

Abstract
Functionality of neurons is dependent on their compartmentalized polarization of dendrites and an axon. The rapid and selective outgrowth of one neurite, relative to the others, to form the axon is critical in initiating neuronal polarity. Axonogenesis is regulated in part by an optimal intracellular calcium concentration. Our investigation of Ca2+-signaling pathways involved in axon formation using cultured hippocampal neurons demonstrates a role for Ca2+/calmodulin kinase kinase (CaMKK) and its downstream target Ca2+/calmodulin kinase I (CaMKI). Expression of constitutively active CaMKI induced formation of multiple axons, whereas blocking CaMKK or CaMKI activity with pharmacological, dominant-negative, or short hairpin RNA (shRNA) methods significantly inhibited axon formation. CaMKK signals via the γ-isoform of CaMKI as shRNA to CaMKIγ, but not the other CaMKI isoforms, inhibited axon formation. Furthermore, overexpression of wild-type CaMKIγ, but not a mutant incapable of membrane association, accelerated the rate of axon formation. Pharmacological or small interfering RNA inhibition of transient receptor potential canonical 5 (TRPC5) channels, which are present in developing axonal growth cones, suppressed CaMKK-mediated activation of CaMKIγ as well as axon formation. We demonstrate using biochemical fractionation and immunocytochemistry that CaMKIγ and TRPC5 colocalize to lipid rafts. These results are consistent with a model in which highly localized calcium influx through the TRPC5 channels activates CaMKK and CaMKIγ, which subsequently promote axon formation.
Introduction
Establishment of morphological and functional polarity of neurons is seminal to formation of neuronal circuits in the CNS. Cultured rat embryonic hippocampal neurons are a well established model system to study the development of neuronal polarity in vitro (Craig and Banker, 1994). In this system, neuronal polarity is initiated by axon formation that occurs within the first 24–48 h. On plating, neurons initially form lamellapodia (stage 1) that develop into multiple short neurites that undergo repeated stochastic episodes of extension and retraction (stage 2) until one neurite extends rapidly to become the axon (stage 3). Amplification of a positive signaling feedback loop in one neurite presumably accelerates its outgrowth, which, when combined with negative-feedback signals from the other neurites, ensures formation of a single axon (Andersen and Bi, 2000). Here, we examined the role of calcium influx-activated signaling via CaM kinases (CaMKs) in the development of neuronal polarity.
Calcium regulates axon outgrowth and growth cone motility in complex ways (Gomez and Spitzer, 2000; Gomez and Zheng, 2006). Calcium transients occur spontaneously in growth cones and are linked to preaxonogenesis events (Bentley et al., 1991; Davenport and Kater, 1992). Transient receptor potential canonical (TRPC) channels are implicated in Ca2+-dependent growth cone dynamics (Li et al., 2005; Shim et al., 2005; Wang and Poo, 2005). TRPC members (TRPC1-7) form homo- or hetero-tetramers that function as nonselective cation channels and are abundantly expressed in embryonic brain (Strübing et al., 2001; Ambudkar and Ong, 2007). Notably, TRPC4- and 5-containing TRPC channels are the most prevalent TRPC channels in rodent brain (Fowler et al., 2007), and TRPC5-containing channels are present in growth cones of stage 2 hippocampal neurons (Greka et al., 2003). Given the increased calcium permeability of TRPC5 homomeric channels (Ambudkar et al., 2006; Beech, 2007), they are an attractive candidate for mediating Ca2+ entry into developing neurons.
Transduction of Ca2+ gradients in axonal growth cones is thought to be dictated by calmodulin-dependent protein kinases (CaMKs) or the CaM-dependent phosphatase, calcineurin (Wen et al., 2004). Previous studies concluded that CaMKII mediates neurite outgrowth since pharmacological inhibitors of CaMKII [KN-62 (1-[N, O-bis(5-isoquinolinesulfonyl)-N-methyl-l-tyrosyl]-4-phenylpiperazine) and KN-93 (2-[N-(2-hydroxyethyl)]-N- (4-methoxybenzenesulfonyl)amino-N-(4-chlorocinnamyl)-N-methylbenzylamine)] (Tokumitsu et al., 1990) generally suppress Ca2+-dependent neurite outgrowth (Zheng et al., 1994; Kuhn et al., 1998). However, we now know these inhibitors are not specific for CaMKII but also inhibit CaMKIV and CaMKI (Mochizuki et al., 1993; Enslen et al., 1994) as well as some Ca2+-activated potassium channels (Ledoux et al., 1999) and voltage-gated Ca2+ channels (Anderson et al., 1998). We previously demonstrated that the Ca2+/calmodulin kinase kinase (CaMKK)/CaMKI cascade (Soderling, 2000), which is present in neurons throughout development (Kamata et al., 2007a,b), regulates various stages of neuronal development including stage 3 axonal growth cone motility and outgrowth (Wayman et al., 2004), stage 4 dendritic arborization (Wayman et al., 2006), and stage 5 spine/synapse formation (Saneyoshi et al., 2008). Using multiple approaches, we demonstrate here that CaMKK/CaMKIγ, activated by Ca2+-permeable TRPC5 channels, promote axon formation in cultured hippocampal neurons.
Materials and Methods
Pharmacological inhibitors and antibodies.
1,8-Naphthoylene benzimidazole-3-carboxylic acid (STO-609) was purchased from Tocris. 1-[β-[3-(4-Methoxyphenyl)propoxy]-4-methoxyphenethyl]-1H-imidazole, HCl (SKF96365), anthra[1,9-cd]pyrazol-6(2H)-one (SP600125), and nifedipine were from Calbiochem. Monoclonal antibody for Tau-1 was from Millipore Bioscience Research Reagents. Monoclonal anti-Flotillin-1 antibody was from BD Biosciences Transduction Laboratories. Monoclonal anti-Transferrin receptor (TfR) antibody was from Zymed. Rat polyclonal anti-TRPC5 and anti-TRPC6 antibodies were from Alomone Labs. E7 monoclonal ascites to detect β-tubulin by Western blotting and immunocytochemistry was purchased from Developmental Studies Hybridoma Bank. Phospho-CaMKI and CaMKIγ antibodies were gifts from our collaborator, Dr. Naohito Nozaki (Kanagawa Dental College, Kanagawa, Japan).
Cell culture and transfection.
Low-density hippocampal neuron cultures were prepared from embryonic day 18 (E18) rat hippocampi and cultured using the glial sandwich method as previously described in detail (Kaech and Banker, 2006; Oliva et al., 2006). For cDNA and plasmid based short hairpin RNA (shRNA) expression, 0.5 × 106 E18 hippocampal neurons were electroporated with 1–5 μg of cDNA or with 2.5 μg of Stealth small interfering RNA (siRNA) using AMAXA nucleofection system before plating [details described by Kaech and Banker (2006)]. Approximately 30–50% of the electroporated neurons (∼0.15–0.25 × 106) were plated on four poly-l-lysine-coated 18 mm glass coverslips that were placed in a 60 mm culture dish with plating media. For pharmacological inhibition of signaling pathways, the inhibitors were added to the plates containing glia (and glial conditioned media) just before introducing the glass coverslips containing the hippocampal neurons after the 2–4 h attachment period. When using plasmid based shRNA knockdown approach, to extend the time window for effective shRNA knockdown of CaMKK/CaMKI pathway, we incubated the neurons with 20 μm SP600125 for the first 48 h in culture. We then released the suppressed stage 2 neurons from the SP600125 block by gently moving the coverslips to new glial plates (with glial conditioned media) without inhibitors. This constituted a washout of SP600125. This technique has been previously described by Oliva et al. (2006). Once transferred to new glial plates, the neurons were kept in culture for an additional 48 h to allow axon formation.
shRNA, siRNA, and plasmids.
The construction, validation, and specificity of the plasmid-based shRNA constructs for knockdown of CaMKK/CaMKI cascade have been described previously (Wayman et al., 2006; Saneyoshi et al., 2008). For selective knockdown of TRPC5 and TRPC6 subunits of TRPC channels we used Block-iT Stealth RNA interference (RNAi) (Invitrogen). siTRPC5 was custom synthesized targeting nucleotide starting position 1900 (CATGCTGATATTGAGTGGAAGTTTG; accession number NM_080898; rat TRPC5), and siTRPC6 was generated by custom synthesis of RNAi against nucleotide starting position 1506 (AGGAATGATATGGGCTGAATGTAAA; accession number NM_053559; rat TRPC6). siTRPC5 and siTRPC6 were validated for knockdown and specificity by cotransfection of Myc-mTRPC5 and HA-mTRPC6 with 20 and 40 nm of corresponding siRNA in COS7 cells.
Constitutively active CaMKI (caCaMKI), dominant-negative CaMKI (dnCaMKI), dnCaMKK, CaMKKins, and enhanced green fluorescent protein (EGFP)-CaMKIIN have been described previously (Wayman et al., 2004, 2006; Saneyoshi et al., 2008). Truncated Kinesin-1 construct used to identify axon(s) has also been described previously (Jacobson et al., 2006). Human CaMKIγ (hCaMKIγ) was cloned from human hippocampal cDNA library from BD Biosciences (primers based on accession number NM_020439) and subsequently subcloned into pCAGGS vector with N-terminal Flag epitope tag using ClaI and XhoI sites. EGFP-hCaMKIγ was constructed by first subcloning hCaMKIγ into pEGFP-C1 followed by subcloning of the EGFP-hCaMKIγ fusion into pCAGGS. To generate truncation mutant CaMKIγDPP, Met407 was mutated to a TAG stop codon using QuickChange Site-Directed Mutagenesis kit (Stratagene). All constructs were verified by sequencing and their expression was tested in transiently transfected COS7 cells and cultured hippocampal neurons before use in experiments.
Immunocytochemistry.
For Tau-1 immunostaining, neurons were fixed at indicated times, in prewarmed 4% paraformaldehyde (PFA) in PHEMS buffer (60 mm PIPES, 25 mm HEPES, 2 mm MgC12, 1 mm EGTA, pH 6.9) for 20–30 min at room temperature (RT). After fixation, neurons were permeabilized with 0.1% Triton X-100 in PBS for 5–10 min and blocked for 1 h at RT with PBS containing 0.5% Fish Gelatin (Sigma-Aldrich). Axons were labeled by overnight incubation (at 4°C) with anti-Tau-1 monoclonal antibody diluted at 1:3000 in blocking buffer followed by brief wash (three times; 5 min each) and incubation with donkey anti-mouse Cy3 secondary antibody (1:3000) for 1 h at RT. After secondary antibody incubation, neurons were washed with PBS (four times; 10 min) and coverslips were mounted using elvanol mounting medium. Endogenous TRPC5 was immunostained using the primary rabbit anti-TRPC5 antibody (Alomone Labs; 1:1000) and donkey anti-rabbit Cy3 (1:5000) secondary antibody. β-Tubulin was immunostained using E7 ascites (1:3000) and Alexa Fluor 350-conjugated anti-mouse IgG (1:1000). Flotillin-1 immunocytochemistry was performed with monoclonal antibody diluted 1:1000 in blocking buffer, followed by incubation with Alexa Fluor 488-conjugated goat anti-mouse (1:1000). Endogenous monoganglioside 1 (GM1) in 1 d in vitro (DIV 1) E18 hippocampal neurons plated at low density was stained live with Alexa Fluor 546-conjugated cholera toxin B subunit (CTxB) (Invitrogen) diluted to a final concentration of 2 μg/ml in prewarmed artificial CSF (ACSF) for 6–8 min at 37°C. Immediately after staining, the coverslips were briefly rinsed (2–4 s) in prewarmed ACSF and fixed with 4% PFA diluted in PHEMS buffer for 20 min at room temperature. For colocalization with EGFP-CaMKIγ, the coverslips were mounted and imaged immediately. Flotillin-1 costaining with CTxB::GM1 was done as described above.
To measure the level of endogenous TRPC5 knockdown by siTRPC5 with immunocytochemistry, neurons were electroporated with control siRNA or siTRPC5 along with soluble EGFP as a transfection marker and were fixed and immunostained with anti-TRPC5 after 48 h as described in Materials and Methods. To quantify the level of TRPC5 knockdown by immunocytochemistry, multiple random fields were captured from both control and siTRPC5 conditions. We measured the pixel intensity (P.I.) (in arbitrary units) of immunofluorescence from siTRPC5-transfected (i.e., EGFP-positive) neuron relative to neighboring nontransfected neuron(s) within the same field that served as internal positive controls.
Microscopy, imaging, and quantification.
