. BP is an iterative algorithm that tries to find a maximum a-posteriori estimate of the label distribution by repeatedly passing local messages between neighboring nodes of the MRF graph. These messages build on the potentials defined above and quantify the local fit of the labels to the data and the prior assumptions. After a stopping criterion is met, so-called “beliefs” are calculated for each node that express the probability of assigning a certain label to a node. Finally, the label with maximum belief is selected for each node. Since the defined graph contains cycles, BP is not guaranteed to converge. However, it usually results in good approximations of the optimum solution [41].
Vector-valued median filter
The scalar median filter [42] is known to efficiently remove salt-and-pepper noise in gray-valued images. Welk [37] introduced a vector-valued version of the median that Lerch [43] later enhanced by weighting factors. The weighted vector-valued median μ of a set S̃ = {x̃1, …,x̃K} of K vectors in a M-dimensional feature space is given by
| (2) |
where ‖.‖2 denotes the Euclidean distance and wk the weight of the k-th spectrum in S̃. The weights wk were chosen from a Gaussian kernel centered at the respective pixels and with variance σ2= 1.5. The convex optimization problem in eq. 2 can be solved with a gradient descent approach [37]. Note that the resulting median μ is not necessarily a member of S̃.
Experiments
Data
Experimental data was acquired from orthotopic human breast cancer xenografts grown in mice from MCF-7, a weakly metastatic and estrogen-sensitive breast cancer cell line. For this study, six parallel tissue slices of the same tumor (entitled S3, S4, S5, S7, S9, S11; the S-slices) were subjected to IMS analysis resulting in a total of six data sets. Slices with odd numbers are equispaced and the distance between S3 and S5 equals ≈ 500μm. Additionally, one slice (entitled T1) from a second tumor of the same cell line, grown in a genetically identical mouse was analyzed. No genetic variation was present in the data. For each tumor slice, an additional Hematoxylin-Eosin (HE) stained parallel slice is available.
Despite some topological differences between the HE-stained and IMS-subjected slices, the stained images can be used as gold standards in the labeling process. Note, however, that they are only an approximation to a ground truth. As can be seen from the label maps in figure 3, five different regions are present in the tissue samples: necrotic tissue (overall 4844 labeled spectra), viable/active tumor (16663), embedding gelatine (6340), tumor interface region (3114) and glass/holes (10373). In the glass/holes area, the tissue was torn in the freeze-drying/microtome cutting process and the glass surface is exposed to the analytical ion beam.
Figure 3.
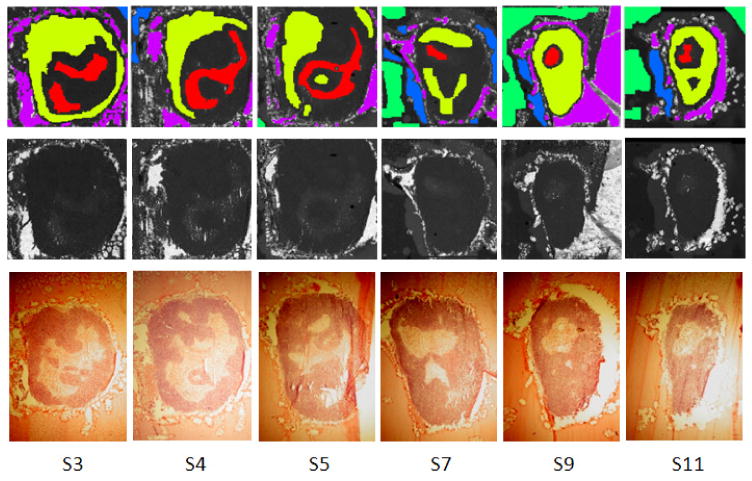
Labels for the six slices S3, S4, S5, S7, S9 and S11: the top row shows the label maps that have been obtained by closely investigating the corresponding stained parallel slices (bottom row) as well as the total ion count images (TIC, middle row). Five regions can be observed: necrotic (red), viable (yellow), gelatine (green), interface (blue) and glass/hole (violet). Black/white indicates that no label is available. Note that after normalization (see Data processing section) the structure in the TIC images is no longer visible.
For data acquisition, a Physical Electronics TRIFT II TOF SIMS equipped with an Au+ liquid metal ion cluster gun was used. The tumor samples were embedded in gelatine, flash-frozen, cryo-sectioned to ≈ 10μm and thaw-mounted on a cold indium tin oxide-coated glass slide. The tissues were not washed prior to SIMS analysis, which was confined to a mass range of 0–2000 Da. The spectral resolution was rebinned to 0.1 Da and the range between 0–400 Da was selected, resulting in 4009 mass channels. Due to the large amount of data processed in this study, short acquisition times of 2 seconds per spot were used. Consequently, the spatial resolution had to be rebinned in order to guarantee a reasonable number of ion counts in each mass spectrum. After rebinning, one pixel spans 35 × 35μm.
Research questions
In our experiments we addressed five research questions. Experiments 1 to 3 concern the performance of the Random Forest algorithm on real-world IMS data, experiment 4 analyzes the effect of post-hoc smoothing and experiment 5 deals with identifying important features for the classification:
Experiment 1: We evaluated if Random Forests are capable of distinguishing different tissue types at all; in this restricted setting, a cross validation over pixels was performed, using all slices from the first mouse.
Experiment 2: We used samples from all but one S-slice for training and evaluated the classifier's performance by predicting the classes for the (labeled) samples of the remaining S-slice (“leave one-slice-out cross validation”). This experiment shows if the classifier generalizes well to different parts of the same tumor in the same individual which were acquired in separate experiments.
