

Abstract
The Akt pathway is very important in both development and cancer. Here we show that expression of Casein kinase I epsilon (CKIε) causes up-regulation of the Akt pathway despite normal protein expression of the pathway inhibitor Phosphata and tensin homologue deleted on chromosome ten (PTEN). Conversly we show that a CKIε/δ-specific inhibitor can inhibit Akt phosphorylation at both Thr308 and Ser473 and drastically reduce phosphorylation of the Akt target Glycogen Synthase Kinase 3β (GSK3β). These conclusions were confirmed between MCF7 cells transiently transfected with CKIε and Hs578T cells which already express endogenous CKIε. The results suggest that CKIε is a new positive regulator of the Akt pathway. Here we propose that, rather than inhibiting PTEN function, CKIε positively regulates Akt possibly by inhibiting Protein Phosphatase 2A (PP2A).
Keywords: CKIε, Akt
Introduction
The Protein Kinase B/Akt pathway is critical for several developmental processes including cell metabolism, protein synthesis, cell cycle control, and cell survival [1]. This pathway has been studied extensively because its over-expression is associated not only with malignant transformation of various tissues [1-2] but also with resistance to chemotherapy [2-3]. Akt can be activated by a variety of stimuli including growth factors, phosphatase inhibitors and cellular stress [2]. The priming phosphorylation at Thr 308, which is performed by phosphoinositide-dependent kinase 1 (PDK1), is necessary for initial activation, but phosphorylation of Akt on Ser 473 is considered critical for full activation [4]. Over the years, several kinases, including Akt itself, have been proposed as candidates for Ser 473 phosphorylation [5]. Recently, Sarbassov and co-workers have provided strong evidence that the rapamycin-insensitive mammalian Target of Rapamycin is the elusive kinase that phosphorylates Akt at Ser 473 [6].
While phosphorylation is critical for activation of the pathway, dephosphorylation is the major mechanism of Akt pathway inhibition. This can be achieved by one of two phosphatases: PTEN or PP2A. PTEN, which dephosphorylates phosphatidylinositol 3-kinase (PI3K) upstream of Akt, is the best known Akt pathway inhibitor and its loss or inhibition has been documented in 50% of sporadic breast cancers [1]. In addition, even though not as well studied, PP2A has also been shown to also inhibit the Akt pathway by inactivating Akt itself through dephosphorylation of both its activation sites [7].
CKIε, but not its closely related isoform delta, was recently suggested to positively regulate PTEN following genotoxic stress in hematopoietic cells, providing a protective advantage against leukemia [8]. This possibility is potentially relevant to disease mechanisms since non-synonymous point mutations in CKIε were identified in more than 10% of breast cancer patients [9].
Since loss of PTEN function has been associated with both leukemia and mammary tumorigenesis, we investigated whether wild-type (WT) CKIε played a role in breast cancer through a mechanism similar to that proposed by Okamura and co-workers for leukemia. This led to our discovery of a different mode of regulation of the Akt pathway by CKIε in mammary tissue, which may help to further elucidate the complex regulation of the Akt pathway in development and cancer.
Materials and Methods
CKIε construct
The full-length cDNA encoding CKIε was obtained in the pRSET vector from Dr David Virshup and cloned into the mammalian expression vector pCDNA-3.1 using the TOPO cloning reaction as outlined in the TOPO cloning kit (Invitrogen), followed by chemical transformation using DH5a-T1 One Shot chemically competent E. coli cells. In this vector the expression of CKIε is under the control of the immediate-early Cytomegalovirus (CMV) promoter. The following primers were used to generate the polymerase chain reaction (PCR) product to be used in the TOPO cloning reaction: 5-CACCATGGAGCTACGTGTGGGG-3; 5-TCACTTCCCGAGATGGTCA-3. The above primers include a stop codon at the end of the message so that the final product is not tagged.