Images were acquired by using a cooled CCD camera (Hamamatsu Photonics) attached to a Zeiss Axioplan 2 (Carl Zeiss) inverted microscope with a 10 or 20× lens for quantification of neuronal polarity and with a 63× oil-immersion lens for imaging immunostaining of endogenous TRPC5, Flotillin-1, β-tubulin, GM1, and for colocalization of EGFP-CaMKIγ and TRPC5. Images were processed using ImageJ and Adobe Photoshop 10. For analyzing numbers of neurons that formed axons under various conditions, 15–30 random fields from two to four individual coverslips (usually representing ∼100–200 neurons) were captured as described above using the filter sets for EGFP (soluble fill as marker for transfection) and Cy3 (Tau-1). Total neurons, neurons expressing Tau-1-positive process (termed axon), and where indicated, number of Tau-1-positive axons per neurons were manually counted using ImageJ (W. S. Rasband, ImageJ, National Institutes of Health, Bethesda, MD; http://rsb.info.nih.gov/ij/; 1997–2008). The percentage of neurons under basal conditions that formed an axon ranged from 60 to 85% because of natural variability in cultures. To facilitate statistical analysis between experiments, the control average from each experiment was set to 100%, and the experimental data (average ± SEM) were normalized relative to the control. Statistical analyses and graphing were performed using unpaired Student's t test using Microsoft Excel or GraphPad Prism 3.0. To measure axonal length, the NeuronJ plugin was used in conjunction with ImageJ. Raw axonal length data from NeuronJ were adjusted to micrometers based on calibration ruler measurements. Data in the graphs are represented as averages plus SEMs from two to five independent experiments.
Lipid raft fractionation and Western blotting.
Lipid raft fractionation was done from postnatal day 0 (P0) rat forebrains with a discontinuous sucrose gradient using protocols adapted from previously described methods (Maekawa et al., 2001; Suzuki et al., 2004). Briefly, we made homogenates from 1.3 g of P0 forebrain material per 10 ml of buffer A (50 mm HEPES, pH 7.4, 1 mm MgCl2, 1 mm NaF plus protease and phosphatase inhibitors) with a Dounce homogenizer. This crude homogenate was centrifuged at 200 × g for 5 min in a SS-34 rotor to pellet larger particulate material. The supernatant from this was centrifuged at 50,000 rpm (179,000 g force) in a 70Ti rotor for 30 min. The resulting supernatant was designated as the crude cytosol and was discarded. The pellet fraction represented the crude membranes. To concentrate the crude membrane fraction before loading on the gradient, the membranes were resuspended in 5 ml of buffer B (buffer A plus 1% Triton X-100) by homogenization with a Dounce homogenizer. After homogenization, this membrane suspension was incubated on ice for 30 min. This membrane fraction was then loaded on a discontinuous sucrose gradient as follows: 0.5 ml of the crude membrane suspension was mixed with 0.5 ml of 90% sucrose (in buffer A). This 1 ml fraction composed of a 50:50 mix of membranes in 45% sucrose was loaded on the bottom of SW41Ti rotor tubes; this was gently overlaid with 8 ml of 35% sucrose in buffer A and finally 3.5 ml of 5% sucrose in buffer A. The gradient was centrifuged at 140,000 × g for 18 h in a SW41Ti rotor at 4°C (low brake setting). One milliliter fractions were collected by puncturing the tubes from the bottom and collecting fraction from bottom to top (bottom-most fraction is designated as fraction 12). Protein concentration was determined using Bradford reagent (Bio-Rad), and fractions were loaded on SDS-PAGE and transferred with polyvinylidene difluoride. Western blotting was with indicated antibodies at the following dilutions: anti-TRPC5, 1:1000; anti-TRPC6, 1:1000; anti-CaMKI, 1:100; anti-Flotillin-1, 1:2000; and anti-TfR, 1:2000. Western blot detection (IR700- and IR800-conjugated secondary antibodies) and quantification were done using the Odyssey Infrared System (LI-COR Biosciences).
Results
Constitutively active CaMKI promotes multiple axons
We have previously demonstrated a role for CaMKK and CaMKI in regulation of axonal extension and growth cone motility in neonatal hippocampal neurons (Wayman et al., 2004). These experiments were conducted using stage 3 (DIV 2–4) cultured neurons that are already polarized with a single extended axon and several short neurites that subsequently become dendrites. Under these conditions, expression of dnCaMKK or dnCaMKI, but not nuclear localized dnCaMKIV or a protein inhibitor of CaMKII, caused growth cone collapse and significantly reduced total axonal length and branching. Conversely, transfection of a caCaMKI stimulated axonal extension in stage 3 neurons. In the current study, we investigated the role of the CaMKK/CaMKI signaling pathway in the early steps involved in the establishment of neuronal polarity. We used a low-density embryonic (E18) hippocampal culture system that is an optimal and well defined model system for studying neuronal polarity (Craig and Banker, 1994; Kaech and Banker, 2006). In these cultures, most neurons do not display an axon at 24 h (stage 2; all neurites are similar length), whereas at 48 h a majority (>70%) have one neurite that is more than twice the length of the others, and is specified as the axon (stage 3 neuron) (Deitch and Banker, 1993). To modulate axon formation with pharmacological inhibitors and transfection protocols, we electroporated neurons before plating and initiated all pharmacological treatments at 2 h after plating.
To investigate the role of the CaMKK/CaMKI cascade in axon formation, we transfected hippocampal neurons with constitutively active CaMKIα that has been previously tested (caCaMKI) (see below). Overexpression of caCaMKI should act as a generic constitutively active CaMKI (Wayman et al., 2004, 2006; Saneyoshi et al., 2008). Approximately 70% of neurons transfected with caCaMKI were polarized by 48 h, and although transfection with caCaMKI did not alter the length of the primary projection (control, 263.9 ± 13.4 μm; caCaMKI, 251.5 ± 12.6; n = 50 neurons), neurons expressing caCaMKI had significantly longer minor processes (Fig. 1A). Most of these secondary projections were in fact axons, as defined by staining for the well established axonal marker, Tau-1 (Mandell and Banker, 1996) (Fig. 1B). In Figure 1B, the left vertical panels show representative images of soluble EGFP cotransfected with empty vector (control, top) or caCaMKI (bottom); the middle vertical panels show Tau-1 staining in one distinct elongated process in control neurons but in multiple elongated processes in neurons expressing caCaMKI. Analyses from five independent experiments revealed that control neurons have an average of 1.12 ± 0.16 axons per neuron (n = 400 neurons) versus 2.7 ± 0.25 (n = 276 neurons) axons in neurons expressing caCaMKI (Fig. 1D).
Figure 1.
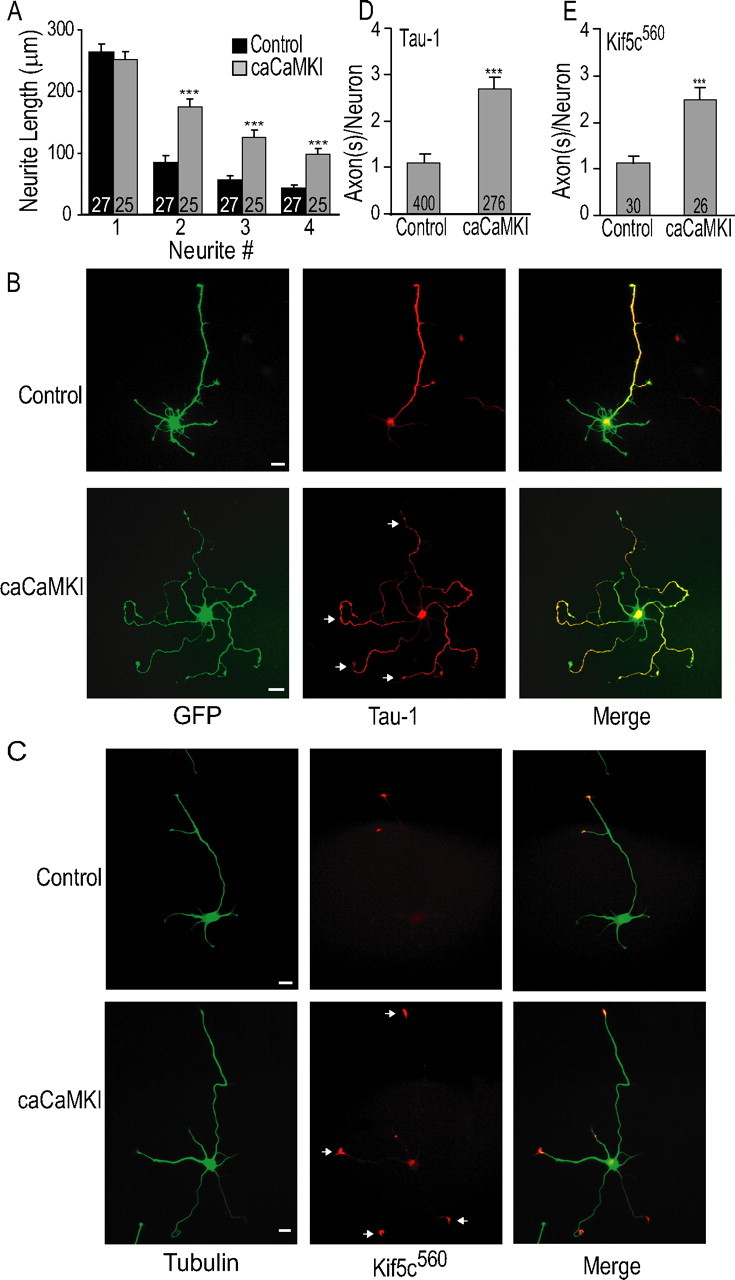
Constitutively active CaMKI induces multiple axon formation in hippocampal neurons. A, Average neurite length at 48 h from neurons transfected either with control plasmid (empty vector) or caCaMKI. B, Low-density E18 hippocampal cultures were electroporated before plating with either pCAGGS (control; top panel) or pCAGGS-caCaMKI (bottom panel) and soluble EGFP (marker for transfection) for 48 h followed by fixation and staining as described in Materials and Methods. Representative immunofluorescent images demonstrate formation of single Tau-1-positive neurite defined as the axon in control cells. In contrast, expression of caCaMKI strongly increases the number of Tau-1-positive axons. C, GFP-tagged Kif5C560 (pseudocolored red) was cotransfected with control or caCaMKI before plating, and neurons were fixed and stained with anti-β-tubulin (pseudocolored green) at 48 h. Representative images are shown. D, E, Quantification (means ± SEM) of number of Tau-1-positive axons per neuron from three independent experiments (D) and GFP-tagged Kif5C560-containing neurite tips from control and caCaMKI (E). Scale bars: 20 μm (all panels). Here and in all subsequent figures, all graphs show means ± SEM (n values are shown as numerical insets within bars of graphs; where indicated, p ≤ 0.05, **p ≤ 0.005, ***p ≤ 0.0005 by Student's t test).
To further validate that these Tau-1-positive projections induced by caCaMKI were axons, we used another confirmed marker of axons, Kinesin-1. It was recently shown that truncated Kinesin-1 selectively accumulates in the emerging axon and is excluded from all other neurites (Jacobson et al., 2006). We cotransfected neurons with an EGFP-tagged truncated Kinesin-1 (Kif5c560) along with empty vector (control) or caCaMKI. Neurons were stained with β-tubulin antibody to identify all projections (pseudocolored green). As shown in Figure 1C, EGFP-Kif5C560 (pseudocolored red) was selectively accumulated in the only in the tips of axonal branches of control cells and was absent from other neurites. However, in caCaMKI-expressing neurons, Kif5C560 was present in tips of multiple projections, demonstrating that expression of caCaMKI shifted multiple minor neurites toward biochemically defined axons. The caCaMKI induced a ∼2.4-fold increase in the number of Kif5C560-containing axons per neuron compared with control neurons (caCaMKI, 2.5 ± 0.09 axons/neuron, vs control, 1.12 ± 0.1 axons/neuron; n = 30 neurons for both conditions) (Fig. 1E).
We also performed live imaging of hippocampal neurons expressing control vector or caCaMKI to visualize axon formation (supplemental movies, available at www.jneurosci.org as supplemental material). As seen in the movies, neurons expressing caCaMKI show enhanced outgrowth of multiple axons compared with a single axon in control transfection. The above data suggest that the CaMKI pathway may play a role in regulating not just axonal outgrowth of stage 3 neurons as reported previously (Wayman et al., 2004) but may regulate axon formation itself. Therefore, we designed additional experiments to specifically test whether modulation of CaMKI pathway alters axon formation and axonal length in low-density embryonic hippocampal cultures.
