

Abstract
We have furthered our understanding of the separative mechanism of a novel CE approach, termed ion-interaction CZE (II-CZE), developed in our laboratory for the resolution of mixtures of cationic peptides. Thus, II-CZE and RP-HPLC were applied to the separation of peptides differing by a single amino acid substitution in 10- and 12-residue synthetic model peptide sequences. Substitutions differed by a wide range of properties or side-chain type (e.g., alkyl side-chains, polar side-chains, etc.) at the substitution site. When carried out in high concentrations (400 mM) of pentafluoropropionic acid (PFPA), II-CZE separated peptides in order of increasing hydrophobicity when the substituted side-chains were of a similar type; when II-CZE was applied to the mixtures of peptides with substitutions of side-chains that differed in the type of functional group, there was no longer a correlation of electrophoretic mobility in II-CZE with relative peptide hydrophobicity, suggesting that a third factor is involved in the separative mechanism beyond charge and hydrophobicity. Interestingly, the hydrophobic PFPA− anion is best for separating peptides that differ in hydrophobicity with hydrophobic side-chains but high concentrations of the hydrophilic H2PO4− anion are best when separating peptides that differ in polar side-chains relative to hydrophobic side-chains. We speculate that differential hydration/dehydration properties of various side-chains in the peptide and the hydration/dehydration properties of the hydrophilic/hydrophobic anions as well as the electrostatic attractions between the peptide and the anions in solution all play a critical role in these solution-based effects.
Keywords: Ion-interaction CZE, Ion-pairing reagents, Reversed-phase HPLC
1 Introduction
CZE is a CE mode based on differences in analyte charge-to-size ratios [1]. Our laboratory [2] and others ([3–7]; see also review [8] and references therein) have previously demonstrated the ability of CE to resolve synthetic peptide diaster-eomers. Specifically, in our case, a series of synthetic 18-residue amphipathic α-helical monomeric peptide analogs was synthesized, where a single site in the center of the hydrophobic face of the α-helix was substituted by 19 L- or D-amino acids [2]. These L- and D-peptide pairs have the same charge-to-mass ratio, amino acid sequence, and intrinsic hydrophobicity, varying only in the stereochemistry of a single residue. CE methods used included CZE (uncoated capillary), micellar EKC (MEKC) (uncoated capillary in the presence of CHAPS), and open-tubular CEC (OT-CEC) (C8-coated capillary in the presence of 25% v/v 2,2,2-tri-fluoroethanol (TFE) or 25% ethanol). Overall, OT-CEC was most effective at separating most of the peptide pairs, particularly for those containing hydrophilic side-chains, while the MEKC method proved most effective for separation of peptide pairs containing hydrophobic or aromatic side-chains at the single substitution site.
We have also previously shown that random-coil peptides (i.e., no secondary structure) varying only subtly in hydrophobicity of alkyl side-chains could be separated using high concentrations (400 mM) of hydrophobic anionic ion-pairing reagents (TFA; pentafluoropropionic acid, PFPA; and heptafluorobutyric acid, HFBA) in uncoated capillaries. The addition of such perfluorinated anionic ion-pairing reagents more commonly employed in (RP-HPLC) mobile phases at low pH [9–18] to the cationic peptides allowed resolution of the peptides through a mechanism based on differences in peptide hydrophobicity [14, 19, 20]. Migration times of the peptides increased with increasing acid concentration and increasing hydrophobicity of the acidic counterion (TFA−< PFPA− <HFBA−). The separations at these high perfluorinated acid concentrations represent a monodimensional separation process based on two unrelated parameters: first, peptides of the same length but of different nominal charge are separated in order of decreasing charge (i.e., +3 peptides migrate the earliest and +1 peptides migrate last) according to a CZE mechanism; and second, peptides within each group of peptides with the same nominal charge are separated in order of increasing hydrophobicity according to an hydrophobically mediated mechanism introduced by the anionic ion-pairing reagent. Thus, we termed this CE method as ion-interaction CZE (II-CZE) [14, 19–21]. In our most recent study, peptides of identical charge-to-mass ratio were resolved using II-CZE [21]. For example, two peptides with a +2 charge (GGVAGLGLGK-amide and GGIGGLGLGK-amide, mass 828) or +3 charge (GGVAKLGLGK-amide and GGIGKLGLGK-amide, mass 899) at pH 2 were separated. Interestingly, the +1 versions of these peptides (Ac-GGVAGLGLGK and Ac-GGIGGLGLGK-amide, mass 870) were not resolved, suggesting that charge density on the peptide is a critical factor in the separative mechanism.
Advantages of II-CZE to gain an understanding of the separative mechanism based on differences in hydrophobicity of the peptide analyte are four-fold: (i) with uncoated capillaries, there is no hydrophobic surface with which hydrophobic ion-pairing reagents can interact; (ii) low pH (pH 2) prevents any ionization of silanols on the silica at the capillary wall; (iii) at high concentrations of the hydrophobic anionic ion-pairing reagents (400 mM ion-pairing reagent), all ionic analytes must be partially paired with counterion and; (iv) the observed separation (e.g., selectivity) must therefore be due to some type of hydrophobic interaction within the BGE. That is, under the conditions of II-CZE it is reasonable to assume that there are no interactions between the analytes to be separated and the capillary wall surface and the separation mechanism would solely involve the BGE. Even so, the separative mechanism can be extremely complex involving a combination of the basic CZE mechanism (charge-to-size), ion-pairing interactions between charged residues in the peptide analyte and the ions in the BGE (these interactions being dependent on pH, acid-base equilibria and concentration of various ions in the BGE) and other physical interactions between the analyte and the BGE that could involve hydrophilic/hydrophobic interactions and/or differential hydration/dehydration of various side-chains in the peptide within the BGE. The investigation of the specificity of the ionic analyte interaction with an aqueous solution (the BGE in CE) in general and with hydrophilic and hydrophobic counterions in particular, is very important not only for the immediate, practical purpose of designing new and more powerful separation methods (as required by genomics, proteomics, metabolomics, etc.) but also for a better and profound overall understanding of the fundamentals of the physicochemical interactions between analyte (peptides/proteins) and solution.