Experiment 3: We trained the algorithm on the six S-slices and tested it on the T1 slice. This experiment shows if the classifier generalizes well if the same tumor is studied in different individuals. This experiment is still limited in that both the tumors and the individuals are clones, i.e. potential variability between different cell lines or individuals is not covered.
Experiment 4: The next research question analyzed the effect of smoothing of the classification results with the Markov Random Field and the vector-valued median filter defined above.
Experiment 5: Finally, we were interested which features are decisive in the classification of the tissue samples.
Evaluation criteria
For Random Forest training we used balanced training sets [44], i.e. we trained the classifier with the same number of training samples for each tissue class. To ensure that a sufficient number of training samples was available from each slice, we further balanced the data sets by training the forest with the same number of labeled samples per class and slice. Ten-fold-cross-validation over pixels was used to evaluate the classifier's performance by means of sensitivity (SE) and positive predictive value (PPV) (see experiment 1 below). For a given class k, the sensitivity (also termed “true positive rate”) measures the ratio of samples correctly classified as k to all samples that really belong to class k, i.e. where TP is the number of true positives and FN is the number of false negatives. The PPV estimates the ratio of samples correctly classified as k among all samples classified as k: where FP is the number of false positives. To ensure a fair comparison, we averaged the results over five training runs to minimize the influence of the random sampling of training points.
Data Processing
In our study, we compare different datasets, each of which corresponds to one mass spectral image. Mass spectral count data are not quantitative due to matrix and ionization effects and possible other variations. Moreover, depending on the acquisition time per spot and other instrument settings, the average intensity can vary, even if the same kind of tissue is imaged. We normalize the data sets by dividing each spectrum by its total ion count (TIC). This way, intensity differences visible in the total ion count images (cf. fig. 3) are removed. It is ensured that the classification is based on different spectral distributions and not on intensity differences. The scaled spectra are baseline-corrected by subtraction of the channel-wise minimum with respect to all spectra in a dataset. We detect the local maxima in the mean spectrum of each dataset and keep the mass channels that correspond to maxima that exceed a given threshold in order to extract features and obtain “feature lists”. To increase the robustness of the quantification, the pertaining intensity value is calculated as the integral over the whole peak width. In our experiments, peak picking on the average spectrum was more robust to noise than peak picking on individual spectra which is in line with the findings of Morris [45]. Moreover, the described procedure is faster.
Further, when training of the classifier is performed with samples from multiple images, the feature sets of the individual images have to be combined. First, the data sets have to be recalibrated to correct for potential peak shifts between sets. This is done by performing hierarchical clustering on the peak positions [46, 38] using an optimized cutoff value of 0.2 Da. After correction, the single feature sets are merged. Whenever a member of the merged feature list does not occur in the feature list of a data set, we assume the corresponding intensity value to be zero.
Experiments were conducted using an in-house implementation of the Random Forest classifier and the vector-valued median algorithm; the in-house belief propagation code is based on Murphy's Bayes Net Toolbox [47]. Iterations were stopped as soon as the maximum change in local beliefs was below a given threshold. The regularization parameter was set to λ = 0.01 to provide good results in all our experiments.
Results
Experiment 1: We randomly chose (labeled) samples from all S-slices for training and testing of the Random Forest. Ten-fold cross validation was used, i.e. nine out of ten subsets were used for training and the remaining one was used for testing. Results can be found in table 1.
Experiment 2: We trained the classifier with samples from all but one S-slice and the labeled samples of the remaining S-slice were used for testing. Tissue sample preparation for IMS analysis was done individually for each slice, potentially decreasing the classifier's performance. Sensitivity and PPV estimates were calculated with respect to the label maps and results are shown in table 2 and figure 4.
Experiment 3: Random Forest performance was tested on the T1 slice after training with samples from all six S-slices. Results are displayed in table 3 and figure 5.
Experiment 4: Results concerning the last research question are given in table 4 and figure 6. Both smoothing methods required approximately the same computation time and resulted in similar SE and PPV estimates. One advantage of the vector-valued median is that in contrast to an MRF with Potts potential as described here, it can be used to directly smooth the probability maps instead of the crisp classification maps. Thus, smoothed versions of the soft and the crisp maps can be obtained.
Experiment 5: Results concerning the feature importance are given in table 5 and figure 7.
Table 1.
Experiment 1: Training and testing has been performed with samples from all six slices and results are shown for different numbers of samples per class. We conducted two experiments with 198 and 1188 training samples per class, respectively. No post-hoc smoothing was performed. SE and PPV values were estimated by ten-fold cross validation. High SE and PPV values are obtained for 198 samples. More samples clearly increase both the sensitivity and positive predictive values.
| tissue class | |||||||
|---|---|---|---|---|---|---|---|
| samples/class | measure | necrotic | viable | gelatine | interface | glass/hole | mean |
| 198 | SE | 88.3 | 92.5 | 95.4 | 97.1 | 94.2 | 93.5 |
| PPV | 94.0 | 90.0 | 97.1 | 94.0 | 95.1 | 93.5 | |
| 1188 | SE | 91.8 | 94.5 | 97.1 | 98.5 | 94.5 | 95.3 |
| PPV | 93.0 | 92.5 | 98.8 | 95.5 | 96.8 | 95.3 | |
Table 2.