The CaMKK inhibitor, STO-609, blocks axon formation and elongation
In the preceding experiments, we used a mutant of CaMKI that is constitutively active, but the physiological upstream activator of CaMKI is CaMKK. To assess the cellular role of CaMKK, we incubated E18 hippocampal neurons with the cell-permeable CaMKK inhibitor STO-609 (Tokumitsu et al., 2002, 2003). In vitro, STO-609 selectively blocks CaMKK with little direct effect on CaMKI, CaMKII, CaMKIV, PKA (protein kinase A), PKC (protein kinase C), MLCK (myosin light chain kinase), or p42/44 MAPK (mitogen-activated protein kinase) (Tokumitsu et al., 2002), and we have shown that STO-609 can selectively block activation of CaMKI in cultured hippocampal neurons without suppressing CaMKII activation (Wayman et al., 2004). Treatment with 5.2 μm STO-609, beginning at 2 h after plating, resulted in only 45.6 ± 3% of neurons developing an axon at 48 h compared with 75% of control neurons (Fig. 2A, B). We performed a time course experiment to determine whether incubation of neurons with STO-609 only delays axon formation or results in a chronic block of axonogenesis. Hippocampal neurons were transfected with soluble EGFP, incubated with 5.2 μm STO-609 for up to 96 h in culture, fixed, and stained with Tau-1 antibody to mark axons. As shown in Figure 2B, incubation with STO-609 for up to 96 h resulted in a significant number of neurons remaining chronically suppressed at stage 2 (average decrease of 51.1 ± 2.8% at 24–96 h). Inhibition of CaMKK by STO-609 selectively suppresses axonal development, since the length of the minor neurites, which were Tau-1 negative, was not significantly altered in the presence of this inhibitor (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). This is consistent with our previous observation that inhibition of CaMKK does not suppress basal (i.e., activity-independent) stage 4 development of dendrites from neurites (Wayman et al., 2006). The suppression of axon formation in hippocampal neurons with STO-609 was reversible (data not shown). These data suggest a regulatory role for CaMKK in axon formation during stage 2 to stage 3 progression.
Figure 2.
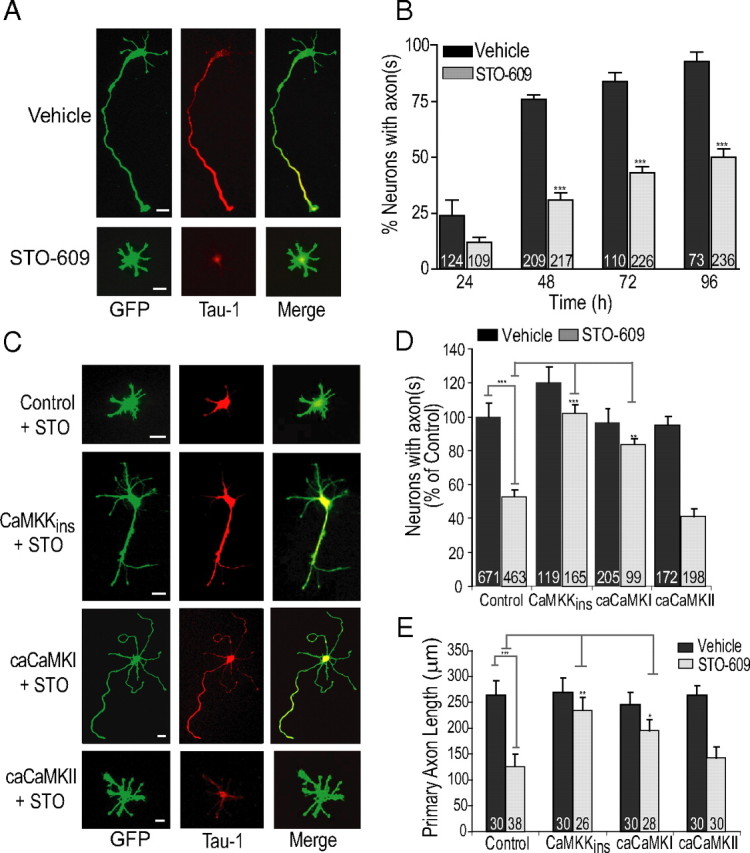
Inhibition of CaMKK by STO-609 chronically suppresses axon formation. A, Low-density hippocampal cultures electroporated before plating with soluble EGFP were incubated with vehicle (control) or 5 μm STO-609 from 2 h until fixation at 48 h. Representative images with axonal marker Tau-1 shows normal formation of a single axon in control neurons with suppressed axon formation in neurons incubated in STO-609. B, STO-609 time course with neurons fixed at 24, 48, 72, and 96 h reveals chronic partial block of axon formation. C–E, Expression of STO-609-insensitive CaMKK (CaMKKins) or caCaMKI, but not caCaMKII, rescue STO-609 inhibition of axon formation and outgrowth. All inhibitor and inhibitor rescue data are shown normalized to control conditions set to 100% [60–90% of neurons polarized under basal (control) conditions] in this and subsequent figures. Representative images from STO-609-insensitive CaMKK, caCaMKI, and caCaMKII are shown in C. Quantification of three independent experiments are shown in D. Average axonal length as measured by length of primary Tau-1-positive axon is shown in graph in E. Scale bars: 20 μm (all panels).
Although STO-609 has strong selectivity for inhibition of CaMKK relative to other protein kinases (Tokumitsu et al., 2002), we wanted to confirm that its inhibitory effect was in fact attributable to suppression of CaMKK. To this end, we used a STO-insensitive CaMKK mutant (designated CaMKKins) that has a single amino acid mutation of CaMKKα (L233F), rendering it 100-fold less sensitive to STO-609 (Tokumitsu et al., 2003). We tested whether expression of this STO-insensitive CaMKK could rescue axon formation in the presence of STO-609. In the presence of STO-609, 52.2% of neurons remained blocked at stage 2 relative to vehicle-treated control neurons (set to 100%) at 48 h. Expression of CaMKKins in the presence of STO-609 showed complete rescue of axon formation and axonal length (Fig. 2C–E). The effects of STO-609 were attributable to inhibition of CaMKK and downstream signaling in neurons, and not the supporting glial cells, because in glial conditioned medium alone STO-609 suppressed axon formation to the same degree as with the neuron-glial sandwich cultures (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material).
By suppressing activity of CaMKK, STO-609 inhibits activation of its downstream targets including CaMKI. Thus, expression of caCaMKI, which bypasses the requirement for activation of CaMKI by CaMKK, should also restore axon formation in STO-609-treated cells. Transfection with caCaMKI significantly reversed the STO-609 phenotype by increasing Tau-1-positive neurons to 83.8% in the presence of STO-609 (compare with 52.8 ± 4% with empty vector) (Fig. 2C, D). Even in the presence of STO-609, a significant number of caCaMKI-expressing neurons still exhibited more than one axon (as in Fig. 1). Expression of a constitutively active kinase poses the risk of promiscuous substrate activation (Wayman et al., 2008), since various CaMKs have overlapping substrate specificity determinants (e.g., an Arg in the P-3 position). CaMKII is the other major neuronal calcium/calmodulin kinase with functional implications in aspects of neuronal development (Wen et al., 2004), so it was important to test whether expression of caCaMKII [H282R mutant (Brickey et al., 1994)] might also reverse the STO-609 phenotype. However, expression of caCaMKII was unable to rescue the suppression of axon formation or length of axon by STO-609 (Fig. 2C–E). Therefore, it is unlikely that the effects of CaMKKins and caCaMKI are attributable to nonspecific substrate phosphorylation.
Since STO-609 produces only a partial block (∼50%) (Fig. 2D) of axon formation, we examined axonal length in those neurons that form a morphologically distinct axon in the presence of STO-609. Axon elongation is a critical component of establishing neuronal polarity in the sense that continuous growth of one neurite permits the establishment of axonal characteristics. The neurons that still formed an axon in the presence of STO-609 had significantly shorter axons (Fig. 2E) (control average length of the primary axon, 263.9 ± 13.4 μm; STO-609-treated cells, 128.6 ± 7.5 μm). Transfection with CaMKKins or caCaMKI, but not caCaMKII, rescued inhibition of axonal outgrowth by STO-609 (Fig. 2E). These data together indicate a specific role of the CaMKK/CaMKI signaling cascade in axon formation.
In studies of axon specification, neuronal polarization is typically defined by the appearance of a projection that is longer than the other neurites and displays hallmark axonal markers. One neuronal polarity model suggests that axon fate is conferred on one neurite because of accumulation of “axon-specifying” components in this neurite, followed by its rapid outgrowth to form the axon. Another polarity model conceptualizes that accelerated growth of one neurite is the critical initial step in axonogenesis, leading to preferential enrichment of key axonal components and consequential acquisition of axonal fate by this neurite (Jiang and Rao, 2005; Winckler, 2007). Because of experimental dependence on appearance of a morphologically discernable axon in our cultured neuron model system, it is difficult to mechanistically differentiate between “specification” and “outgrowth” as both are requisite steps. The fact that transfection with caCaMKI promoted formation of multiple axons may indicate it has a role in axon “specification.” However, our other experiments cannot differentiate between specification and outgrowth, so we have used a more general term “axon formation” that is commonly used in this field.
Dominant-negative CaMKK and CaMKI block axon formation
To establish the physiological role of a signaling pathway, it is essential to use multiple, independent protocols to modulate the functionality of that pathway (for review, see Wayman et al., 2008). In addition, to further investigate the potential role of endogenous CaMKII in axon development, we expressed CaMKIIN, a highly selective endogenous protein inhibitor of CaMKII (Chang et al., 1998). Transfection of CaMKIIN completely blocks activation of CaMKII, even on depolarization with KCl, without effect on axonal outgrowth of stage 3 neurons (Wayman et al., 2004). Expression of dnCaMKK mimicked the effects of STO-609 such that only 56.3 ± 3.5% of neurons had Tau-1-positive axons (Fig. 3A,B). Of the dnCaMKK-expressing neurons that did develop axons, their axonal length was significantly reduced (control, 210.3 ± 19.1 μm, vs dnCaMKK, 124 ± 12 μm) (Fig. 3C). The dnCaMKI had a similar effect with 69.7 ± 5.8% of neurons developing axons compared with control. We previously showed that nuclear-restricted dnCaMKIV (dnCaMKIVNLS) had no effect on stage 3 axonal outgrowth (Wayman et al., 2004). Here, in our experiments with the stage 1 to stage 2 neurons in the low-density culture system, expression of dnCaMKIVNLS resulted in significant toxicity, thereby making the results uninterpretable. Overexpression of CaMKIIN did not significantly suppress axon formation or affect axonal length. These data corroborate the results from the preceding experiments indicating that CaMKK and CaMKI, but not CaMKII, play a modulatory role in axon formation of E18 hippocampal neurons in our system.
Figure 3.

dnCaMKK and dnCaMKI inhibit axon formation and decrease axonal length. A, E18 hippocampal neurons were coelectroporated with control (empty vector), dnCaMKK, dnCaMKI, or CaMKIIN containing plasmid and soluble EGFP before plating. A shows representative images of neurons fixed at 48 h and stained with the axonal marker Tau-1. B, Quantification of axon formation as measured by number of neurons with Tau-1-positive projections from three to four independent experiments. C, Quantification of average axonal length as measured by the length of the primary Tau-1-positive neurite. Scale bars: 20 μm (all panels).
shRNA knockdown of CaMKK and CaMKI decreases axonogenesis
Since ectopic expression of dnCaMKK and dnCaMKI may result in unknown regulation of other signaling pathways, we verified these results by acutely knocking down the expression of endogenous CaMKK and CaMKI using plasmid-based shRNA approach. Both CaMKK isoforms (α and β) and CaMKI subfamily members that include four isoforms (α, β, γ, and δ) are present in the hippocampus at varying levels during early development (Nishimura et al., 2003; Sakagami et al., 2005; Kamata et al., 2007a,b). To determine which CaMKI isoform(s) regulates axon formation in neurons, we used CaMKI isoform-specific shRNAs that have been previously validated for knockdown efficacy and specificity in HEK293 cells (Wayman et al., 2006) as well as shRNAs for α and β CaMKK (Saneyoshi et al., 2008). As shown in Figure 2B, axon formation in these cultures occurs precipitously between 24 h (24 ± 7% stage 3 neurons) and 48 h (76 ± 2% stage 3 neurons), but with the plasmid-based shRNA approach it is difficult to achieve substantial reductions of proteins within 24 h. To increase the duration of shRNA expression to allow sufficient knockdown of CaMKK and CaMKI before axon formation initiates, we arrested neuronal development at stage 2 with 20 μm SP600124 from 2 to 48 h in culture, followed by release from block to initiate axon formation for another 48 h. SP600125 is a Jun kinase inhibitor that reversibly blocks axon formation without affecting neuronal viability (Oliva et al., 2006). To release hippocampal neurons from SP600125 block, the coverslips were transferred to new plates containing glia and their glial-conditioned media. Incubating neurons with SP600125 for 48 h suppressed axon formation in 98.5 ± 1% of neurons—this block was almost completely reversible, with 90 ± 2% of neurons developing axons after the 48 h washout (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material).