In the present study, we set out to further our understanding of the II-CZE separative mechanism, e.g., to determine whether the migration order during II-CZE peptide separations represents a measure of peptide relative hydrophobicity as measured by RP-HPLC. Thus, we applied this method to the separation of peptides that differed by a single amino acid substitution in 10- and 12-residue synthetic model peptide sequences. We compared our II-CZE method at pH 2 with RP-HPLC at pH 2 in the presence of the same hydrophobic perfluorinated anionic ion-pairing reagent, PFPA. In addition, to assist in elucidation of the mechanism, we also separated the same peptides by II-CZE in 0.8 M phosphoric acid at pH 2, where the counterion to the cationic peptides is hydrophilic (H2PO4−) rather than hydrophobic in the case of PFPA.
2 Materials and methods
2.1 Reagents
TFA, PFPA, HFBA, phosphoric acid, lithium hydroxide, and HPLC-grade ACN were purchased from Sigma–Aldrich (St. Louis, MO, USA). The coating solution, INSTCoating was obtained from Biotaq (Gaithersburg, MD, USA) and used as received. Deionized water was purified by an E-pure water filtration device from Barnstead/Thermolyne (Dubuque, IA, USA).
2.2 Solutions and synthetic peptides
The BGEs were prepared from the corresponding acids at different concentrations adjusted to pH 2 with lithium hydroxide. The peptides were synthesized by standard solid-phase synthesis methodology, purified and characterized as described previously [22].
2.3 RP-HPLC
Analytical RP-HPLC runs were carried out on an Agilent 1100 Liquid Chromatograph System (Agilent Technologies, Little Falls Site, DE, USA) using a Zorbax 300SB-C8 column (150 mm × 1 mm id; 3.5 μm particle size, 30 nm pore size) from Agilent Technologies. Run conditions: linear AB gradient (0% B to 35% B over 140 min for a gradient rate of 0.25% B/min) at the flow rate of 100 μL/min and 25°C, where eluent A is 20 mM aq. PFPA and eluent B is 20 mM PFPA in ACN.
2.4 CE
All CE runs were carried out on a Beckman–Coulter P/ACE MDQ Capillary Electrophoresis System (Fullerton, CA, USA) controlled by 32 Karat software (Version 5.0). Uncoated capillaries, 60.2 cm (50 cm) × 50 μm id and 375 μm od, were provided by Beckman–Coulter. In all cases, capillaries were thermostated at 15°C and used with the shorter rectangular aperture (100 μm in width × 200 μm in length). Samples were introduced into the capillary by pressure injection (0.5 psi [34.5 kPa] for 3–10 s). Peptides were detected at 195 nm by UV absorption with a photodiode detector (DAD). In all cases the applied voltage was 25 kV (normal polarity, e.g., the anode on the injection side) with 10-min voltage ramp; the current was less than 220 μA. The washing protocol consisted of a prerun and postrun wash with BGE for 5 min each at 80 psi (551.6 kPa) pressure. The coating procedure consisted of the addition of two prerun washing steps to the previous washing protocol: (i) (INSTCoating) × 20 psi (137.9 kPa) × 5 min; (ii) (BGE) × 80 psi (551.6 kPa) × 5 min. The next normal washing step (e.g., BGE × 80 psi (551.6 kPa) × 5 min) was performed from a different BGE vial.
3 Results and discussion
3.1 Synthetic model peptides
As noted above, we previously separated a mixture of 27 ten-residue peptides consisting of three groups of nine peptides that differ in positive charge (+1, +2, and +3); within each group of nine peptides of the same charge, there was only a subtle difference in hydrophobicity between adjacent peptide pairs. The separation of the three peptide groups (+1, +2, and +3 group) occurs due to a CZE mechanism (differences in charge-to-size ratio); within each group of peptides with the same nominal charge the peptides are separated in order of increasing hydrophobicity according to a hydrophobically mediated mechanism introduced by the hydrophobic anionic ion-pairing reagent. In the present study, we wanted to determine if this mixed mode effect was solely based on these two parameters or was more complex. We have shown previously that, in RP-HPLC, the relative hydrophobicity of 17 side-chains (Gly, Ala, Cys, Pro, Val, nVal, Leu, nLeu, Ile, Met, Tyr, Phe, Trp, Ser, Thr, Asn, and Gln) at a single substitution site in 10-residue peptides was unaffected by pH (2, 5, and 7), ion-pairing reagents, the presence or absence of salts, or whether C8 or C18 RP columns were used [23]. In contrast, the relative hydrophobicity of potentially charged side-chains (Orn, Lys, His, Arg, Asp, and Glu) was dependent on pH and buffer conditions, including the type of salt or ion-pairing reagent [23]. Thus, if the separation of peptides is different between RP-HPLC and II-CZE, then we would know that other factors are also involved in the II-CZE separative mechanism besides the CZE and the hydrophobically mediated mechanisms.