Experiment 2: Training has been performed with samples from five slices and the remaining one was used for testing. We trained the forest with 198 samples per class and slice and no post-hoc smoothing was performed. The dash indicates that no labels are available for a certain slice/class combination. Average SE and PPV values are between 80 and 95 percent. We see that a Random Forest trained with samples from five out of the six slices generalizes reasonably well and can be used to classify the test slice.
| tissue class | |||||||
|---|---|---|---|---|---|---|---|
| slice | measure | necrotic | viable | gelatine | interface | glass/hole | mean |
| S3 | SE | 92.8 | 82.7 | - | 89.7 | 89.5 | 88.7 |
| PPV | 57.6 | 96.8 | - | 41.9 | 98.6 | 73.7 | |
| S4 | SE | 64.7 | 98.3 | - | 89.9 | 97.1 | 87.4 |
| PPV | 95.6 | 85.7 | - | 98.3 | 86.3 | 91.4 | |
| S5 | SE | 94.7 | 91.3 | 86.7 | - | 98.1 | 92.7 |
| PPV | 86.7 | 96.8 | 100 | - | 99.4 | 95.8 | |
| S7 | SE | 99.4 | 75.4 | 99.1 | 99.6 | 89.1 | 92.5 |
| PPV | 30.4 | 99.7 | 99.5 | 94.8 | 99.5 | 84.8 | |
| S9 | SE | 81.0 | 96.2 | 84.0 | 96.4 | 97.2 | 90.1 |
| PPV | 54.0 | 97.3 | 99.4 | 60.0 | 97.0 | 81.5 | |
| S11 | SE | 96.4 | 90.4 | 87.4 | 99.1 | 91.0 | 92.8 |
| PPV | 40.0 | 98.7 | 99.4 | 68.3 | 99.7 | 81.2 | |
Figure 4.
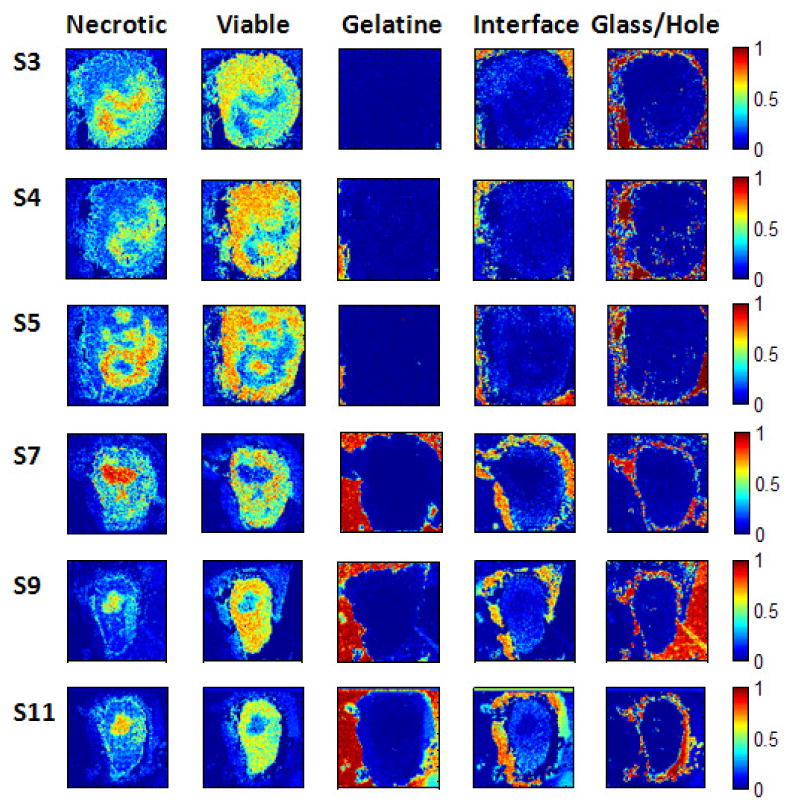
Experiment 2: Soft classification maps for the S-slices. The probability maps (128 × 128 pixels in size) show the distribution of the five different tissue classes/regions and are nicely correlated with the label maps in fig. 3. See text for details.
Table 3.
Experiment 3: Training has been performed with samples from all six slices of the first tumor (S3,S4,S5,S7,S9,S11), whereas testing was done on the T1-slice of the second tumor. We chose 198 samples per class and slice for training and no post-hoc smoothing was performed. The results indicate that the classifier is reasonably accurate even if training and testing is performed on the same tumor type in different individuals.
| tissue class | |||||||
|---|---|---|---|---|---|---|---|
| slice | measure | necrotic | viable | gelatine | interface | glass/hole | mean |
| T1 | SE | 71.9 | 91.6 | 99.9 | 90.3 | 99.6 | 90.4 |
| PPV | 94.4 | 84.0 | 97.6 | 93.4 | 93.4 | 92.5 | |
Figure 5.
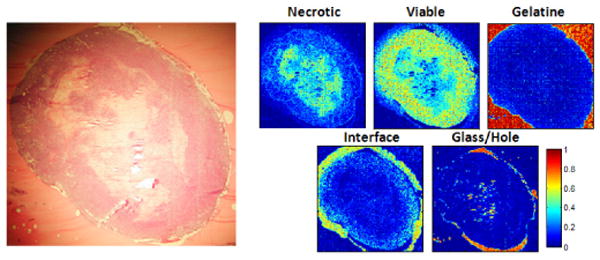
Experiment 3: Classification of the T1 set after training on the S-slices. The similarity between the (parallel) stained slice (left) and the classification maps (right) is apparent. The necrotic part in the middle is well detected by the classifier. As expected, the interface part surrounds the viable part and is itself surrounded by gelatine. The holes are also visible in the stained slice.
Table 4.