Transfection of E18 hippocampal neurons with a mixture of shRNAs for both CaMKK isoforms (α and β) decreased the number of neurons exhibiting Tau-1-positive axons to only 63 ± 4.3% of control values (control, 100%). Of the four distinct isoforms of CaMKI (α, β, γ, δ), shCaMKIγ significantly blocked axon formation (only 58.4 ± 6.9% neurons formed Tau-1-positive axons) to the same degree as shCaMKK (Fig. 4A,B). shCaMKIα also had a modest but statistically significant effect on axon formation (83.1 ± 7.3% formed an axon). As shown in Figure 4C, the neurons that did develop an axon in the presence of shCaMKK and shCaMKIγ had significantly shorter axonal lengths (52.2% decrease with shCaMKK and 40.1% decrease with shCaMKIγ) relative to control. Thus, the shRNAs phenocopied the inhibitory effects of STO-609, dnCaMKK, and dnCaMKI.
Figure 4.

Knockdown of CaMKK and CaMKIγ protein expression inhibits axonogenesis and decreases axonal length. A, E18 hippocampal neurons were electroporated before plating with pMU6pro alone (control) or pMU6pro containing shCaMKK, shCaMKIα, shCaMKIβ, shCaMKIδ, or shCaMKIγ. Between 2 and 48 h in culture, low-density cultures of electroporated neurons were incubated with 20 μm SP600125 to suppress intrinsic neuronal polarization to achieve effective knockdown with the plasmid based shRNA (see Results). At 48–72 h in culture, electroporated neurons were released from SP600125 block by transfer of coverslips to new glial culture plates for an additional 48 h. Representative images of neurons fixed and stained with Tau-1 at 96 h are shown. The third from top panel shows staining from CaMKIα shRNA. This image is also representative of neurons transfected with shCaMKIβ or shCaMKIδ. B, Summary of three to five independent experiments. C, Average axonal length as measured by length of primary process containing Tau-1 staining from three independent experiments. Scale bars: 20 μm (all panels).
To ensure that the effects of shCaMKIγ are specific, we rescued the effect by cotransfection with human CaMKIγ. Experiments in HEK293 cells showed that the shCaMKIγ, designed against the rat CaMKIγ nucleotide sequence, did not suppress expression of hCaMKIγ (data not shown). In Figure 5A, the representative images demonstrate that coexpression of hCaMKIγ with shCaMKIγ significantly rescued the inhibition of both axon formation and length. Quantification of these data (Fig. 5B,C) demonstrate that only 63 ± 5% of shCaMKIγ-expressing neurons formed axons with average axonal length of 130.8 ± 15 μm versus 86 ± 7% of shCaMKIγ plus hCaMKIγ-expressing neurons that formed an axon with average axonal length of 200.6 ± 26.5 μm. Furthermore, if CaMKIγ regulates axon formation, transient overexpression of CaMKIγ might increase percentage of neurons that exhibit an axon. To test this hypothesis, we transfected hippocampal neurons with wild-type EGFP-CaMKIγ, and fixed neurons at 24 h when most control neurons do not exhibit an axon (Fig. 2B). Hippocampal neurons expressing EGFP-CaMKIγ showed a dramatic acceleration of axon formation at 24 h in culture. At 24 h, only 24.3 ± 2.5% of control neurons had a defined axon compared with 56.4 ± 2.2% of neurons expressing EGFP-CaMKIγ (Fig. 5D,E). These axons were also significantly longer (171.8 ± 8.6 μm) than control neurons (85.3 ± 5.5 μm) (Fig. 5F). These data strongly support a role for CaMKIγ in axon formation.
Figure 5.

CaMKIγ expression promotes axon formation and increases axonal length. A–C, Coexpression of shRNA-insensitive human CaMKIγ rescues the effect of shCaMKIγ. Representative images are shown in A. Quantification of percentage of neurons exhibiting an axon and total axonal length from shCaMKIγ and shCaMKIγ plus rescue plasmid containing hCaMKIγ are shown in B and C, respectively. D, Representative images of EGFP-wtCaMKIγ induced accelerated axon formation at 24 h in culture. Controls contained soluble EGFP. E, F, Quantification of percentage of neurons exhibiting an axon at 24 h and total axonal length of neurons polarized at 24 h, respectively. Scale bars: 20 μm (all panels).
TRPC5 channel plays a role in axon formation
Calcium plays multiple and complex roles in determining neuronal development both in vitro and in vivo (Mattson, 1992, 1999; Dotti and Poo, 2003; Jin et al., 2005; Zheng and Poo, 2007). The spatiotemporal frequency and amplitude of calcium oscillations trigger varying responses, mediated in part by CaM kinases and CaM-dependent protein phosphatases, which are critical for axon formation, outgrowth, and growth cone guidance as well dendritic arborization and synaptogenesis (Mattson and Kater, 1987; Gomez and Spitzer, 2000; Spitzer et al., 2000; Gomez and Zheng, 2006; Zheng and Poo, 2007). Our pharmacological, dominant-negative, and shRNA data establish a role for CaMKK and CaMKIγ in axon formation of cultured hippocampal neurons. Since the only known mechanism for activation of both of these enzymes is an increase in intracellular calcium, we investigated the source of this calcium upstream of CaMKK and CaMKIγ in axon development. L- and N-type voltage-gated calcium channels are present in hippocampal neurons during early developmental stages (Pravettoni et al., 2000). Multiple recent studies also implicate the TRPC cation channel in regulation of growth cone morphology (Greka et al., 2003), neurite outgrowth (Wu et al., 2007), spinogenesis (Zhou et al., 2008), and as the source of calcium influx during chemotropic turning of growth cones in response to neurotrophins, netrin-1, and BDNF (Gomez, 2005; Li et al., 2005; Shim et al., 2005; Wang and Poo, 2005).
To determine the source of intracellular calcium elevation in neurites of stage 2 hippocampal neurons, we used pharmacological inhibitors. As shown in Figure 6A, the L-type channel blocker nifedipine (10 μm) had no effect on the rate of axon formation in control neurons or those expressing caCaMKI. However, inhibition of TRPC channels with 3 μm SKF96365 markedly blocked axon formation in control cells (34 ± 4% polarized compared with 100% of control), and caCaMKI partially rescued axon formation (71 ± 3% polarized) in the presence of SKF96365.
Figure 6.
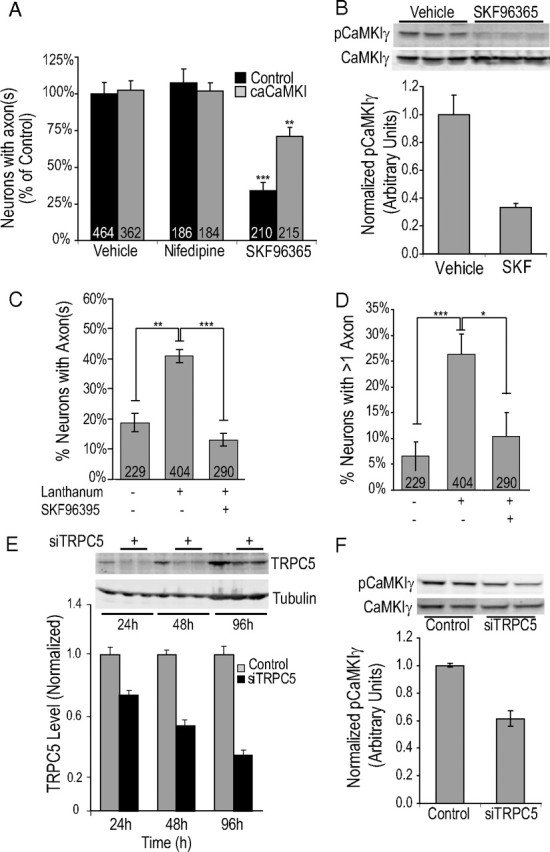
Regulation of CaMKIγ and axon formation by TRPC channel. A, Low-density hippocampal neurons electroporated before plating were incubated for 46 h (2 h after plating until fixation at 48 h) in culture with vehicle, the L-type channel blocker nifedipine (10 μm), or the TRPC blocker SKF96395 (3 μm). Neurons fixed at 48 h were stained with the axonal marker Tau-1. Quantification of results from three independent experiments is shown. B, Representative blot showing regulation of CaMKIγ by TRPC antagonist, SKF96365. P0 hippocampal neurons were electroporated with Flag-CaMKIγ before plating and 24 h later exposed to 50 μm SKF96365 for 60 min. Lysates were immunoblotted with anti-phospho-CaMKI antibody (top blot) or anti-Flag antibody to detect total CaMKIγ (bottom blot). The bottom graph shows average normalized phospho-CaMKIγ. C, D, Graphs depict quantification of axon formation (C) and induction of multiple axons (D) in E18 neurons that were incubated for 20 h (from 2 to 22 h) with growth medium containing 100 μm lanthanum chloride, a selective activator of TRPC4 and -5 channels. E, High-density hippocampal neurons were electroporated with 2.5 μg of siTRPC5 and harvested at the indicated times with SDS sample buffer. Western blots show the time course of reduction of endogenous TRPC5 (top blot) and β-tubulin (bottom blot) with siTRPC5. The bottom graph shows quantification of TRPC5 levels relative to tubulin at 24, 48, 72 h (with control at 24 h set to arbitrary unit of 1). F, Flag-CaMKIγ and control siRNA or siTRPC5 were coelectroporated in P0 hippocampal neurons and harvested after 48–72 h in SDS sample buffer. Phospho-CaMKIγ and total CaMKIγ (Flag) were detected by multiplexed Western blotting and the bottom graph represents summary of ratio of pCaMKIγ/total CaMKIγ from four independent experiments.
We next ascertained whether inhibition of TRPC channels also suppressed activation of CaMKIγ. Because of its low abundance in young cultures, endogenously phosphorylated CaMKIγ was not detected by our phosphospecific CaMKI antibody. However, in neurons as well as heterologous systems, we are able to detect phosphorylation of transiently transfected CaMKIγ. To monitor the phosphorylation of CaMKIγ, we electroporated neurons with Flag-tagged CaMKIγ and subsequently treated them 50 μm SKF96365 or vehicle for 1 h. The activation state of Flag-CaMKIγ by CaMKK was determined by Western blotting with anti-phospho-CaMKI antibody. As shown in Figure 6B (top, representative blot; bottom, quantification), acute inhibition of TRPC channels strongly suppressed phospho-CaMKIγ, but total Flag-CaMKIγ remained constant. Thus, CaMKIγ activation in stage 2 hippocampal neurons was dependent on TRPC channel activity.
The TRPC channel family is composed of seven members (TRPC1-7) that assemble into homomeric or heteromeric channels with diverse properties including mode of activation, calcium conductance, and signaling partners (Clapham et al., 2001; Ambudkar, 2006; Ambudkar and Ong, 2007; Beech, 2007). Based on subunit assembly, TRPC channels are divided into distinct groups—TRPC3/6/7 heteromers, TRPC1/4/5 heteromers, and TRPC subunit homomers. It has been recently established that, by acting at extracellular sites close to the pore, TRPC4/5 heteromers and TRPC5 homomers are distinctively potentiated by the lanthanum (La3+) in the micromolar range (10–100 μm) (Jung et al., 2003; Plant and Schaefer, 2003; Beech, 2007), a concentration that typically is used to suppress other TRPC and other calcium-permeable channels (Beedle et al., 2002). Because of this unique property of TRPC5 channels to be potentiated by La3+, we used it as an experimental tool to test the role of calcium influx by TRPC5-containing TRPC channels in axon formation while inhibiting other calcium channels. When E18 neurons were incubated in media containing 100 μm La3+, there was an approximately twofold increase in neurons exhibiting axon(s) at 20–24 h in culture (Fig. 6C) as well as an approximately threefold increase in neurons with more than one axon (Fig. 6D). Both the La3+-stimulated axon formation and multiple axon effects were strongly blocked by coincubation with the TRPC inhibitor, SKF96365. These data are similar to the effect of transfection with caCaMKI (Fig. 1). This observation with La3+ is supportive of a hypothesis that TPRC4 or -5 channels are a relevant source of Ca2+ influx during axon formation.