Cell Culture
All cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) with 10 % Fetal Bovine Serum (FBS). Prior to transfection, MCF-7 cells were plated in FBS media without antibiotics. 500 ng DNA were transiently transfected using ExpressFect (Denville Scientific, Inc, Metuchen, MJ) according to manufacturer's instructions, and non-transfected MCF-7 cells were used as a negative control. All cells were plated into 6-well plates and after they reached 80 % confluence they were serum-starved overnight. The following day the Akt pathway was stimulated by addition of 10 μg/ml insulin-containing media, or 10 ng/ml Epidermal Growth Factor (EGF) -containing media for Hs578T, for 20-60 minutes prior to lysing. Cells were harvested in 100 μl lysis buffer. Where indicated, 20-40 μM IC261 (Calbiochem) was added 2 hours prior to harvest. DMSO was added to CKIε-transfected cells as negative drug control. To increase drug solubility, IC261 was first incubated in FuGENE 6 (Roche) at 1 μl/200 μl media according to manufacturer protocol and added drop wise to the cells.
Antibodies and other reagents
Antibodies against the following proteins were used for either Western Blot (WB) or immunoprecipitation (IP) as described below: anti-CKIε (C-20; Santa Cruz Biotechnology, Santa Cruz, CA), anti-PTEN, anti-phospho PTEN S380/T382/383, anti-phospho PTEN Ser 370, anti-phospho Akt Ser 473, anti-phospho Akt Thr 308, anti-Akt, anti-phospho GSK3 α/β S21/9, anti-GSK3β and anti-actin (Santa Cruz Biotechnology). Unless otherwise specified, all primary antibodies were purchased from Cell Signaling Technology, Inc., Danvers, MA.
Western Blot Analysis
20 μg protein was separated in 10 % Sodium Dodecyl Sulfate (SDS) gel and transferred onto Polyvinylidene fluoride (PDVF) membrane. Membrane was activated in methanol and equilibrated 30 minutes in cold transfer buffer prior to transfer. Membrane was blocked in 5 % milk, or Bovine Serum Albumin (BSA) for phospho antibodies, for one hour. With the exception of total AKT, PTEN and actin, which were incubated at room temperature for 2 hours, all other primary antibodies were incubated overnight at 4°C in 5% BSA (antibody dilution range between 1:750 and 1:1000). ECL Plus reagent (Amersham) was used for chemiluminescent detection. Band intensities were quantified using the Gel Analysis tool from Image J software and results reported as the ratio between the experimental and control value.
Immunoprecipitation
250 μg total cell lysate was first pre-incubated with 1 μg of goat IgG and sepharose for 30 minutes and subsequently with primary antibody at 4 °C for 3 hours, followed by addition of protein A/G Sepharose PLUS (Santa Cruz Biotech) overnight on a shaker at 4 °C. After centrifugation, immuno-precipitates were washed four times with lysis buffer. Pellets were resuspended in 40 μl 2X SDS and boiled for 3 minutes. Samples were centrifuged 5 minutes to pellet beads before analyzing them by WB as described previously. Cell lysate was run next to IP samples as positive control for WB analysis.
Phosphorylation site Prediction
The human homologue of each target protein was identified in the SWISS-PROT database and the corresponding ID number was then used to scan each protein by ID using the SCANSITE Program from MIT. Each protein sequence was searched using the individual motif for Casein Kinase 1 at low stringency. If sites were identified at low stringency, the scan was repeated at medium stringency. Otherwise the scan was repeated for Acidophilic Ser/Thr kinase motifs.
Results
CKIε expression leads to activation of the Akt pathway
MCF7 cells, which do not express endogenous CKIε, were transiently transfected with constructs encoding either WT CKIε or kinase-dead mutant (DN) CKIε and incubated in insulin for 60 minutes to activate the Akt pathway. The effect on the Akt pathway was initially measured by phosphorylation of Akt at Ser 473, which is required for full activation of Akt. Non-transfected cells which had not been treated with insulin were compared to insulin-treated cells that were transfected with constructs encoding either WT or DN CKIε. Expression of functional CKIε led to much higher levels of Akt phosphorylation at Ser 473 compared to either DN or non-transfected MCF7 cells (Fig. 1A), showing that the kinase activity of WT CKIε is responsible for the increase in Akt phosphorylation. This conclusion was confirmed when cells exposed to insulin-containing media were treated with the CKIε/δ-specific inhibitor IC261, which prevented Akt phosphorylation at both Thr 308 and S473, compared to CKIε-transfected MCF7 cells treated with insulin +/- DMSO (vehicle control) (Fig. 1B). IC261 decreased Akt phosphorylation not only at Ser 473 but also at Thr 308, showing that the effect of CKIε is not limited to Akt phosphorylation at Ser 473.