Two synthetic model peptide series, with single amino acid substitutions in both series of peptides, were now employed: Series 1 has the 10-residue sequence Ac-XLGAK-GAGVG-amide, where position X is substituted by 1 of 19 of the naturally occurring amino acids (Cys being the sole exception) plus ornithine (O), norvaline (nV), and norleucine (nL); Series 2 has the 12-residue sequence Ac-XLGLGGGLGLGK-OH, where position X is substituted by the same amino acids as Series 1; Ac denotes Nα-acetyl, amide denotes Cα-amide, and −OH denotes Cα-carboxyl. Peptides within the two series differ not only in side-chain hydrophilicity/hydrophobicity but also in the type of side-chain at the substitution site: aromatic side-chains (Y, F, W); alkyl side-chains (G, A, V, nV, L, nL); acidic side-chains (D, E); basic side-chains (O, K, H, R); polar side-chains (S, T, N, Q); a sulfur-containing side-chain (M); and a cyclic side-chain (P). Peptides are denoted by the amino acid residue substituted at position X in both Series 1 (Figs. 1, 2, and 3) and Series 2 (Figs. 4, 5, 6, 7, and 8). It should be noted that the presence of O, K, H, or R at the substitution site of either peptide series results in a +2 charge on the peptides (denoted +2 group) compared to the remainder of the peptides with a charge of +1 (denoted +1 group) from the single lysine residue in both peptide series. Finally, statements concerning relative hydrophilicity/hydrophobicity of amino acid side-chains are based on the work of Kovacs et al. [23] who determined the intrinsic hydrophilicity/hydrophobicity of amino acid side-chains in the absence of nearest neighbor or conformational effects. In addition, the RP-HPLC runs shown in Figs. 5–7 also represent relative orders of peptide (and consequently substituted side-chain) hydrophilicity/hydrophobicity.
Figure 1.

Effect of phosphoric acid concentration on CE separation of random coil peptides in an uncoated capillary. Conditions: BGE, various concentrations of phosphoric acid adjusted to pH 2 with LiOH. The peptide sequence is Ac-XLGAKGAGVG-amide, where position X is substituted by amino acids indicated by their one letter code.
Figure 2.
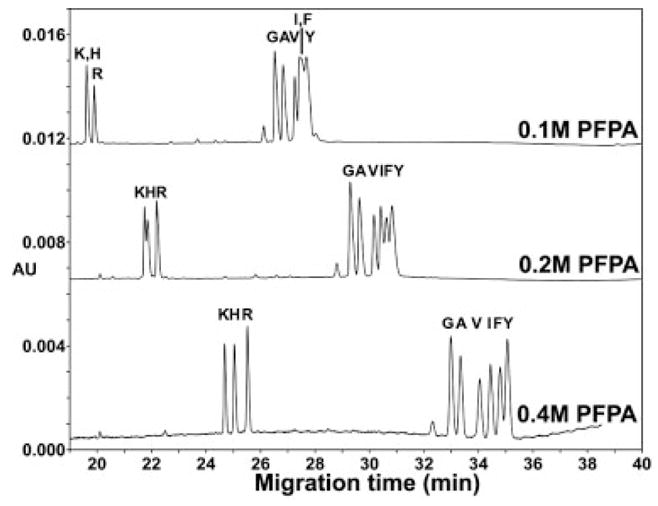
Effect of PFPA concentration on CE separation of random coil peptides in an uncoated capillary. Conditions: same as Fig. 1, except for various concentrations of PFPA in the BGE. Peptide sequence: same as Fig. 1.
Figure 3.

Effect of anionic ion-pairing reagents (H3PO4, TFA, PFPA, and HFBA) on the CE separation of random coil peptides in an uncoated capillary. Conditions: BGE, with approximately the same concentration of salt (obtained by acid adjustment to pH 2 with LiOH). Peptide sequence: same as Fig. 1.
Figure 4.

Effect of capillary coating on run reproducibility. Conditions: BGE, 400 mM PFPA adjusted to pH 2 with LiOH. Coating procedure: prerun wash with INSTCoating × 20 psi (137.9 kPa) × 5 min followed by BGE × 80 psi (551.6 kPa) × 5 min. After each run the capillary is washed with water × 80 psi (551.6 kPa) × 5 min. The peptide sequence is Ac-XLGLGGGLGLGK-OH, where position X is substituted by amino acids indicated by their one-letter code.
Figure 5.

Comparison of CE and RP-HPLC separation of random coil peptides differing by a single substitution of hydrophobic amino acids. CE conditions: capillary coated with INSTCoating solution; BGE, 400 mM PFPA adjusted to pH 2 with LiOH. RP-HPLC conditions: linear AB gradient (0.25% B/min) at the flow rate of 0.1 mL/min on a microbore C8 column (1 mm id × 150 mm) and 25°C, where eluent A is 20 mM aq. PFPA and eluent B is 20 mM PFPA in ACN. Peptide sequence: same as Fig. 4.
Figure 6.

Comparison of CE and RP-HPLC separation of random coil peptides which differ in the polar/charged amino acid at the substitution site. CE conditions: same as Fig. 5. RP-HPLC conditions: same as Fig. 5. Peptide sequence: same as Fig. 4.
Figure 7.

Comparison of CE and RP-HPLC separation of random coil peptides where the peptides differ in the hydrophobicity of the amino acid at the substitution site. CE conditions: same as Fig. 5. RP-HPLC conditions: same as Fig. 5. Peptide sequence: same as Fig. 4.