To further investigate the role of TRPC5 channels, we used siRNA techniques. TRPC5 channels are of particular interest in neuronal development as these channels are present in growth cones of stage 2 hippocampal neurons grown in culture and in hippocampus of intact brain (Okada et al., 1998; Strübing et al., 2001; Greka et al., 2003; Fowler et al., 2007) and are the most abundant TRPC channel in the hippocampus (Chung et al., 2006; Fowler et al., 2007). We specifically tested the involvement of TRPC5-containing TRPC channels in activation of CaMKIγ and subsequently axon formation with selective knockdown of this subunit with RNAi. As initially validated in COS7 cells, siTRPC5 specifically knocked down expression of Myc-TRPC5 without nonspecific targeting of related channel subunit TRPC6 (expressed as HA-TRPC6) (supplemental Fig. 1E, available at www.jneurosci.org as supplemental material). To determine the extent of endogenous TRPC5 knockdown, we nucleofected hippocampal neurons with siTRPC5 and harvested neurons at 24, 48, and 96 h after nucleofection. Immunoblotting with anti-TRPC5 antibody and anti-β-tubulin (loading control) revealed a time-dependent reduction of TRPC5 (percentage reduction: 24 h, 28%; 48 h, 46%; 96 h, 65%) (Fig. 6E). To specifically test the role of TRPC5 in activation of CaMKIγ, control siRNA or siTRPC5 and Flag-CaMKIγ were coelectroporated into cultured hippocampal neurons before plating and harvested on DIV 3. In Figure 6F, expression of siTRPC5 markedly reduced the level of pCaMKIγ by 39.5 ± 5.5% compared with vector control. The extent of inhibition of CaMKI activation (40%) closely matched with the extent of TRPC5 suppression at 48 h (46%). These data support the results attained with the pharmacological inhibition of TRPC channels with SKF96365.
TRPC5 channels form functional channels by homo- or hetero-tetrameric assembly of individual subunits. It has been recently reported that the first ankyrin domain within the N terminus of TRPC5 is critical for the assembly of functional tetrameric channels (Schindl et al., 2008). Furthermore, Schindl et al. demonstrated that N-terminal fragments containing the first ankyrin repeat functioned as a dominant-negative species on TRPC4/TRPC5 heteromeric and homomeric channels by disrupting channel assembly resulting in downregulation of channel currents. We generated a dominant-negative TRPC5 construct based on these findings that included the N-terminal 330 aa containing the ankyrin repeats of mTRPC5, designated as TRPC5 NT330. Transfection of this dominant-negative TRPC5 modestly but significantly reduced axon formation (72.5 ± 8% formed an axon compared with 100% for control) in hippocampal neurons at 48 h (supplemental Fig. 1D, available at www.jneurosci.org as supplemental material). We further evaluated the role of TRPC5 in the developmental progression from stage 2 to stage 3 hippocampal neurons by expressing RNAi to the TRPC5 subunit. We validated the knockdown of TRPC5 by immunocytochemistry with TRPC5-specific antibodies (as shown by Western blotting in Fig. 6E). As shown in Figure 7A, left panels, siTRPC5, but not control siRNA, reduced the anti-TRPC5 staining intensity throughout the neuron compared with neighboring nontransfected cell. The level of knockdown was variable between individual neurons, ranging from reductions of 30–70%, with an average reduction to 56.5 ± 5.8% of the control level. These data confirmed the specificity of the anti-TRPC5 antibody used for immunocytochemistry.
Figure 7.
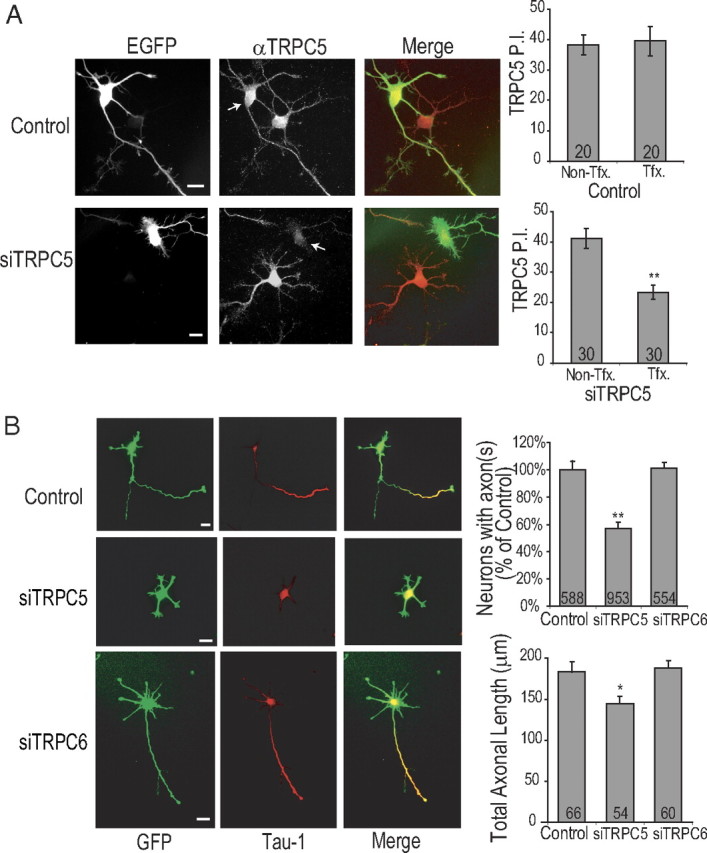
Regulation of axon formation by TRPC5 channels. A, E18 Hippocampal neurons were electroporated before plating with control siRNA or siTRPC5 plus soluble EGFP. Neurons were fixed at 48 h after electroporation and stained with anti-TRPC5 polyclonal antibody and imaged as described in Materials and Methods. Representative images from control siRNA (top) and siTRPC5 (bottom) are shown in left panel and the graphs summarizing quantification (y-axis shows TRPC5 P.I. in arbitrary units) are shown on right. B, E18 neurons were electroporated before plating with control siRNA, siTRPC5, or siTRPC6. Neurons were fixed at 48 h and evaluated for axon formation by staining with anti-Tau-1 antibody (see Materials and Methods). Representative images are shown in left panel(s) and quantification of percentage of polarized neurons (top graph) and length of (bottom graph) in the neurons that did polarize is shown on right. Scale bars: 20 μm (all panels).
Next, we evaluated the effect of siTRPC5 knockdown on axon formation. As shown in Figure 7B, transfection with siTRPC5 before plating revealed a significant reduction in the percentage of polarized neurons (siTRPC5, 57.2 ± 4%, compared with control set at 100%). siTRPC5 also produced a small but significant decrease in axonal length of the remaining neurons that polarized in the presence of siTRPC5 (Fig. 7B, right bottom graph). A recent report indicates that TRPC6 channels are involved in synapse formation in mature (DIV 21) hippocampal neurons (Zhou et al., 2008). TRPC6 channel immunoreactivity peaks around P14 in hippocampus and is not detectable in E17 brain (Zhou et al., 2008) or DIV 1 hippocampal neurons (data not shown), so it is not likely to play a major role in axonogenesis. Indeed, siRNA to TRPC6, which was effective against TRPC6 but not TRPC5 in transfected COS7 cells (supplemental Fig. 1E, available at www.jneurosci.org as supplemental material), had no significant effect on axon formation under our conditions (Fig. 7B).
Endogenous CaMKIγ and TRPC5 reside in lipid rafts
CaMKIγ has a unique C-terminal extension containing multiple cysteines, not present in other CaMKI isoforms, that undergo prenylation and palmitoylation to target the γ-isoform to plasma membranes, Golgi and sarcoplasmic reticulum compartments (Takemoto-Kimura et al., 2003, 2007). Of the various TRPC channel members, TRPC1, TRPC3, and TRPC4 have recently been reported to localize within detergent-insoluble, lipid raft compartment in various non-neuronal cell lines (Remillard and Yuan, 2006). This compartmentalization modulates the functional properties of TRPC1. TRPC1 is a store-operated channel when in lipid rafts and a receptor-operated channel in nonraft plasma membranes (Alfonso et al., 2008; Alicia et al., 2008). These functional differences are likely attributable to selective protein–protein interactions that are compartment specific. However, to date, there are very little data on the submembrane distribution of endogenous TRPC5 in developing brain.
We postulated that endogenous CaMKIγ and TRPC5 channels may colocalize in a membrane microdomain to generate efficient coupling between calcium influx, activation of kinase, and highly localized downstream signaling to promote axonogenesis. Fractionation of P0 rat forebrains and Western blotting revealed endogenous CaMKK and CaMKIγ were identified both in the lipid raft fraction 4 (marked by Flotillin-1) and nonraft membrane fraction 12 (marked by TfR) (Fig. 8A). The other isoforms, which are soluble, were not detectable in either fraction 4 or 12. Moreover, we found strong enrichment of TRPC5 in the lipid raft fraction (fraction 4), compared with the nonraft membrane fraction (fraction 12). In striking contrast with this result, TRPC6 is located almost completely in fraction 12. These are the first data to reveal distinct compartmentalization of TRPC5- and TRPC6-containing TRPC channels, which may allow distinct signaling of the different channel subtypes within different microdomains of the neurons. We performed immunocytochemistry to determine whether TRPC5 colocalized with the neuronal lipid raft marker, Flotillin-1. In cultured hippocampal neurons, TRPC5 (pseudocolored red) partially colocalizes with Flotillin-1 (pseudocolored green) particularly in the central region of the growth cone, with lesser colocalization along the length of the neurite. We also evaluated colocalization of EGFP-CaMKIγ with the well established lipid raft marker GM1 as detected with CTxB. CTxB selectively binds the GM1 and subsequently clusters GM1, thus permitting live labeling of the lipid raft marker (Harder et al., 1998; Nichols et al., 2001; Hullin-Matsuda and Kobayashi, 2007). We observed significant colocalization between EGFP-CaMKIγ and the raft marker, GM1, particularly in the axonal growth cone (Fig. 8C, arrows). We also tested whether the GM1-containing lipid rafts colocalized with the raft marker, Flotillin-1. We found overlap of Flotillin-1 and CTxB throughout the cell and particularly in and the axonal growth cone (arrow) of the nascent axon in the stage 2+ and stage 3 hippocampal neurons (supplemental Fig. 1F, available at www.jneurosci.org as supplemental material). Since CaMKIγ and TRPC5 biochemically cofractionate in the low-density raft fraction, we also confirmed their colocalization in cultured hippocampal neurons. Since we do not have an antibody specific for detecting endogenous CaMKIγ by immunocytochemistry, we electroporated hippocampal neurons with EGFP-CaMKIγ, before plating, and fixed at 24 h, followed by immunostaining with anti-TRPC5 antibody and anti-β-tubulin antibodies. We observed prominent colocalization of EGFP-CaMKIγ and endogenous TRPC5 in growth cones of stage 2 neurons as well as in the axonal growth cone of stage 3 neurons (Fig. 8D; insets show growth cone). These data confirm the biochemical fractionation results, suggesting CaMKIγ and TRPC5 may reside in close proximity in the submembrane, lipid raft domain.
Figure 8.

CaMKIγ and TRPC5 colocalize with lipid raft markers in rat brain and cultured hippocampal neurons. A, Endogenous CaMKK, CaMKIγ, and TRPC5 are present in the lipid raft fraction (fractions 4) as confirmed by enrichment of the raft marker Flotillin-1 in fraction 4 of a discontinuous sucrose density gradient (fractions 1–4, 5% sucrose; fractions 5–11, 35% sucrose; fraction 12, 50% sucrose) from P0 rat forebrains. The input is the crude homogenate before any centrifugation steps separating cytosol and membranes. B, Immunofluorescent staining of endogenous TRPC5 (pseudocolored red) and endogenous Flotillin-1 (pseudocolored green) in axonal growth cone of E18 hippocampal neuron. Scale bar: 5 μm. C, Colocalization of Alexa Fluor 546-CTxB labeled raft marker GM1 (as described in Materials and Methods) and EGFP-CaMKIγ in DIV 1 hippocampal neurons. Scale bar: D, 20 μm. Immunofluorescent staining of endogenous β-tubulin, TRPC5 with electroporated EGFP-CaMKIγ in stage 2 (top row) and stage 3 (bottom row) neurons shows extensive colocalization of TRPC5 and EGFP-CaMKIγ. In final column, the images are a merge of all three channels, with β-tubulin pseudocolored blue, EGFP-CaMKIγ pseudocolored green, and endogenous TRPC5 pseudocolored red in stage 2 and stage 3 hippocampal neurons. The insets show additional magnification of largest growth cones. Scale bars: β-tubulin (bottom) panel, 10 μm; EGFP-CaMKIγ (bottom inset showing growth cone), 5 μm. E, Quantification of neurons with Tau-1-positive axons expressing wild-type CaMKIγ, CaMKIγDPP (mutant lacking prenylation and palmitoylation sites), or CaMKIα fixed at 24 h to evaluate acceleration of axonogenesis. F, Quantification of Tau-1-positive axons in neurons expressing wild-type CaMKIγ alone or wild-type CaMKIγ plus siTRPC5 at 24 h. Note, in D and E, neurons are fixed at 24 h to test mutant CaMKIγ and siTRPC5 in acceleration of axon formation assay. G, Quantification of rescue experiment from neurons expressing control, siTRPC5, or siTRPC5 plus rescue with caCaMKIγ.