Experiment 4: The table shows the classification results obtained for slices S4 and S7 after training of the Random Forest with samples from all other S-slices. Post-hoc smoothing of the classification maps with Markov Random Fields (MRF) and vector-valued median filtering (VVM) clearly improves the classification result by more than 5% in sensitivity and positive predictive value. However, great care has to be taken when smoothing is applied, see text for details.
| tissue class | ||||||||
|---|---|---|---|---|---|---|---|---|
| slice | smoothing | measure | necrotic | viable | gelatine | interface | glass/hole | mean |
| S4 | none | SE | 64.7 | 98.3 | - | 89.9 | 97.1 | 87.4 |
| PPV | 95.6 | 85.7 | - | 98.3 | 86.3 | 91.4 | ||
| MRF | SE | 83.6 | 100 | - | 93.6 | 98.8 | 94.0 | |
| PPV | 100 | 93.4 | - | 100 | 92.5 | 96.4 | ||
| VVM | SE | 84.5 | 100 | - | 94.4 | 99.0 | 94.5 | |
| PPV | 100 | 95.1 | - | 100 | 96.6 | 97.2 | ||
| S7 | none | SE | 99.4 | 75.4 | 99.1 | 99.6 | 89.1 | 92.5 |
| PPV | 30.4 | 99.7 | 99.5 | 94.8 | 99.5 | 84.8 | ||
| MRF | SE | 100 | 92.7 | 99.7 | 99.8 | 94.8 | 97.4 | |
| PPV | 58.9 | 99.7 | 99.5 | 97.8 | 100 | 91.2 | ||
| VVM | SE | 100 | 91.3 | 100 | 100 | 95.8 | 97.4 | |
| PPV | 55.2 | 99.9 | 99.9 | 98.4 | 100 | 90.1 | ||
Figure 6.
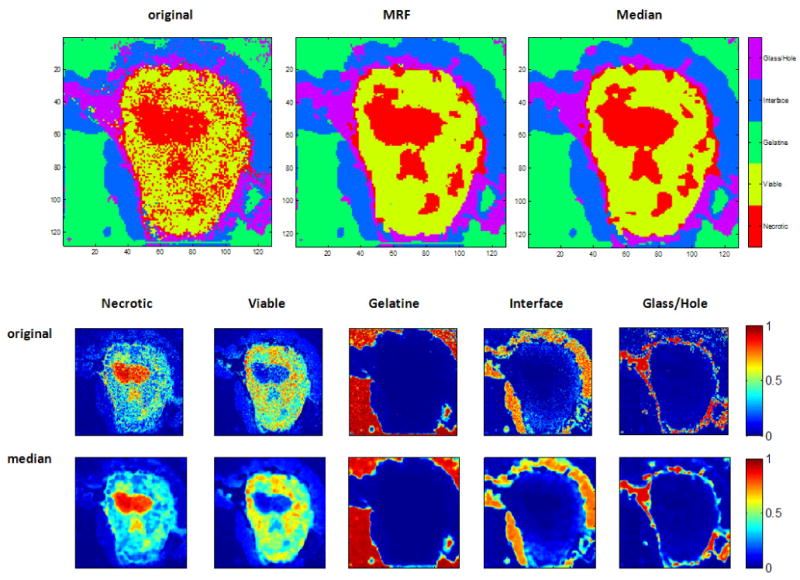
Experiment 4: classification results for slice S7 after training with all other slices. The top row shows the crisp classification results: on the left the Random Forest result is shown, in the middle the result after post-hoc smoothing with MRFs (λ = 0.01) and on the right the result after applying the vector-valued median. The classification results are very close to the respective label maps, shown in figure 3. The smoothing efficiently removes the salt-and-pepper noise. Smoothed versions of the soft classification maps can be obtained with vector-valued median-filtering and results are shown in the bottom rows.
Table 5.
Experiments 5: For each tissue class the five most important features with respect to the permutation accuracy criterion are listed by their m/z-position in Da and their corresponding importance score (given in brackets). In addition, the overall most important features and their interpretation (if available) are given (also cf. fig. 7).
| tissue class | |||||||
|---|---|---|---|---|---|---|---|
| rank | necrotic | viable | gelatine | interface | glass/hole | overall | interpretation |
| 1 | 23.3 (0.092) | 39.0 (0.052) | 70.0 (0.144) | 23.3 (0.131) | 112.9 (0.102) | 114.9 (0.058) | indium |
| 2 | 114.9 (0.080) | 23.3 (0.050) | 58.1 (0.097) | 184.0 (0.099) | 114.9 (0.087) | 23.3 (0.056) | sodium |
| 3 | 39.0 (0.049) | 114.9 (0.032) | 184.0 (0.091) | 114.9 (0.078) | 118.0 (0.070) | 70.0 (0.056) | |
| 4 | 184.0 (0.035) | 70.0 (0.031) | 44.1 (0.045) | 39.0 (0.051) | 246.8 (0.064) | 184.0 (0.050) | phosphocholine |
| 5 | 44.1 (0.032) | 58.1 (0.027) | 104.1 (0.044) | 58.1 (0.048) | 136.8 (0.042) | 58.1 (0.041) | |
Figure 7.