Figure 8.

CE separation (in phosphoric acid) of random coil peptides where the peptides differ dramatically at substitution site in their side-chain properties. CE conditions: same as Fig. 5, except for 0.8 M H3PO4 in the BGE. Peptide sequence: same as Fig. 4.
3.2 CE of Series 1 peptides (Ac-XLGAKGAGVG-amide)
Figure 1 illustrates the effect of phosphoric acid concentration on the CE separation of a mixture of Series 1 peptides. The pH of the BGE was adjusted to pH 2 with LiOH for all concentrations of phosphoric acid; thus, there is a significant concentration of the anion H2PO4− at each acid concentration. At a low concentration of phosphoric acid (25 mM) the only separation is between the +1 group of peptides (denoted G, A, V, I, F, and Y) and the +2 group of peptides (denoted K, R, and H), but no separation of peptides within each charged group. That is, the group separation is based on a charge difference according to a CZE mechanism (charge-to-size). At a concentration of 100 mM phosphoric acid, a partial separation within the +1 group of peptides is observed. The G/A peptides are partially separated in order of their hydrophobicity, i.e., Gly first followed by the Ala peptide. The more hydrophobic peptides, V/I/F, are well resolved from the G/A peptides but unresolved from each other. The Tyr peptide, which is less hydrophobic than the Phe peptide, nevertheless migrates slower than the latter, suggesting that the mechanism for the separation is more complex than hydrophobicity alone. The +2 group of peptides (K, R, and H) is unresolved under these conditions. At a concentration of 400 mM phosphoric acid, peptides comprising the +2 peptide group are baseline-resolved. Within the +1 group of peptides the G/A peptides comigrate and are well resolved from the more hydrophobic peptides (V/I/F group) which comigrate. Interestingly, the Tyr peptide is now baseline resolved from the Phe peptide, but in the reverse order of their intrinsic hydrophobicity. At a concentration of 800 mM phosphoric acid the resolution of the +2 peptides is greatly improved compared to the separation achieved at 400 mM phosphoric acid. However, the +1 peptides (G, A, V, I, and F) are more poorly resolved at 800 mM phosphoric acid compared to 400 mM and the order of migration of the G/A peptides is now reversed, i.e., the Ala peptide migrates faster than the Gly peptide under these conditions. The resolution is further improved between the Phe and Tyr peptide compared to lower acid concentrations. In summary, increase in the concentration of phosphoric acid from 25 to 800 mM has a dramatic effect on the resolution of the +2 peptides, with the 800 mM conditions providing the best separation. Similarly, the Phe and Tyr peptides are best resolved in the presence of 800 mM phosphoric acid. On the other hand, the separation of the +1 peptides, with just subtle changes in hydrophobicity of the alkyl side-chain (G vs. A and V vs. I), is basically not resolved in this concentration range of phosphoric acid.
Figure 2 shows the effect of PFPA concentration on the CE separation of the same Series 1 peptide mixture shown in Fig. 1. The pH was adjusted to pH 2 with LiOH for all concentrations of PFPA; thus, the majority of the acid is in its ionized form (PFPA−). From Fig. 2, the increase in concentration of the hydrophobic counterion (PFPA) from 0.1 to 0.4 M dramatically improves the resolution of the +1 (G, A, V, I, F, Y) group and the +2 (K, H, R) group of peptides with best resolution at 400 mM PFPA. The +1 peptides differing in their alkyl side-chain at the substitution site (G, A, V, I) are all baseline resolved in the presence of 0.4 M PFPA, with the order of migration corresponding to their increasing hydrophobicity (G<A<V<I) due to the increase in number of methylene groups in the side-chain. In addition, this result clearly shows that these 10-residue peptides which differ by just a single methylene group (G vs. A peptide or V vs. I peptide) can be readily resolved by CE. These latter results would suggest that the mechanism for the separation is based solely on hydrophobicity. However, the migration order for the separation of the Phe and Tyr peptides is in the reverse order of their intrinsic hydrophobicity. Thus, the Tyr peptide would be expected to migrate faster than the Phe peptide if the separation was based solely on hydrophobicity but the observed reverse order of migration suggests that the separation mechanism is more complex than hydrophobicity alone.
Figure 3 now compares the resolution of two groups of Series 1 peptides (+1 and +2 groups) using the hydrophilic counterion (H2PO4−) compared to three hydrophobic counterions (TFA, PFPA, and HFBA). Clearly, the separation of the +1 group of peptides, differing in the hydrophobicity of their side-chains, is poor in the presence of the hydrophilic counterion and much improved in the presence of hydrophobic counterions, with 0.4 M PFPA providing the best overall resolution compared to 0.4 M TFA or HFBA. The migration order in the presence of all three hydrophobic counterions is the same for both the +1 and +2 peptide groups. These results confirm our previous findings that the phosphate counterion performs poorly in the separation of peptides differing subtly in hydrophobicity of alkyl side-chains [20]. All three of the +2 peptides (K, R, H) were baseline resolved under all conditions, albeit the separation improved with increasing hydrophobicity of perfluorinated acid (TFA<PFPA<HFBA).