Localization of transfected EGFP-CaMKIγ in lipid raft fractions in cultured cortical neurons was dependent on the prenylation/palmitoylation of its unique C terminus—mutational deletion of these lipid modification sites ablated the incorporation of transfected CaMKIγ into lipid rafts and its ability to enhance dendritic outgrowth (Takemoto-Kimura et al., 2007). If the lipid modification-mediated targeting of CaMKIγ to plasma membrane and submembrane compartments such as lipid rafts, is important for its role in axon formation, deletion of these lipid modification sites should likewise obviate the ability of transfected CaMKIγ to accelerate axonogenesis (Fig. 5E,F). Therefore, we truncated CaMKIγ at residue M407 to remove its unique C terminus containing the prenylation/palmitoylation sites (CaMKIγΔPP) (supplemental Fig. 1G, available at www.jneurosci.org as supplemental material). When hippocampal neurons were transfected with CaMKIγΔPP, there was no increase in the formation of axons in contrast to wild-type CaMKIγ (Fig. 8E). This result leads to the conclusion that membrane localization of CaMKIγ, possibly in the lipid raft fraction, is important for axon formation.
Wild-type CaMKIγ requires binding of Ca2+/CaM for full activation. Therefore, we also tested the ability of wild-type CaMKIγ to accelerate axon formation with reduced calcium contribution of TRPC5-containing channels in activation of CaMKIγ, by cotransfecting siTRPC5 with CaMKIγ and fixing neurons at 24 h. As shown in Figure 8F, reduction of TRPC5 levels by siRNA significantly reduced the ability of CaMKIγ to accelerate axon formation, suggesting that the calcium influx from TRPC5 is important for activation of CaMKIγ during this process. In contrast, a caCaMKIγ construct with a mutant CaM binding domain that renders its catalytic activity independent of Ca2+/CaM binding was able to rescue the suppression of axon formation by siTRPC5 (Fig. 8G). The simplest interpretation of these data is that the calcium influx mediated by TRPC5 channels in developing hippocampal neurons is required for full activation of CaMKIγ and its subsequent acceleration of axon formation. Since CaMKIγ and TRPC5 seem to biochemically codistribute and colocalize to detergent-resistant submembrane (lipid raft) compartment, together they may efficiently control signaling dynamics in a highly restricted way to facilitate axon formation.
Discussion
Despite extensive evidence for a the role of calcium signaling in axonal outgrowth and guidance in cultured neurons, relatively little is known about signaling downstream of calcium during the initial steps of neuronal polarization defined by formation of the axon. In the current study, we demonstrate that activation of CaMKK and its downstream target CaMKIγ promote axon formation in cultured hippocampal neurons. Treatment with the CaMKK pharmacological blocker, STO-609, and suppression of CaMKK or CaMKI by expression of dominant-negative constructs and shRNA resulted in 40–50% reduction in the number of embryonic hippocampal neurons that elaborated a morphologically and biochemically defined axon. Moreover, modest overexpression of CaMKIγ resulted in pronounced acceleration of axon formation.
CaMKIγ appears to be involved in several aspects of neuronal development as we previously reported a role for CaMKIγ in activity-dependent dendritic arborization of hippocampal neurons (Wayman et al., 2006). This conclusion was extended to demonstrate a role CaMKIγ in BDNF-stimulated dendritic growth of cortical neurons (Takemoto-Kimura et al., 2007). Takemoto-Kimura et al. did not observe any effect of CaMKIγ knockdown by shRNA on axonal length in high-density cortical neurons and they did not measure the percentage of polarized stage 2/3 neurons. Measurement of neuronal polarity and axonal length of individual neurons would likely be challenging in these high-density cultures because of the extent of their axonal arborization. We used the low-density E18 hippocampal (“glial sandwich”) culture system that is optimal for investigation of neuronal polarity (Craig and Banker, 1994; Bradke and Dotti, 2000; Kaech and Banker, 2006). This allowed us to measure the percentage of polarized neurons as well as measure their axonal length with a high degree of confidence because of the clear separation of neurons. Furthermore, our conclusion that CaMKIγ is involved in axon formation is based on multiple, independent techniques.
We also document a novel role for TRPC channels during axon formation. In our experiments, incubation with the TRPC inhibitor SKF96365 blocked activation of CaMKIγ and strongly suppressed axon formation—this latter effect was rescued by caCaMKI. As we (Fig. 8D) and others (Greka et al., 2003) demonstrate, TRPC5 channels are expressed in the cell body and axonal growth cones of stage 2 and stage 3 cultured hippocampal neurons. These results suggest that under basal conditions TRPC5 channels, which can exhibit significant spontaneous activity (Yamada et al., 2000), may mediate calcium influx that activates CaMKK and subsequently CaMKIγ. Furthermore, micromolar La3+, known to inhibit most Ca2+-permeable channels including TRPC channels except for TRPC4 and 5, strongly stimulated axon formation (Fig. 6B,C). It has been previously shown that short-term neurite outgrowth (1–3 h duration) was stimulated by 10 μm La3+, and this effect required extracellular calcium (Mattson and Kater, 1987). Last, expression of dominant-negative N-terminal fragment of TRPC5, which disrupts functional channel assembly at plasma membrane, as well siRNA suppression of TRPC5, but not TRPC6, before plating caused a decrease (∼42%) in the number of neurons that formed an axon and reduced axonal length in the remaining neurons. Recently, Greka et al. (2003) showed that transfection of a dominant-negative TRPC5 (dnTRPC5-EGFP) construct at DIV 1 resulted in increased neurite and filopodial length (Greka et al., 2003). It is unclear whether Greka et al. evaluated neuronal polarity or measured axonal (vs neurite) length in stage 2/3 hippocampal neurons. There may be differences in the signaling responses to TRPC5-mediated localized calcium influx on the outgrowth of neurites versus the axon in stage 2 or stage 3 neurons. For instance, in NG108 neuroblastoma cells and primary cerebellar granule neurons, early stages of neurite initiation and differentiation require calcium influx via TRPC5 channels; however, after differentiation, both cell types exhibited a sharp decline in TRPC5 levels (Wu et al., 2007). Our data using multiple approaches implicate a role for TRPC5 channels in the development of axons in low-density cultured hippocampal neurons.
We also show, by biochemical fractionation, the colocalization of CaMKK, CaMKIγ, and TRPC5 in the low-density lipid raft fraction, identified the specific marker Flotillin-1. These biochemical data were supported by immunostaining for endogenous TRPC5 and Flotillin-1 in DIV 1 hippocampal neurons. Another lipid raft marker, GM1, also colocalized with EGFP-CaMKIγ in these neurons. Targeting of signaling molecules to lipid rafts results in microdomains that serve to enrich and spatially restrict the signaling from membrane receptors/ion channels to the downstream effectors, thereby creating localized signal amplification. Various studies indicate that the segregation of signaling molecules to rafts may be an important mechanism during axon guidance and growth (Kamiguchi, 2006). TRPC5 channels in lipid rafts may mediate spatially restricted calcium entry into the growth cone to locally activate CaMKK/CaMKIγ during axon formation. Consistent with this model, transfection with the CaMKIγ truncation mutant that lacked the lipid modification sites necessary for its plasma membrane and lipid raft targeting, did not accelerate axon formation similar to wild-type CaMKIγ.
Recently, Bezzerides et al. (2004) demonstrated that treatment of DIV 1 hippocampal neurons with NGF or BDNF significantly increased the number of functionally available surface TRPC5 channels by membrane insertion from an intracellular vesicular pool. This increase in TRPC5 surface expression was dependent on PI3 kinase and Rac1 activity. In addition, Wu et al. (2007) showed that integrin receptor-mediated calcium influx via activation of TRPC5 channels was required for differentiation of NG108 cells and cerebellar granule neurons. In this case, TRPC5 channel activation and subsequent neurite outgrowth were also dependent on phospholipase Cγ (PLCγ) and PI3 kinase signaling. Neurotrophins such as BDNF and NGF, and guidance factors such as Netrin-1 that are coupled with PLCγ, PI3 kinase, and Rac1 signaling, are well established modulators of neuronal polarization in vivo and in vitro (Estrach et al., 2002; Da Silva et al., 2005; Barnes et al., 2007; Shelly et al., 2007). This suggests a model in which asymmetric presentation of axonal guidance or growth cues, such as BDNF or Netrin-1, may increase the activity of existing surface TRPC5 channels via PLCγ/DAG (diacylglycerol) (Beech, 2007) and/or also enhance the number of surface TRPC5 channels by membrane insertion (Bezzerides et al., 2004). In cultured neuron model systems, because of the presumed uniform extracellular milieu, the symmetry breaking event that is required for neuronal polarization is thought to occur stochastically (Andersen and Bi, 2000; Fivaz et al., 2008). Therefore, in cultured hippocampal neurons, TRPC5 channel activation in the growth cone of one neurite may be stochastically enhanced by increased surface expression or by higher spontaneous channel activity. The subsequent increase in spatially restricted intracellular calcium and activation of CaMKK/CaMKIγ may alter cytoskeletal or other signaling dynamics favoring accelerated growth of this neurite and eventual axon formation.
Because of the lack of a phosphospecific antibody that only detects the CaMKK-phosphorylated γ-isoform of CaMKI, we were unable to demonstrate enrichment of phospho-CaMKIγ in a single neurite of a fixed stage 2 neuron with the assumption that this neurite is the presumptive future axon. This conclusion has been made for several other signaling proteins that are putative “specificity determinants.” However, interpretation of such observations in fixed stage 2 cells is questionable. There are only two examples of live-cell imaging of such proteins in hippocampal neurons during stage 2 and their subsequent progression into stage 3—Kif5c and shootin1. For both, Kif5C (Jacobson et al., 2006) and shootin1 (Toriyama et al., 2006), it is clear that these “specificity proteins” do not stably localize in one neurite but show fluctuating accumulation in multiple stage 2 neurites before eventually stably associating with the axon in stage 3 (see movie S6, www.cell.com/neuron/supplemental/S0896-6273(06)00095-X, and video 2 in http://jcb.rupress.org/cgi/content/full/jcb.200604160/DC1). Thus, based on these findings, we conclude that that demonstrating selective enrichment of a signaling protein in one neurite of a fixed stage 2 neuron is not a definitive or predictive measurement of the future axon. Only repetitive imaging beginning at stage 2 and transitioning through stage 3 can identify which neurite will become the axon. However, the development of the necessary tools and technology to image activation of the CaMKK/KI cascade goes beyond the scope of this paper.
There are multiple potential downstream targets of CaMKI that might regulate axon formation. One possibility is the small G-protein Rac1. Activation of Rac1 may be critical for the localized suppression of RhoA and for activation of Pak1, both signaling pathways previously shown to be important for determination of neuronal polarity (Nishimura et al., 2005; Mertens et al., 2006; Watabe-Uchida et al., 2006; de Curtis, 2008). It was recently proposed that CaMKIγ may regulate Rac1 as shRNA knockdown of CaMKIγ reduced GTP loading of Rac1 in 3-d-old cortical neurons (Takemoto-Kimura et al., 2007). The Rac1-guanine exchange factor (GEF) βPIX can be activated by CaMKK/CaMKI signaling (Saneyoshi et al., 2008), and Tiam1 and Stef, other Rac1 GEFs, are also potential mediators (Takemoto-Kimura et al., 2007). Therefore, it is possible that increased localized activation of CaMKIγ because of calcium influx via the TRPC5 channel will allow for RacGEF-mediated modulation of Rac1 signaling in the growth cone of one neurite that is essential for elongation of this neurite. Microtubules and local microtubule stabilization in one neurite are suggested to be a physiological signal that specifies initial neuronal polarity (Witte et al., 2008). Microtubule polymerization is weakened by sequestration of tubulin because of binding of depolymerizing factors such as stathmin family members. Interaction of stathmin with tubulin is suppressed by phosphorylation of Ser16 by CaMKIV, thereby favoring microtubule polymerization (Melander Gradin et al., 1997). In cotransfected HEK293 cells or in vitro with purified proteins, CaMKI can also phosphorylate Ser16 (M. A. Davare and G. A. Wayman, unpublished observation), so CaMKIγ may regulate microtubule assembly by this mechanism.