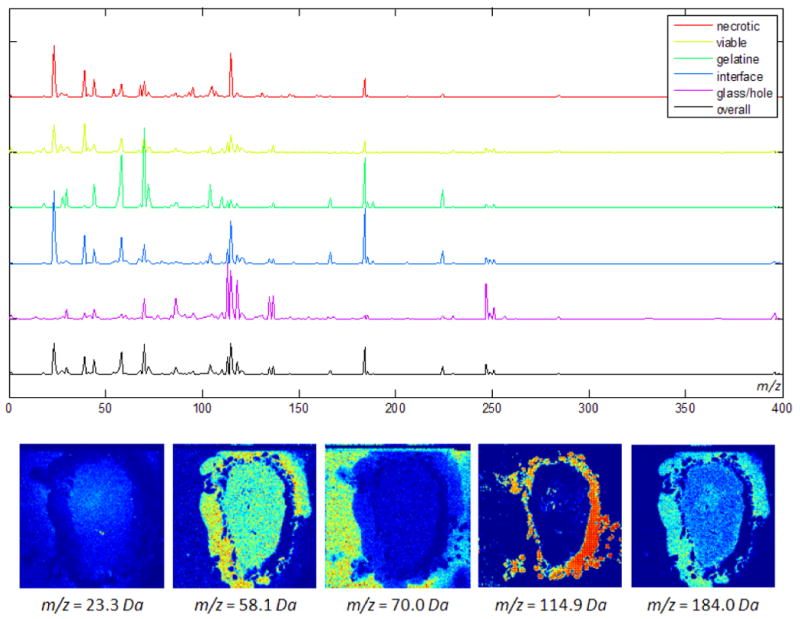
Experiment 5: The top row shows the permutation accuracy feature importance scores for the five tissue classes as well as the overall score. The five most important features are listed in table 5 and include indium (m/z = 114.9Da) and phosphocholine (m/z = 184.0Da). The corresponding channels for tissue sample S11 are plotted in the bottom row.
Discussion
Experiment 1: We first used 198 samples per class corresponding to 33 samples per class and slice, for which we obtained estimates for sensitivity and positive predictive value slightly above 90% (see table 1). With a total of 1188 samples per class (i.e. 198 per class and slice), we gained a further increase of roughly 2%. Note that especially for the gelatine and interface regions, only few labeled samples are available (see Data section and label maps in figure 3) and that these samples might have to be replicated in order to obtain a balanced training set when training is done with many samples [44]. In this scenario, the classifier is prone to overfitting to the training data. However, the results obtained after training with only 33 samples per class and slice (which is significantly below the number of available samples) and the results for classes for which hundreds of labeled points exist (see for example viable, glass/hole) are reasonably good. Most problems occurred in separating necrotic and viable tissue, probably because these classes are more similar to each other than to gelatine or glass/hole. Nevertheless, we still obtained good classification results for these tissue types.
-
Experiment 2: We note that the sensitivity and PPV rates were slightly lower than in experiment 1 where also some samples from the test slice had been used in the training. However, the sensitivity was still at about 90% and the PPV at ≈ 85%. This experiment shows that Random Forests can be robust with respect to experimental variability witnessed when different slices are considered which were experimentally prepared and processed separately (“technical repeats”).
The soft classification maps obtained with the Random Forest classifier (see fig. 4) provide further insight into the composition of a tissue sample. Crisp classification maps fail to give insight on the information regarding the ambiguity or amount of uncertainty of a prediction at a pixel. In situations where a tissue class is dominated by another tissue class, the information on the distribution of the weaker class(es) is lost in the crisp classification map. In some areas, the (crisp) class assignment is obvious, e.g. in regions where the glass slide is visible. In contrast, the class decision in the lower right part of slice S3 is less clear since both necrotic and viable tumor have high probabilities. Since the spatial transition between different stages of tumor development can be continuous, this information can be highly relevant in the treatment of the cancer. Imposing a hard threshold would suppress the subtleties of the true distribution, and a small shift of the threshold might lead to widely differing crisp classification maps which is undesirable.
Experiment 3: We trained the classifier with 198 samples per class and slice from all six S-slices. When classifying T1, we obtained reasonably accurate SE and PPV estimates close to 90% (see tab. 3), indicating that the classifier is reliable even in situations where training and testing is performed with samples from different tumors. The slightly reduced sensitivity value for the necrotic class results from the fact that the classification result for this class is speckled. It is unclear if this has biological reasons or has to be considered a misclassification. Apart from that, the visual comparison between the stained slice and the classification result in figure 5 clearly underlines the good performance of the algorithm.
-
Experiment 4: With respect to the gold standard, post-hoc smoothing with the proposed MRF and the vector-valued median algorithm significantly improved the classification results. We note however that in general, manual labeling builds on an underlying assumption of homogeneity; in addition, overly smooth labels may be obtained in situations where it is challenging for a human expert to mark every single differentiation that can be seen in a tissue sample. In our comparison, this favors the postprocessing step. The salt-and-pepper noise was efficiently removed leading to a better highlighting of the shapes of the different tissue types (see figure 6). This observation is confirmed by the sensitivity and PPV estimates displayed in table 2. The classification results in figure 6 are well correlated with the stained images and label maps in figure 3.
Removing salt-and-pepper noise is beneficial if these structures arise from artifacts of the acquisition process like low signal to noise ratios at certain spatial locations. In these cases, isolated misclassifications can be efficiently corrected by the proposed methods leading to a clearer visualization of the main structures in the data. However, if these structures do indeed have a biological significance, i.e. different content in the tissue sample, oversmoothing is a problem as it may lead to a loss of valuable information. The decision of whether smoothing should be applied depends on the research question as well as on the quality of the data. In our experiments, smoothing led to increased sensitivity and PPVs with respect to our gold standard label maps.
-
Experiment 5: The Random Forest was trained as described in experiment 3. Among the most important features (mass channels) that were identified by the permutation accuracy criterion are m/z = 114.9Da (indium) and m/z = 184.0Da (phosphocholine). The former is identified to be important for classifying samples of the glass region. This is plausible since the glass slides were indium-coated prior to IMS analysis (see Data section) and indium is dominant in these areas. The latter has previously been found to play an important role in the discrimination of necrotic and viable tissue and the interface region [12] as well as in the metabolism of breast cancer cells in general [48, 49]. Note that a high score for a given feature and class combination does not necessarily imply that the respective mass channel shows high intensities for that class. A particularly high (or low) score rather indicates that the respective feature is important for the classification of samples belonging to that class (i.e. not belonging to other classes). As a consequence, some markers occur in the top lists of multiple tissue types. As can be seen from the channel images in figure 7 the mass channels with high importance scores show informative spatial distributions which are nicely correlated with the different areas in the label maps (cf. fig. 3).