Interestingly, the order of migration of the +2 peptides is K<R<H in 0.8 M phosphoric acid but is changed in the presence of hydrophobic counterions where the order of migration is K<H<R. Also of note is the observation that the Tyr and Phe peptides are best resolved in phosphoric acid compared to the hydrophobic counterions (TFA, PFPA, and HFBA.) In all four conditions, the Tyr peptide migrates slower than the Phe peptide and the migration order is the reverse of their hydrophobicities, i.e., the peptide with the more hydrophilic side-chain substitution, Tyr, migrates slower. Finally, with the perfluorinated acids, especially HFBA, there were significant differences in the migration rates of the +1 peptides due to changes in the EOF. However, slight variations in the magnitude of the EOF do not change the migration order as demonstrated below in Fig. 4.
3.3 CE of Series 2 peptides (Ac-XLGLGGGLGLGK-OH)
Figure 4 illustrates the effect of capillary coating on the CE separation of a mixture of Series 2 peptides. From Fig. 4, in an uncoated capillary, there is a significant drift in migration times for replicate runs, albeit the separation is essentially identical. However, by coating the capillary with INST Coating solution prior to each run, the repeatability is significantly improved. Further, by replacing the BGE after each run in addition to coating the capillary, the repeatability is now excellent between runs. Thus, the critical observation from Fig. 4 is that the variability in the migration time between runs is attributed to variability in the EOF which can be overcome by coating the capillary and replacing the BGE between runs. Such conditions were now employed for subsequent CE separations.
In order to understand the mechanism for the II-CZE separation of peptides in the presence of the anionic ion-pairing reagent PFPA, we now investigated the separation of pairs of peptides that differed subtly in hydrophobicity at the substitution site (Gly vs. Ala, Val vs. Ile, Val vs. Leu, Pro vs. Met, Ile vs. Leu, Ile vs. nLeu, and Val vs. nVal) and compared the separation of the same peptide pairs by RP-HPLC. Though the counterion (PFPA−) is identical in both the II-CZE and RP-HPLC separations, the concentrations used are dramatically different (20 mM in RP-HPLC and 400 mM in II-CZE). In addition, characteristics of RP-HPLC include a hydrophobic stationary phase (C8, packing in the present case) and an organic solvent (ACN) in the mobile phase, both of which are absent in II-CZE, where the separation is dependent on the composition of the BGE. Nevertheless, the separation in RP-HPLC is based on the overall hydrophobicity of the peptides and the idea was to determine if the II-CZE separation of these peptide pairs under these unique conditions in the BGE during II-CZE was also based on hydrophobicity differences as observed in RP-HPLC.
From Fig. 5, all peptide pairs are separated by both II-CZE and RP-HPLC, i.e., the migration/elution order is the same in both systems (the more hydrophobic peptide migrates/is eluted later), albeit the separations are considerably better with the RP-HPLC runs. Arg and Tyr peptides were included in the CE runs (Figs. 5–8) and the Phe peptide in the RP-HPLC runs (Figs. 5–7) as internal standards to ensure run-to-run repeatability and valid comparison of different separations. From Fig. 5, the II-CZE approach is able to separate the 12-residue peptides that differ by a single methylene group at the substitution site (e.g., Val vs. Ile, Val vs. Leu). Even the isosteric substitutions can be resolved by II-CZE, though not to the baseline (e.g., Ile vs. nLeu). Interestingly, peptides of identical charge-to-mass ratio (I/nL, V/nV, I/L) are separated by both II-CZE and RP-HPLC. In the case of II-CZE, this result clearly invalidates the CZE interpretation that peptides are separated solely by differences in charge-to-size ratio. An explanation for such separations in both systems is that the apparent hydrophobicity of the side-chains of the I/nL, V/nV, and I/L peptide pairs is dependent on their structures and distance from the polypeptide backbone. Thus, although the number of carbon atoms is identical within these three peptide pairs, in each case a methyl group has been moved from the β-carbon position (Ile, Val) to an extended position further away from the polypeptide backbone (nLeu, Leu, nVal), i.e., the methyl group(s) is now more exposed to its environment and, hence, more readily able to express fully its hydrophobicity. The apparent hydrophobicity of Leu and nLeu will thus be greater than that of Ile and that of nVal will be greater than Val, leading to the migration (II-CZE) and elution (RP-HPLC) orders seen in Fig. 5 (I<L, I<nL, and V<nVal). Overall, the results shown in Fig. 5 suggest that the hydrophobic mechanism is similar in both systems for these peptides although a better resolution is obtained in RP-HPLC.
Figure 6 now compares separations by II-CZE and RP-HPLC of pairs of peptides with polar or charged amino acid substitutions. For each peptide pair, there is a difference of one methylene group prior to the polar or charged side-chain between the substituted amino acids such that, in order of increasing hydrophobicity, T>S, Q>N, E>D, and K>O. From Fig. 6, these peptide pairs are separated in order of increasing hydrophobicity in both systems, i.e., the more hydrophobic peptide migrates later (II-CZE) or is eluted later (RP-HPLC). Interestingly, II-CZE is generally better than RP-HPLC at separating these peptide pairs. Indeed, the peptides N/Q peptide pair are coeluted during RP-HPLC but are baseline resolved in II-CZE. The results of Figs. 5 and 6 would suggest that the hydrophobically mediated mechanism proposed by Popa et al. [20] is in fact occurring for a wide range of side-chain types.