Over the past few years, significant effort has led to the identification of multiple signaling molecules, including PI3 kinase, the Par3/Par6/aPKC complex, CRMP2 and LKB1 kinases, which regulate axon specification (Yoshimura et al., 2006; Barnes et al., 2007; Shelly et al., 2007). Our study is one of the first to demonstrate a calcium-dependent signaling pathway that directly affects axon formation in cultured embryonic hippocampal neurons. It will be of interest to understand whether and how the CaMKK cascade impinges on or has positive cross talk with any of the previously described pathways regulating axon formation. Our future effort will be directed at determining the downstream targets and signaling pathways modulated by CaMKγ that are responsible for the acceleration of axon formation.
Footnotes
This study was supported by National Institutes of Health Grants GM41292 (T.R.S.) and NS17112 (G.B.). We thank Dr. Naohito Nozaki for CaMKI antibodies and Drs. David Clapham and Katsuhiko Mikoshiba for TRPC constructs.
References
- Alfonso S, Benito O, Alicia S, Angélica Z, Patricia G, Diana K, Vaca L, Luis V. Regulation of the cellular localization and function of human transient receptor potential channel 1 by other members of the TRPC family. Cell Calcium. 2008;43:375–387. doi: 10.1016/j.ceca.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Alicia S, Angélica Z, Carlos S, Alfonso S, Vaca L. STIM1 converts TRPC1 from a receptor-operated to a store-operated channel: moving TRPC1 in and out of lipid rafts. Cell Calcium. 2008;44:479–491. doi: 10.1016/j.ceca.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Ambudkar IS. Ca2+ signaling microdomains: platforms for the assembly and regulation of TRPC channels. Trends Pharmacol Sci. 2006;27:25–32. doi: 10.1016/j.tips.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Ambudkar IS, Ong HL. Organization and function of TRPC channelosomes. Pflugers Arch. 2007;455:187–200. doi: 10.1007/s00424-007-0252-0. [DOI] [PubMed] [Google Scholar]
- Ambudkar IS, Bandyopadhyay BC, Liu X, Lockwich TP, Paria B, Ong HL. Functional organization of TRPC-Ca2+ channels and regulation of calcium microdomains. Cell Calcium. 2006;40:495–504. doi: 10.1016/j.ceca.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Andersen SS, Bi GQ. Axon formation: a molecular model for the generation of neuronal polarity. Bioessays. 2000;22:172–179. doi: 10.1002/(SICI)1521-1878(200002)22:2<172::AID-BIES8>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Anderson ME, Braun AP, Wu Y, Lu T, Wu Y, Schulman H, Sung RJ. KN-93, an inhibitor of multifunctional Ca++/calmodulin-dependent protein kinase, decreases early afterdepolarizations in rabbit heart. J Pharmacol Exp Ther. 1998;287:996–1006. [PubMed] [Google Scholar]
- Barnes AP, Lilley BN, Pan YA, Plummer LJ, Powell AW, Raines AN, Sanes JR, Polleux F. LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell. 2007;129:549–563. doi: 10.1016/j.cell.2007.03.025. [DOI] [PubMed] [Google Scholar]
- Beech DJ. Canonical transient receptor potential 5. Handb Exp Pharmacol. 2007:109–123. doi: 10.1007/978-3-540-34891-7_6. [DOI] [PubMed] [Google Scholar]
- Beedle AM, Hamid J, Zamponi GW. Inhibition of transiently expressed low- and high-voltage-activated calcium channels by trivalent metal cations. J Membr Biol. 2002;187:225–238. doi: 10.1007/s00232-001-0166-2. [DOI] [PubMed] [Google Scholar]
- Bentley D, Guthrie PB, Kater SB. Calcium ion distribution in nascent pioneer axons and coupled preaxonogenesis neurons in situ. J Neurosci. 1991;11:1300–1308. doi: 10.1523/JNEUROSCI.11-05-01300.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzerides VJ, Ramsey IS, Kotecha S, Greka A, Clapham DE. Rapid vesicular translocation and insertion of TRP channels. Nat Cell Biol. 2004;6:709–720. doi: 10.1038/ncb1150. [DOI] [PubMed] [Google Scholar]
- Bradke F, Dotti CG. Establishment of neuronal polarity: lessons from cultured hippocampal neurons. Curr Opin Neurobiol. 2000;10:574–581. doi: 10.1016/s0959-4388(00)00124-0. [DOI] [PubMed] [Google Scholar]
- Brickey DA, Bann JG, Fong YL, Perrino L, Brennan RG, Soderling TR. Mutational analysis of the autoinhibitory domain of calmodulin kinase II. J Biol Chem. 1994;269:29047–29054. [PubMed] [Google Scholar]
- Chang BH, Mukherji S, Soderling TR. Characterization of a calmodulin kinase II inhibitor protein in brain. Proc Natl Acad Sci U S A. 1998;95:10890–10895. doi: 10.1073/pnas.95.18.10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung YH, Sun Ahn H, Kim D, Hoon Shin D, Su Kim S, Yong Kim K, Bok Lee W, Ik Cha C. Immunohistochemical study on the distribution of TRPC channels in the rat hippocampus. Brain Res. 2006;1085:132–137. doi: 10.1016/j.brainres.2006.02.087. [DOI] [PubMed] [Google Scholar]
- Clapham DE, Runnels LW, Strübing C. The TRP ion channel family. Nat Rev Neurosci. 2001;2:387–396. doi: 10.1038/35077544. [DOI] [PubMed] [Google Scholar]
- Craig AM, Banker G. Neuronal polarity. Annu Rev Neurosci. 1994;17:267–310. doi: 10.1146/annurev.ne.17.030194.001411. [DOI] [PubMed] [Google Scholar]
- Da Silva JS, Hasegawa T, Miyagi T, Dotti CG, Abad-Rodriguez J. Asymmetric membrane ganglioside sialidase activity specifies axonal fate. Nat Neurosci. 2005;8:606–615. doi: 10.1038/nn1442. [DOI] [PubMed] [Google Scholar]
- Davenport RW, Kater SB. Local increases in intracellular calcium elicit local filopodial responses in Helisoma neuronal growth cones. Neuron. 1992;9:405–416. doi: 10.1016/0896-6273(92)90179-h. [DOI] [PubMed] [Google Scholar]
- de Curtis I. Functions of Rac GTPases during neuronal development. Dev Neurosci. 2008;30:47–58. doi: 10.1159/000109851. [DOI] [PubMed] [Google Scholar]
- Deitch JS, Banker GA. An electron microscopic analysis of hippocampal neurons developing in culture: early stages in the emergence of polarity. J Neurosci. 1993;13:4301–4315. doi: 10.1523/JNEUROSCI.13-10-04301.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotti CG, Poo MM. Neuronal polarization: building fences for molecular segregation. Nat Cell Biol. 2003;5:591–594. doi: 10.1038/ncb0703-591b. [DOI] [PubMed] [Google Scholar]
- Enslen H, Sun P, Brickey D, Soderling SH, Klamo E, Soderling TR. Characterization of Ca2+/calmodulin-dependent protein kinase IV. Role in transcriptional regulation. J Biol Chem. 1994;269:15520–15527. [PubMed] [Google Scholar]
- Estrach S, Schmidt S, Diriong S, Penna A, Blangy A, Fort P, Debant A. The human Rho-GEF trio and its target GTPase RhoG are involved in the NGF pathway, leading to neurite outgrowth. Curr Biol. 2002;12:307–312. doi: 10.1016/s0960-9822(02)00658-9. [DOI] [PubMed] [Google Scholar]
- Fivaz M, Bandara S, Inoue T, Meyer T. Robust neuronal symmetry breaking by Ras-triggered local positive feedback. Curr Biol. 2008;18:44–50. doi: 10.1016/j.cub.2007.11.051. [DOI] [PubMed] [Google Scholar]
- Fowler MA, Sidiropoulou K, Ozkan ED, Phillips CW, Cooper DC. Corticolimbic expression of TRPC4 and TRPC5 channels in the rodent brain. PLoS One. 2007;2:e573. doi: 10.1371/journal.pone.0000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez T. Neurobiology: channels for pathfinding. Nature. 2005;434:835–838. doi: 10.1038/434835a. [DOI] [PubMed] [Google Scholar]
- Gomez TM, Spitzer NC. Regulation of growth cone behavior by calcium: new dynamics to earlier perspectives. J Neurobiol. 2000;44:174–183. [PubMed] [Google Scholar]
- Gomez TM, Zheng JQ. The molecular basis for calcium-dependent axon pathfinding. Nat Rev Neurosci. 2006;7:115–125. doi: 10.1038/nrn1844. [DOI] [PubMed] [Google Scholar]
- Greka A, Navarro B, Oancea E, Duggan A, Clapham DE. TRPC5 is a regulator of hippocampal neurite length and growth cone morphology. Nat Neurosci. 2003;6:837–845. doi: 10.1038/nn1092. [DOI] [PubMed] [Google Scholar]
- Harder T, Scheiffele P, Verkade P, Simons K. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J Cell Biol. 1998;141:929–942. doi: 10.1083/jcb.141.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hullin-Matsuda F, Kobayashi T. Monitoring the distribution and dynamics of signaling microdomains in living cells with lipid-specific probes. Cell Mol Life Sci. 2007;64:2492–2504. doi: 10.1007/s00018-007-7281-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson C, Schnapp B, Banker GA. A change in the selective translocation of the Kinesin-1 motor domain marks the initial specification of the axon. Neuron. 2006;49:797–804. doi: 10.1016/j.neuron.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Jiang H, Rao Y. Axon formation: fate versus growth. Nat Neurosci. 2005;8:544–546. doi: 10.1038/nn0505-544. [DOI] [PubMed] [Google Scholar]
- Jin M, Guan CB, Jiang YA, Chen G, Zhao CT, Cui K, Song YQ, Wu CP, Poo MM, Yuan XB. Ca2+-dependent regulation of rho GTPases triggers turning of nerve growth cones. J Neurosci. 2005;25:2338–2347. doi: 10.1523/JNEUROSCI.4889-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Mühle A, Schaefer M, Strotmann R, Schultz G, Plant TD. Lanthanides potentiate TRPC5 currents by an action at extracellular sites close to the pore mouth. J Biol Chem. 2003;278:3562–3571. doi: 10.1074/jbc.M211484200. [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kamata A, Sakagami H, Tokumitsu H, Owada Y, Fukunaga K, Kondo H. Spatiotemporal expression of four isoforms of Ca2+/calmodulin-dependent protein kinase I in brain and its possible roles in hippocampal dendritic growth. Neurosci Res. 2007a;57:86–97. doi: 10.1016/j.neures.2006.09.013. [DOI] [PubMed] [Google Scholar]
- Kamata A, Sakagami H, Tokumitsu H, Sanda M, Owada Y, Fukunaga K, Kondo H. Distinct developmental expression of two isoforms of Ca2+/calmodulin-dependent protein kinase kinases and their involvement in hippocampal dendritic formation. Neurosci Lett. 2007b;423:143–148. doi: 10.1016/j.neulet.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Kamiguchi H. The region-specific activities of lipid rafts during axon growth and guidance. J Neurochem. 2006;98:330–335. doi: 10.1111/j.1471-4159.2006.03888.x. [DOI] [PubMed] [Google Scholar]
- Kuhn TB, Williams CV, Dou P, Kater SB. Laminin directs growth cone navigation via two temporally and functionally distinct calcium signals. J Neurosci. 1998;18:184–194. doi: 10.1523/JNEUROSCI.18-01-00184.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledoux J, Chartier D, Leblanc N. Inhibitors of calmodulin-dependent protein kinase are nonspecific blockers of voltage-dependent K+ channels in vascular myocytes. J Pharmacol Exp Ther. 1999;290:1165–1174. [PubMed] [Google Scholar]
- Li Y, Jia YC, Cui K, Li N, Zheng ZY, Wang YZ, Yuan XB. Essential role of TRPC channels in the guidance of nerve growth cones by brain-derived neurotrophic factor. Nature. 2005;434:894–898. doi: 10.1038/nature03477. [DOI] [PubMed] [Google Scholar]
- Maekawa S, Morii H, Kumanogoh H, Sano M, Naruse Y, Sokawa Y, Mori N. Localization of neuronal growth-associated, microtubule-destabilizing factor SCG10 in brain-derived raft membrane microdomains. J Biochem. 2001;129:691–697. doi: 10.1093/oxfordjournals.jbchem.a002908. [DOI] [PubMed] [Google Scholar]