Note however that these results should only be considered as first indicators for potential biomarkers. Besides standardized tissue sample preparation and stable acquisition conditions, a robust detection of biomarkers also requires a more diverse data set that comprises genetic variability. We plan to analyze such data in future work.
The methodology described in this paper can be applied to IMS data of different resolution and quality. Given sufficiently high resolved data and an adequate number of training examples, the Random Forest classifier could also be used on the cell level. This would enable classification, i.e. digital staining, of single cells, providing even further insight into the composition of tissue samples.
Conclusion
We have introduced post-processed Random Forests for the classification of IMS data. Experiments on an animal model of human breast cancer grown in mice suggest that this classifier is well suited for automated annotation of IMS data. With the proposed methodology, we were able to separate necrotic tissue, viable tumor, gelatine, tumor interface and glass/hole areas under the following experimental conditions: High sensitivity rates (≈ 90%) and positive predictive values (≈ 85%) have been obtained when training and testing was performed with samples from different slices of a single tumor. Similar performance was observed when samples from two different tumors of the same cell line were used for training and testing. Further experiments are required in which we will evaluate the presented methods on data featuring genetic variation.
The soft classification output of the Random Forest classifier can give further, valuable insight in the composition of tissue samples and the permutation accuracy criterion yields discriminative features for the classification. We have demonstrated that spatially smoothing the crisp and soft classification maps with Markov Random Fields and vector-valued median filtering significantly improves the classification result, increasing sensitivity by approximately 3% in the examples shown.
Our study gives clear evidence that digital staining may be a powerful complement to chemical staining techniques.
Acknowledgments
We gratefully acknowledge financial support by the DFG under grant no. HA4364/2-1 (M.H, B.Y.R.), the Robert Bosch GmbH (F.A.H.), and the National Institutes of Health of the USA under grant no. NIH R01 CA134695 (K.G., R.M.A.H.). The financial support of the Netherlands BSIK program Virtual Laboratory for e-science is gratefully acknowledged by R.M.A.H. and E.R.A. The experimental data were acquired as part of the research program of the “Stichting voor Fundamenteel Onderzoek der Materie” (FOM), which is financially supported by the “Nederlandse organisatie voor Wetenschappelijk Onderzoek” (NWO). We furthermore like to thank Rahul Nair (University of Heidelberg) for implementing the permutation accuracy criterion, Ivo Klinkert and Gert Eijkel (both FOM-AMOLF, Amsterdam) for providing data conversion tools as well as Tiffany R. Greenwood (Johns Hopkins University School of Medicine, Baltimore) for cutting the tissue sections. Finally, we like to thank our referees and the editor for their constructive criticism.
Footnotes
note that whenever we refer to a physical sample we use the term tissue sample
References
- 1.Uhlen M, Björling E, Agaton C, Szigyarto CA, Amini B, Andersen E, Andersson AC, Angelidou P, Asplund A, Asplund C, Berglund L, Bergström K, Brumer H, Cerjan D, Ekström M, Elobeid A, Eriksson C, Fagerberg L, Falk R, Fall J, Forsberg M, Björklund MG, Gumbel K, Halimi A, Hallin I, Hamsten C, Hansson M, Hedhammar M, Hercules G, Kampf C, Larsson K, Lindskog M, Lodewyckx W, Lund J, Lundeberg J, Magnusson K, Malm E, Nilsson P, Odling J, Oksvold P, Olsson I, Oster E, Ottosson J, Paavilainen L, Persson A, Rimini R, Rockberg J, Runeson M, Sivertsson A, Sköllermo A, Steen J, Stenvall M, Sterky F, Strömberg S, Sundberg M, Tegel H, Tourle S, Wahlund E, Waldn A, Wernrus H, Wan J, Westberg J, Wester K, Wrethagen U, Xu LL, Hober S, Pontn F. A human protein atlas for normal and cancer tissues. Mol Cell Proteomics. 2005;4(12):1920–32. doi: 10.1074/mcp.M500279-MCP200. [DOI] [PubMed] [Google Scholar]
- 2.Caprioli RM, Farmer TB, Gile J. Molecular imaging of biological samples: Localization of peptides and proteins using MALDI-TOF MS. Anal Chem. 1997;69(23):4751–4760. doi: 10.1021/ac970888i. [DOI] [PubMed] [Google Scholar]
- 3.McDonnell LA, Heeren RMA. Imaging mass spectrometry. Mass Spectrometry Reviews. 2006;26:606–643. doi: 10.1002/mas.20124. [DOI] [PubMed] [Google Scholar]
- 4.Seeley EH, Caprioli RM. Molecular imaging of proteins in tissues by mass spectrometry. PNAS. 2008;105(47):18126–18131. doi: 10.1073/pnas.0801374105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chaurand P, Schwartz SA, Caprioli RM. Imaging mass spectrometry: a new tool to investigate the spatial organization of peptides and proteins in mammalian tissue sections. Current Opinion in Chemical Biology. 2002;6(5):676–681. doi: 10.1016/s1367-5931(02)00370-8. [DOI] [PubMed] [Google Scholar]