For the peptide pairs shown in Figs. 5 and 6, differences in peptide hydrophobicity can explain the separations of pairs of peptides which, though differing at the substitution site in side-chain hydrophobicity, are nevertheless substituted by similar classes of amino acids, i.e., substitution of an amino acid with a nonpolar alkyl side-chain (e.g., Val) by another amino acid with a similar side-chain (e.g., Leu) or substitution of an amino acid with a polar side-chain (e.g., Ser) by another amino acid with a similar side-chain (Thr). Thus, in RP-HPLC, the separation is occurring due to inter-action of the peptides with the hydrophobic matrix (C8 alkyl chains) and, thus, peptides with small differences in hydrophobicity can be readily resolved; in II-CZE, the separation is occurring totally in the BGE and the same small differences in hydrophobicity can also be readily resolved. In the latter case, the hydrophobically mediated mechanism is a result of the high concentration of the anionic hydrophobic counterion (PFPA) in the aqueous medium. However, Fig. 7 now illustrates some examples of II-CZE separations where the observed migration orders cannot be explained solely on the basis of differences in peptide hydrophobicity, as expressed by their retention behavior in RP-HPLC. Specifically, when we separate peptides that differ by type of functional group on the side-chain at the substitution site, we observe that there is no longer a correlation of electrophoretic mobility in II-CZE with relative peptide hydrophobicity. Thus, from Fig. 7, the orders of peptide hydrophobicities within the peptide mixtures are W>F≫Y, F≫V≫E, F>I>M, and W>F≫Y. However, from the II-CZE runs in Fig. 7, the Trp peptide comigrates with the Tyr peptide, despite the considerably greater hydrophobicity of the former; the Val and Glu peptides are poorly resolved despite the considerably greater hydrophobicity of the former (indeed, the Val peptide migrates slightly faster than the less hydrophobic Glu peptide); the Met and Ile peptides comigrate despite the greater hydrophobicity of the latter peptide (note also that the least hydrophobic peptide, Tyr, migrates longest); and the Phe and Tyr peptides are only poorly resolved, despite the considerably greater hydrophobicity of the former peptide (also, the Phe peptide migrates slightly faster than the less hydrophobic Tyr peptide). Such results suggest that, in II-CZE, the polar side-chains (e.g., Glu and Tyr) at the substitution site have slower electrophoretic mobility than predicted based on hydrophobicity in RP-HPLC, suggesting that an additional mechanism is present during II-CZE separations in addition to that based on peptide hydrophobicity differences where the more hydrophobic peptide migrates slower. Specifically, it appears there is some interaction between the polar side-chains and the BGE that decreases their electrophoretic mobility relative to the hydrophobic side-chains (e.g., Val and Phe).
To investigate further the observations from Fig. 7 that the mechanism involved in the separation of peptides using high concentrations of acid in the BGE during II-CZE cannot be solely explained by hydrophobicity differences between peptides, Fig. 8 now shows the separation of peptides in the presence of a considerably more hydrophilic anionic ion-pairing reagent compared to PFPA, i.e., 0.8 M phosphoric acid (comprising ~0.4 M H2PO4− and 0.4 M H3PO4 at pH 2). From Fig. 8, peptides within each mixture are separated in their reversed order of hydrophobicity when compared to RP-HPLC: the Tyr peptide migrates slower than the considerably more hydrophobic Trp peptide (note that these peptides comigrated under the PFPA CE conditions of Fig. 7); the order of migration of V<Y<E<D is in order of decreasing hydrophobicity (i.e., the reverse of RP-HPLC) (note also the reversal of the D, E, and Y peptides compared to the CE conditions of Fig. 6); and the order of migration of I<M<Y is in order of decreasing hydophobicity. Thus, in the case of the separations shown in Fig. 8, it appears that the separation in 0.8 M phosphoric acid is based on increasing hydrophilicity of the side-chains at the substitution site, i.e., the more hydrophilic the peptide, the slower the migration rate). Significantly, the excellent separations shown in Fig. 8 using the hydrophilic anion (H2PO4−) are not observed for peptides substituted with nonpolar side-chains (W, F, V, I, M), despite their wide range of hydrophobicities. Thus, it seems that the hydrophobic anion is best when separating peptides that differ in hydrophobicity with hydrophobic side-chains but the hydrophilic anion is best when separating peptides that differ in polar side-chains relative to hydrophobic side-chains. Finally, if the separation of the peptides in Fig. 8 was based solely on a CZE mechanism (charge-to-size), for peptides with the same charge, the peptide with the larger mass/size should migrate slower. However, this is clearly not the case as observed in Fig. 8 for the following peptide pairs, where the peptides with greater mass/size are migrating faster (W<Y; Y<E, D; F<E, D; and E<D).
3.4 Separative mechanism in II-CZE
The results presented above clearly show that high concentrations (400 mM) of perfluorinated hydrophobic anionic ion-pairing reagents (TFA, PFPA, HFBA) can separate peptides by II-CZE based on differences in hydrophilicity/hydrophobicity of peptides at a single substitution site even though the peptides have identical net charge and polypeptide chain length. In these II-CZE experiments, the pH is maintained at pH 2 by titration with LiOH. Due to the extremely low pKa values of TFA, PFPA, and HFBA (i.e., pKa values in the range of 0.12–0.17) at pH 2 [24], the concentration of the respective anions approaches 400 mM. In the case of 800 mM phosphoric acid adjusted to pH 2 with LiOH, the concentration of the negatively charged (H2PO4−) anion is also approximately 400 mM since the pKa is close to the pH of the solution. Eighteen of the peptide analytes have a single Lys residue (i.e., +1 net charge) and four peptides have a net charge of +2 due to the substitution of Lys, His, Arg, or Orn at the substitution site. Due to the high pKa relative to pH 2 of the side-chains of these residues, all peptides are fully charged cations. In addition, due to the extremely high excess of the negatively charged anion in the BGE compared to the peptide cation, one could consider the peptide cation to be fully ion-paired during CE. However, II-CZE theory clearly states that if there is 100% ion-pairing, the peptides would have a net charge of zero and would not migrate in an electric field [25, 26]. That is, fully ion-paired peptides would have no electrophoretic mobility and would comigrate with the EOF marker. Clearly, this is not the situation in our experiments, suggesting that the peptides are not fully ion-paired even at 400 mM concentrations of these ion-pairing reagents.