- Mandell JW, Banker GA. A spatial gradient of tau protein phosphorylation in nascent axons. J Neurosci. 1996;16:5727–5740. doi: 10.1523/JNEUROSCI.16-18-05727.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Calcium as sculptor and destroyer of neural circuitry. Exp Gerontol. 1992;27:29–49. doi: 10.1016/0531-5565(92)90027-w. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Establishment and plasticity of neuronal polarity. J Neurosci Res. 1999;57:577–589. [PubMed] [Google Scholar]
- Mattson MP, Kater SB. Calcium regulation of neurite elongation and growth cone motility. J Neurosci. 1987;7:4034–4043. doi: 10.1523/JNEUROSCI.07-12-04034.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melander Gradin H, Marklund U, Larsson N, Chatila TA, Gullberg M. Regulation of microtubule dynamics by Ca2+/calmodulin-dependent kinase IV/Gr-dependent phosphorylation of oncoprotein 18. Mol Cell Biol. 1997;17:3459–3467. doi: 10.1128/mcb.17.6.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens AE, Pegtel DM, Collard JG. Tiam1 takes PARt in cell polarity. Trends Cell Biol. 2006;16:308–316. doi: 10.1016/j.tcb.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Mochizuki H, Ito T, Hidaka H. Purification and characterization of Ca2+/calmodulin-dependent protein kinase V from rat cerebrum. J Biol Chem. 1993;268:9143–9147. [PubMed] [Google Scholar]
- Nichols BJ, Kenworthy AK, Polishchuk RS, Lodge R, Roberts TH, Hirschberg K, Phair RD, Lippincott-Schwartz J. Rapid cycling of lipid raft markers between the cell surface and Golgi complex. J Cell Biol. 2001;153:529–541. doi: 10.1083/jcb.153.3.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura H, Sakagami H, Uezu A, Fukunaga K, Watanabe M, Kondo H. Cloning, characterization and expression of two alternatively splicing isoforms of Ca2+/calmodulin-dependent protein kinase I gamma in the rat brain. J Neurochem. 2003;85:1216–1227. doi: 10.1046/j.1471-4159.2003.01760.x. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Yamaguchi T, Kato K, Yoshizawa M, Nabeshima Y, Ohno S, Hoshino M, Kaibuchi K. PAR-6-PAR-3 mediates Cdc42-induced Rac activation through the Rac GEFs STEF/Tiam1. Nat Cell Biol. 2005;7:270–277. doi: 10.1038/ncb1227. [DOI] [PubMed] [Google Scholar]
- Okada T, Shimizu S, Wakamori M, Maeda A, Kurosaki T, Takada N, Imoto K, Mori Y. Molecular cloning and functional characterization of a novel receptor-activated TRP Ca2+ channel from mouse brain. J Biol Chem. 1998;273:10279–10287. doi: 10.1074/jbc.273.17.10279. [DOI] [PubMed] [Google Scholar]
- Oliva AA, Jr, Atkins CM, Copenagle L, Banker GA. Activated c-Jun N-terminal kinase is required for axon formation. J Neurosci. 2006;26:9462–9470. doi: 10.1523/JNEUROSCI.2625-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant TD, Schaefer M. TRPC4 and TRPC5: receptor-operated Ca2+-permeable nonselective cation channels. Cell Calcium. 2003;33:441–450. doi: 10.1016/s0143-4160(03)00055-1. [DOI] [PubMed] [Google Scholar]
- Pravettoni E, Bacci A, Coco S, Forbicini P, Matteoli M, Verderio C. Different localizations and functions of L-type and N-type calcium channels during development of hippocampal neurons. Dev Biol. 2000;227:581–594. doi: 10.1006/dbio.2000.9872. [DOI] [PubMed] [Google Scholar]
- Remillard CV, Yuan JX. Transient receptor potential channels and caveolin-1: good friends in tight spaces. Mol Pharmacol. 2006;70:1151–1154. doi: 10.1124/mol.106.029280. [DOI] [PubMed] [Google Scholar]
- Sakagami H, Kamata A, Nishimura H, Kasahara J, Owada Y, Takeuchi Y, Watanabe M, Fukunaga K, Kondo H. Prominent expression and activity-dependent nuclear translocation of Ca2+/calmodulin-dependent protein kinase Idelta in hippocampal neurons. Eur J Neurosci. 2005;22:2697–2707. doi: 10.1111/j.1460-9568.2005.04463.x. [DOI] [PubMed] [Google Scholar]
- Saneyoshi T, Wayman G, Fortin D, Davare M, Hoshi N, Nozaki N, Natsume T, Soderling TR. Activity-dependent synaptogenesis: regulation by a CaM-kinase kinase/CaM-kinase I/betaPIX signaling complex. Neuron. 2008;57:94–107. doi: 10.1016/j.neuron.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindl R, Frischauf I, Kahr H, Fritsch R, Krenn M, Derndl A, Vales E, Muik M, Derler I, Groschner K, Romanin C. The first ankyrin-like repeat is the minimum indispensable key structure for functional assembly of homo- and heteromeric TRPC4/TRPC5 channels. Cell Calcium. 2008;43:260–269. doi: 10.1016/j.ceca.2007.05.015. [DOI] [PubMed] [Google Scholar]
- Shelly M, Cancedda L, Heilshorn S, Sumbre G, Poo MM. LKB1/STRAD promotes axon initiation during neuronal polarization. Cell. 2007;129:565–577. doi: 10.1016/j.cell.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Shim S, Goh EL, Ge S, Sailor K, Yuan JP, Roderick HL, Bootman MD, Worley PF, Song H, Ming GL. XTRPC1-dependent chemotropic guidance of neuronal growth cones. Nat Neurosci. 2005;8:730–735. doi: 10.1038/nn1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderling TR. CaM-kinases: modulators of synaptic plasticity. Curr Opin Neurobiol. 2000;10:375–380. doi: 10.1016/s0959-4388(00)00090-8. [DOI] [PubMed] [Google Scholar]
- Spitzer NC, Lautermilch NJ, Smith RD, Gomez TM. Coding of neuronal differentiation by calcium transients. Bioessays. 2000;22:811–817. doi: 10.1002/1521-1878(200009)22:9<811::AID-BIES6>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Strübing C, Krapivinsky G, Krapivinsky L, Clapham DE. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron. 2001;29:645–655. doi: 10.1016/s0896-6273(01)00240-9. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Numakawa T, Shimazu K, Koshimizu H, Hara T, Hatanaka H, Mei L, Lu B, Kojima M. BDNF-induced recruitment of TrkB receptor into neuronal lipid rafts: roles in synaptic modulation. J Cell Biol. 2004;167:1205–1215. doi: 10.1083/jcb.200404106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto-Kimura S, Terai H, Takamoto M, Ohmae S, Kikumura S, Segi E, Arakawa Y, Furuyashiki T, Narumiya S, Bito H. Molecular cloning and characterization of CLICK-III/CaMKIgamma, a novel membrane-anchored neuronal Ca2+/calmodulin-dependent protein kinase (CaMK) J Biol Chem. 2003;278:18597–18605. doi: 10.1074/jbc.M300578200. [DOI] [PubMed] [Google Scholar]
- Takemoto-Kimura S, Ageta-Ishihara N, Nonaka M, Adachi-Morishima A, Mano T, Okamura M, Fujii H, Fuse T, Hoshino M, Suzuki S, Kojima M, Mishina M, Okuno H, Bito H. Regulation of dendritogenesis via a lipid-raft-associated Ca2+/calmodulin-dependent protein kinase CLICK-III/CaMKIgamma. Neuron. 2007;54:755–770. doi: 10.1016/j.neuron.2007.05.021. [DOI] [PubMed] [Google Scholar]
- Tokumitsu H, Chijiwa T, Hagiwara M, Mizutani A, Terasawa M, Hidaka H. KN-62, 1-[N, O-bis(5-isoquinolinesulfonyl)-N-methyl-l-tyrosyl]-4-phenylpiperazine, a specific inhibitor of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 1990;265:4315–4320. [PubMed] [Google Scholar]
- Tokumitsu H, Inuzuka H, Ishikawa Y, Ikeda M, Saji I, Kobayashi R. STO-609, a specific inhibitor of the Ca2+/calmodulin-dependent protein kinase kinase. J Biol Chem. 2002;277:15813–15818. doi: 10.1074/jbc.M201075200. [DOI] [PubMed] [Google Scholar]
- Tokumitsu H, Inuzuka H, Ishikawa Y, Kobayashi R. A single amino acid difference between alpha and beta Ca2+/calmodulin-dependent protein kinase kinase dictates sensitivity to the specific inhibitor, STO-609. J Biol Chem. 2003;278:10908–10913. doi: 10.1074/jbc.M213183200. [DOI] [PubMed] [Google Scholar]
- Toriyama M, Shimada T, Kim KB, Mitsuba M, Nomura E, Katsuta K, Sakumura Y, Roepstorff P, Inagaki N. Shootin1: a protein involved in the organization of an asymmetric signal for neuronal polarization. J Cell Biol. 2006;175:147–157. doi: 10.1083/jcb.200604160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GX, Poo MM. Requirement of TRPC channels in netrin-1-induced chemotropic turning of nerve growth cones. Nature. 2005;434:898–904. doi: 10.1038/nature03478. [DOI] [PubMed] [Google Scholar]
- Watabe-Uchida M, John KA, Janas JA, Newey SE, Van Aelst L. The Rac activator DOCK7 regulates neuronal polarity through local phosphorylation of stathmin/Op18. Neuron. 2006;51:727–739. doi: 10.1016/j.neuron.2006.07.020. [DOI] [PubMed] [Google Scholar]
- Wayman GA, Kaech S, Grant WF, Davare M, Impey S, Tokumitsu H, Nozaki N, Banker G, Soderling TR. Regulation of axonal extension and growth cone motility by calmodulin-dependent protein kinase I. J Neurosci. 2004;24:3786–3794. doi: 10.1523/JNEUROSCI.3294-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Impey S, Marks D, Saneyoshi T, Grant WF, Derkach V, Soderling TR. Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron. 2006;50:897–909. doi: 10.1016/j.neuron.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Wayman GA, Lee YS, Tokumitsu H, Silva A, Soderling TR. Calmodulin-kinases: modulators of neuronal development and plasticity. Neuron. 2008;59:914–931. doi: 10.1016/j.neuron.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z, Guirland C, Ming GL, Zheng JQ. A CaMKII/calcineurin switch controls the direction of Ca2+-dependent growth cone guidance. Neuron. 2004;43:835–846. doi: 10.1016/j.neuron.2004.08.037. [DOI] [PubMed] [Google Scholar]
- Winckler B. BDNF instructs the kinase LKB1 to grow an axon. Cell. 2007;129:459–460. doi: 10.1016/j.cell.2007.04.021. [DOI] [PubMed] [Google Scholar]
- Witte H, Neukirchen D, Bradke F. Microtubule stabilization specifies initial neuronal polarization. J Cell Biol. 2008;180:619–632. doi: 10.1083/jcb.200707042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Lu ZH, Obukhov AG, Nowycky MC, Ledeen RW. Induction of calcium influx through TRPC5 channels by cross-linking of GM1 ganglioside associated with alpha5beta1 integrin initiates neurite outgrowth. J Neurosci. 2007;27:7447–7458. doi: 10.1523/JNEUROSCI.4266-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada H, Wakamori M, Hara Y, Takahashi Y, Konishi K, Imoto K, Mori Y. Spontaneous single-channel activity of neuronal TRP5 channel recombinantly expressed in HEK293 cells. Neurosci Lett. 2000;285:111–114. doi: 10.1016/s0304-3940(00)01033-8. [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Arimura N, Kaibuchi K. Signaling networks in neuronal polarization. J Neurosci. 2006;26:10626–10630. doi: 10.1523/JNEUROSCI.3824-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng JQ, Poo MM. Calcium signaling in neuronal motility. Annu Rev Cell Dev Biol. 2007;23:375–404. doi: 10.1146/annurev.cellbio.23.090506.123221. [DOI] [PubMed] [Google Scholar]
- Zheng JQ, Felder M, Connor JA, Poo MM. Turning of nerve growth cones induced by neurotransmitters. Nature. 1994;368:140–144. doi: 10.1038/368140a0. [DOI] [PubMed] [Google Scholar]
- Zhou J, Du W, Zhou K, Tai Y, Yao H, Jia Y, Ding Y, Wang Y. Critical role of TRPC6 channels in the formation of excitatory synapses. Nat Neurosci. 2008;11:741–743. doi: 10.1038/nn.2127. [DOI] [PubMed] [Google Scholar]