- 6.Simpkins F, Czechowicz JA, Liotta L, Kohn EC. SELDI-TOF mass spectrometry for cancer biomarker discovery and serum proteomic diagnostics. Pharmacogenomics. 2005;6(6):647–653. doi: 10.2217/14622416.6.6.647. [DOI] [PubMed] [Google Scholar]
- 7.Yanagisawa K, Shyr Y, Xu B, Massion P, Larsen P, White B, Roberts J, Edgerton M, Gonzalez A, Nadaf S, Moore JH, Caprioli RM, Carbone DP. Proteomic patterns of tumour subsets in non-small-cell lung cancer. The Lancet. 2003;362(9382):433–439. doi: 10.1016/S0140-6736(03)14068-8. [DOI] [PubMed] [Google Scholar]
- 8.Rohner TC, Staab D, Stoeckli M. MALDI mass spectrometric imaging of biological tissue sections. Mechanisms of Ageing and Development. 2005;126(1):177–185. doi: 10.1016/j.mad.2004.09.032. [DOI] [PubMed] [Google Scholar]
- 9.Belu AM, Davies MC, Newton JM, Patel N. TOF-SIMS characterization and imaging of controlled-release drug delivery systems. Anal Chem. 2000;72(22):5625–5638. doi: 10.1021/ac000450+. [DOI] [PubMed] [Google Scholar]
- 10.Cornett DS, Frappier SL, Caprioli RM. MALDI-FTICR imaging mass spectrometry of drugs and metabolites in tissue. Anal Chem. 2008;80(14):5648–5653. doi: 10.1021/ac800617s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Broersen A, van Liere R, Heeren RMA. Comparing three pca-based methods for the 3d visualization of imaging spectroscopy data. Proc of the 5th IASTED Intern Conf on Visualization, Imaging, and Image Processing, pages. 2005:540–545. [Google Scholar]
- 12.Hanselmann M, Kirchner M, Renard BY, Amstalden ER, Glunde K, Heeren RMA, Hamprecht FA. Concise representation of mass spectrometry images by probabilistic latent semantic analysis. Analytical Chemistry. 2008;80(24):9649–9658. doi: 10.1021/ac801303x. [DOI] [PubMed] [Google Scholar]
- 13.Deininger SO, Ebert MP, Fütterer A, Gerhard M, Röcken C. MALDI imaging combined with hierarchical clustering as a new tool for the interpretation of complex human cancers. Journal of Proteome Research. 2008;7(12):5230–5236. doi: 10.1021/pr8005777. [DOI] [PubMed] [Google Scholar]
- 14.Schölkopf B, Smola A. Learning with Kernels: Support Vector Machines, Regularization, Optimization, and Beyond. MIT Press; 2002. [Google Scholar]
- 15.Breiman L. Random forests. Machine Learning. 2001;45:5–32. [Google Scholar]
- 16.Sanders ME, Dias EC, Xu BJ, Mobley JA, Billheimer D, Roder H, Grigorieva J, Dowsett M, Arteaga CL, Caprioli RM. Differentiating proteomic biomarkers in breast cancer by laser capture microdissection and MALDI MS. Journal of Proteome Research. 2008;7(4):1500–1507. doi: 10.1021/pr7008109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taguchi F, Solomon B, Gregorc V, Roder H, Gray R, Kasahara K, Nishio M, Brahmer J, Spreafico A, Ludovini V, Massion PP, Dziadziuszko R, Schiller J, Grigorieva J, Tsypin M, Hunsucker SW, Caprioli RM, Hirsch RM, Duncan FR, Bunn PA, Carbone DP. Mass spectrometry to classify non-small-cell lung cancer patients for clinical outcome after treatment with epidermal growth factor receptor tyrosine kinase inhibitors: A multicohort cross-institutional study. Journal of the National Cancer Institute. 2007;99(11):838–846. doi: 10.1093/jnci/djk195. [DOI] [PubMed] [Google Scholar]
- 18.Schwamborn K, Krieg RC, Reska M, Jakse G, Knuechel R, Wellmann A. Identifying prostate carcinoma by MALDI-imaging. International Journal of Molecular Medicine. 2007;20:155–159. [PubMed] [Google Scholar]
- 19.Gerhard M, Deininger SO, Schleif FM. Statistical classification and visualization of MALDI-imaging data. Symp on Computer-Based Medical Systems. 2007:403–405. 20-22. [Google Scholar]
- 20.Groseclose MR, Massion PP, Chaurand P, Caprioli RM. High-throughput proteomic analysis of formalin-fixed paraffin-embedded tissue microarrays using MALDI imaging mass spectrometry. Proteomics. 2008;8:3715–3724. doi: 10.1002/pmic.200800495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwartz SA, Weil RJ, Thompson RC, Shyr Y, Moore JH, Toms SA, Johnson MD, Caprioli RM. Proteomic-based prognosis of brain tumor patients using direct-tissue matrix-assisted laser desorption ionization mass spectrometry. Cancer Research. 2005;65(17):7674. doi: 10.1158/0008-5472.CAN-04-3016. [DOI] [PubMed] [Google Scholar]
- 22.Barrett JH, Cairns DA. Application of the random forest classification method to peaks detected from mass spectrometric proteomic profiles of cancer patients and controls. Statistical Applications in Genetics and Molecular Biology. 2008;7(2) doi: 10.2202/1544-6115.1349. article 4. [DOI] [PubMed] [Google Scholar]
- 23.Ge G, Wong GW. Classification of premalignant pancreatic cancer mass-spectrometry data using decision tree ensembles. BMC Bioinformatics. 2008;9:275–287. doi: 10.1186/1471-2105-9-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ulintz PJ, Zhu J, Qin ZS, Andrews PC. Improved classification of mass spectrometry database search results using newer machine learning approaches. Molecular and Cellular Proteomics. 2006;5:497–509. doi: 10.1074/mcp.M500233-MCP200. [DOI] [PubMed] [Google Scholar]