Based on the present results, what can we conclude about the separative mechanism in II-CZE? The majority of the peptides in this study have a net charge of +1 and the substitution site in these peptides is distant from the ε-amino group on the Lys side-chain (Ac-XLGLGGGLGLGK-OH). We do not believe that the subtle changes in hydrophilicity/hydrophobicity at the substitution site could affect the pKa value of the ε-amino group of Lys (the only positively charged center in the peptides) or affect the association constant between the ion-pairing reagent and the charged ε-amino group of the Lys side-chain. Thus, this leaves us to conclude that the differences in electrophoretic mobility observed between peptides with subtle differences in hydrophilicity/hydrophobicity at the substitution site are due to interactions of the peptides and the bulk BGE. In other words, we are defining these solution-based effects as non-specific interactions or nonspecific molecular associations between the surface of the peptide and the bulk BGE. Clearly, these solution-based effects are dependent on the concentration and hydrophilicity/hydrophobicity of the counterion in solution. We speculate that differential hydration/dehydration properties of various side-chains in the peptide and the hydration/dehydration properties of the hydrophilic/hydrophobic anions as well as the electrostatic attractions between the peptide and the anions in solution all play a critical role in these solution-based effects, concomitantly providing additional selectivity effects not observed in the chromatographic methods.
II-CZE has some interesting analogies to separations occurring in chromatography. In RP-HPLC, peptides with subtle differences in hydrophobicity can be easily resolved due to a combination of effects from the peptide partitioning between the polar mobile phase and the nonpolar stationary phase. In hydrophilic interaction chromatography (HILIC) [27, 28], peptides with subtle differences in hydrophobicity can be easily resolved due to a combination of effects from the peptide partitioning between the nonpolar mobile phase and the polar stationary phase. The order of elution between the two methods is reversed; the most hydrophobic peptide is eluted last in RP-HPLC and the most hydrophobic peptide is eluted first in HILIC [27, 28]. Peptides with the same subtle differences in hydrophobicity can also be resolved without the stationary phase in II-CZE, with the order of migration (solution base effects only) dependent on the composition of the counterion (hydrophilic vs. hydrophobic). With a hydrophobic counterion like the PFPA− anion, the migration order is analogous to RP-HPLC within the same side-chain type, whereas, with a hydrophilic counterion like the H2PO4− anion, the migration order is analogous to HILIC. The closest chromatography analogy to II-CZE is mixed-mode hydrophilic interaction–cation exchange chromatography (HILIC-CEX) [28–34]. In this method, the hydrophilic stationary phase has covalently attached sulfonate anions (SO3−). Thus, the cationic peptide is attracted to the anions on the matrix for the separation based on net positive charge (an increasing salt gradient is required for peptide elution) and with subsequent hydrophilic separation based on peptide partitioning between the nonpolar mobile phase and the polar stationary phase (a decreasing gradient of ACN is required for peptide elution). Thus, the combination of a decreasing gradient of ACN and the increasing salt gradient is both required in the mobile phase. However, in II-CZE, only the hydrophilic H2PO4− anion in solution is required along with CZE charge separation to mimic HILIC-CEX results. The CZE separation is analogous to the CEX separation which requires both the negatively charged matrix and the increasing salt gradient. The attraction between the positively charged residues in the peptide and the negatively charged anions in solution in II-CZE, along with the interactions within the BGE, are responsible for the hydrophilic separation within a positive charge group, analogous to partitioning between the hydrophilic matrix and the mobile phase in HILIC/CEX.
4 Concluding remarks
Understanding the fundamentals of the interactions between the peptide surface and the solution is critical in designing new and more powerful separation methods. CE, specifically II-CZE in the present study, has an excellent potential to serve as a convenient tool to study solution-based interactions of biomolecules in general. Indeed, II-CZE is an extremely simple solution-based method that mimics the more complex chromatography systems where both a stationary phase and mobile phase are required and provides additional selectivity effects not observed in the chromatographic methods.
Acknowledgments
This work was supported by an NIH grant to Robert S. Hodges (RO1GM 61855) and the John Stewart Chair in Peptide Chemistry to R.S.H.