- 25.Datta S, DePadilla LM. Feature selection and machine learning with mass spectrometry data for distinguishing cancer and non-cancer samples. Statistical Methodology. 2006;3(1):79–92. [Google Scholar]
- 26.Hand DJ. Breast cancer diagnosis from proteomic mass spectrometry data: A comparative evaluation. Statistical Applications in Genetics and Molecular Biology. 2008;7(2) doi: 10.2202/1544-6115.1435. article 15. [DOI] [PubMed] [Google Scholar]
- 27.Geurts P, Fillet M, de Seny D, Meuwis MA, Malaise M, Merville MP, Wehenkel L. Proteomic mass spectra classification using decision tree based ensemble methods. Bioinformatics. 2005;21(14):3138–3145. doi: 10.1093/bioinformatics/bti494. [DOI] [PubMed] [Google Scholar]
- 28.Izmirlian G. Application of the random forest classification algorithm to a seldi-tof proteomics study in the setting of a cancer prevention trial. Annals of the New York Academy of Sciences. 2005;1020:154–174. doi: 10.1196/annals.1310.015. [DOI] [PubMed] [Google Scholar]
- 29.Gislason PO, Benediktsson JA, Sveinsson JR. Random forests for land cover classification. Pattern Recognition Letters. 2006;27:294–300. [Google Scholar]
- 30.Gao D, Zhang YX, Zhao YH. Random forest algorithm for classification of multiwavelength data. Res Astron Astrophys. 2009;9:220–226. [Google Scholar]
- 31.Menze BH, Kelm BM, Weber MA, Bachert P, Hamprecht FA. Mimicking the human expert: pattern recognition for an automated assessment of data quality in mr spectroscopic images. Magn Reson Med. 2008;59(6):1457–66. doi: 10.1002/mrm.21519. [DOI] [PubMed] [Google Scholar]
- 32.Diaz-Uriate R, Alvarez de Andrs S. Gene selection and classification of microarray data using random forest. BMC Bioinformatics. 2006;7(3):1–13. doi: 10.1186/1471-2105-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abu-Nimeh S, Nappa D, Wang X, Nair S. A comparison of machine learning techniques for phishing detection. ACM Proceedings of the anti-phishing working groups 2nd annual eCrime researchers summit. 2007;8:60–69. [Google Scholar]
- 34.Pal M. Random forest classifier for remote sensing classification. Int Journal for Remote Sensing. 2005;26(1):217–222. [Google Scholar]
- 35.Geman S, Geman D. Stochastic relaxation, Gibbs distributions and the Bayesian restoration of images. IEEE Trans on Pattern Analysis and Machine Intelligence. 1984;6(6):721–741. doi: 10.1109/tpami.1984.4767596. [DOI] [PubMed] [Google Scholar]
- 36.Bishop CM. Pattern Recognition and Machine Learning. Springer; 2007. [Google Scholar]
- 37.Welk M, Feddern C, Burgeth B, Weickert J. Median filtering of tensor-valued images. Pattern Recognition Lecture Notes in Computer Science. 2003;2781(1724) [Google Scholar]
- 38.Hastie T, Tibshirani R, Friedman J. The Elements of Statistical Learning. Springer; 2001. [Google Scholar]
- 39.Winkler G. Image Analysis, Random Fields and Markov Chain Monte Carlo Methods: A Mathematical Introduction. Springer; 2003. [Google Scholar]
- 40.Pearl J. Probabilistic Reasoning in Intelligent Systems: Networks of Plausible Inference. Morgan Kaufmann; 1988. [Google Scholar]
- 41.Komodakis N, Tziritas G. Image completion using efficient belief propagation via priority scheduling and dynamic pruning. IEEE Trans on Image Processing. 2007;16(11):2649–2661. doi: 10.1109/tip.2007.906269. [DOI] [PubMed] [Google Scholar]
- 42.Jähne Bernd. Digital image processing (3rd ed): concepts, algorithms, and scientific applications. Springer; 1995. [Google Scholar]
- 43.Lerch K. Master's thesis. University of Heidelberg; Germany: 2006. Discontinuity preserving filtering of spectral images. [Google Scholar]
- 44.Chen C, Liaw A, Breiman L. Using random forest to learn imbalanced data. Technical report. 2004 [Google Scholar]
- 45.Morris JS, Coombes KR, Koomen J, Baggerly KA, Kobayashi R. Feature extraction and quantification for mass spectrometry in biomedical applications using the mean spectrum. Bioinformatics. 2005;21(9):1764–1775. doi: 10.1093/bioinformatics/bti254. [DOI] [PubMed] [Google Scholar]
- 46.Tibshirani R, Hastie T, Narasimhan B, Soltys S, Shi G, Koong A, Le QT. Sample classification from protein mass spectrometry, by peak probability contrasts. Bioinformatics. 2004;20(17):3034–3044. doi: 10.1093/bioinformatics/bth357. [DOI] [PubMed] [Google Scholar]
- 47.Murphy K. Bayes net toolbox for MATLAB. [October 2008]; http://www.cs.ubc.ca/murphyk/Software/BNT/bnt.html 1997-2002.
- 48.Glunde K, Jie C, Bhujwalla ZM. Mechanisms of indomethacin-induced alterations in the choline phospholipid metabolism of breast cancer cells. Neoplasia. 2006;8(9):758–771. doi: 10.1593/neo.06187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Glunde K, Ackerstaff E, Natarajan K, Artemov D, Bhujwalla ZM. Real-time changes in 1H and 31P NMR spectra of malignant human mammary epithelial cells during treatment with the anti-inflammatory agent indomethacin. Magn Reson Med. 2002;48:819–825. doi: 10.1002/mrm.10295. [DOI] [PubMed] [Google Scholar]