Abbreviations
- HFBA
heptafluorobutyric acid
- II-CZE
ion-interaction CZE
- PFPA
pentafluoropropionic acid
References
- 1.Messana I, Rossetti DV, Cassiano L, Misiti F, et al. J Chromatogr B. 1997;699:149–171. doi: 10.1016/s0378-4347(97)00198-9. [DOI] [PubMed] [Google Scholar]
- 2.Popa TV, Mant CT, Hodges RS. Electrophoresis. 2004;25:94–107. doi: 10.1002/elps.200305654. [DOI] [PubMed] [Google Scholar]
- 3.Wan H, Blomberg LG. J Chromatogr A. 1997;758:303–311. [Google Scholar]
- 4.Sabah S, Scriba GKE. J Chromatogr A. 1998;822:137–145. [Google Scholar]
- 5.Koval D, Kasicka V, Jirácek J, Collinsová M. J Pept Sci. 2003;26:653–660. [Google Scholar]
- 6.Kasicka V. Electrophoresis. 2006;27:142–175. doi: 10.1002/elps.200500527. [DOI] [PubMed] [Google Scholar]
- 7.Scriba GKE. Electrophoresis. 2006;27:222–230. doi: 10.1002/elps.200500446. [DOI] [PubMed] [Google Scholar]
- 8.Czerwenka C, Lindner W. Anal Bioanal Chem. 2005;382:599–638. doi: 10.1007/s00216-005-3091-x. [DOI] [PubMed] [Google Scholar]
- 9.Bennett HPJ, Browne CA, Solomon S. J Liq Chromatogr. 1980;3:1353–1365. [Google Scholar]
- 10.Schaaper WMM, Voskamp D, Olieman C. J Chromatogr. 1980;195:181–186. [Google Scholar]
- 11.Harding DRK, Bishop CA, Tartellin MF, Hancock WS. Int J Pept Protein Res. 1981;18:314. doi: 10.1111/j.1399-3011.1981.tb02060.x. [DOI] [PubMed] [Google Scholar]
- 12.Guo D, Mant CT, Hodges RS. J Chromatogr. 1987;386:205–222. doi: 10.1016/s0021-9673(01)94598-4. [DOI] [PubMed] [Google Scholar]
- 13.Chen Y, Mehok AR, Mant CT, Hodges RS. J Chromatogr A. 2004;1043:9–18. doi: 10.1016/j.chroma.2004.03.070. [DOI] [PubMed] [Google Scholar]
- 14.Popa TV, Mant CT, Hodges RS. Electrophoresis. 2004;25:1219–1229. doi: 10.1002/elps.200305889. [DOI] [PubMed] [Google Scholar]
- 15.Shibue M, Mant CT, Hodges RS. J Chromatogr A. 2005;1080:49–57. doi: 10.1016/j.chroma.2005.02.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shibue M, Mant CT, Hodges RS. J Chromatogr A. 2005;1080:58–67. doi: 10.1016/j.chroma.2005.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shibue M, Mant CT, Hodges RS. J Chromatogr A. 2005;1080:68–75. doi: 10.1016/j.chroma.2005.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mant CT, Hodges RS. J Chromatogr A. 2006;1125:211–219. doi: 10.1016/j.chroma.2006.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Popa TV, Mant CT, Hodges RS. Electrophoresis. 2003;24:4197–4208. doi: 10.1002/elps.200305576. [DOI] [PubMed] [Google Scholar]
- 20.Popa TV, Mant CT, Hodges RS. J Chromatogr A. 2006;1111:192–199. doi: 10.1016/j.chroma.2005.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Popa TV, Mant CT, Hodges RS. J Chromatogr A. 2004;1043:113–122. doi: 10.1016/j.chroma.2004.04.029. [DOI] [PubMed] [Google Scholar]
- 22.Sereda TJ, Mant CT, Quinn AM, Hodges RS. J Chromatogr. 1993;646:17–30. doi: 10.1016/s0021-9673(99)87003-4. [DOI] [PubMed] [Google Scholar]
- 23.Kovacs JM, Mant CT, Hodges RS. Biopolymers (Pept Sci) 2006;84:283–297. doi: 10.1002/bip.20417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henne AL, Fox GJ. J Am Chem Soc. 1951;73:2323–2325. [Google Scholar]
- 25.Robinson RA, Stokes RH. Electrolyte Solutions. 2. Chapter 14 Butterworths; London, UK: 1959. [Google Scholar]
- 26.Descroix S, Varenne A, Geiser L, Cherkaoui S, et al. Electrophoresis. 2003;24:1577–1586. doi: 10.1002/elps.200305372. [DOI] [PubMed] [Google Scholar]
- 27.Alpert AJ. J Chromatogr. 1990;499:177–196. doi: 10.1016/s0021-9673(00)96972-3. [DOI] [PubMed] [Google Scholar]
- 28.Zhu BY, Mant CT, Hodges RS. J Chromatogr. 1991;548:13–24. doi: 10.1016/s0021-9673(01)88590-3. [DOI] [PubMed] [Google Scholar]
- 29.Zhu BY, Mant CT, Hodges RS. J Chromatogr. 1992;594:75–86. [Google Scholar]
- 30.Mant CT, Hodges RS. In: Encyclopedia of Separation Science. Wilson ID, Adland TR, Poole CF, Cook M, editors. Academic Press; New York: 2000. pp. 3615–3626. [Google Scholar]
- 31.Hartmann E, Chen Y, Mant CT, Jungbauer A, Hodges RS. J Chromatogr A. 2003;1009:61–71. doi: 10.1016/s0021-9673(03)00620-4. [DOI] [PubMed] [Google Scholar]
- 32.Hodges RS, Chen Y, Kopecky E, Mant CT. J Chromatogr A. 2004;1053:161–172. [PubMed] [Google Scholar]
- 33.Lindner H, Sarg B, Hoertnagl B, Helliger J. J Biol Chem. 1998;273:13324–13330. doi: 10.1074/jbc.273.21.13324. [DOI] [PubMed] [Google Scholar]
- 34.Lindner H, Sarg B, Grunicke H, Helliger J. J Cancer Res Clin Oncol. 1999;125:182–186. doi: 10.1007/s004320050261. [DOI] [PMC free article] [PubMed] [Google Scholar]