Fig. 1. Akt phosphorylation requires functional CKIε.

MCF7 cells were transiently transfected with CKIε and incubated in insulin media to activate the PI3K pathway. Pathway activation was measured by phospho Akt levels. Total Akt or actin were used as basal control. Western analysis results were quantified using Image J software, and data was represented graphically as pAkt/control. (A) WT CKIε was much more efficient in phosphorylating Akt than the DN CKIε. (B, C) MCF7 cells transfected with WT CKIε and treated with the CKIε/δ inhibitor IC261 had a significant decrease in phospho Akt levels compared to cells undergoing the same treatment except for no drug (-) or DMSO alone as the vehicle control for IC261. CKIε inhibition by IC261 was able to block phosphorylation at both T308 and S473 (C).
Since phosphorylation of Akt on Ser 473 is critical for full activation of the kinase, we wanted to confirm that CKIε induces full activation, by examining direct targets of Akt. Active Akt phosphorylates, among other targets, GSK3β on Ser 9. This phosphorylation event inactivates GSK3β and reverses its inhibition of cell cycle control, thus promoting cell survival [3]. When cells were treated with the CKIε/δ inhibitor IC261, GSK3β phosphorylation was significantly impaired despite the presence of insulin (Fig. 2), suggesting that functional CKIε contributes to Akt signaling.
Fig. 2. CKIε inhibition prevents phosphorylation of the Akt target GSK3β.
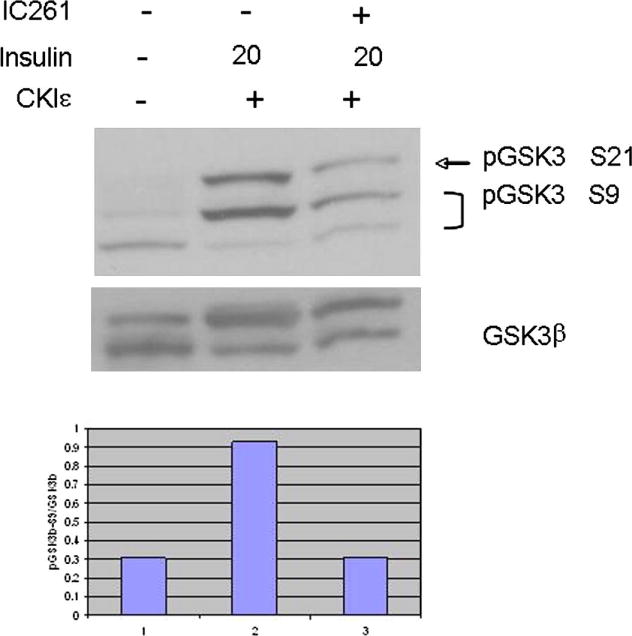
MCF7 cells were transiently transfected with CKIε and/or incubated with insulin as shown below. Pathway activation was measure by phosphorylation of GSK3β at Ser 9 (bottom 2 bands). Inhibition of CKIε by IC261 resulted in GSK3β phosphorylation levels similar to those in the absence of insulin, suggesting that both may be required for maximum signal strength.
CKIε does not interact with PTEN
Okamura and co-workers proposed PTEN as a novel substrate for CKIε based on co-immunoprecipitation experiments [8], but they did not explain how this interaction could affect PTEN function. Even though PTEN regulation is complex and not fully understood, several mechanisms have been proposed, including phosphorylation [10]. We therefore tested whether CKIε inhibits PTEN function by phosphorylating it at the best known inhibitory sites using an antibody that detects phospho-PTEN at S380/T382/T383. There was no detectable difference in phospho-PTEN levels between CKIε-transfected and non-transfected MCF7 cells (Fig. 3A), suggesting that the effect of CKIε is not mediated through PTEN phosphorylation at any of these sites. We also found no effect of CKIε on PTEN phosphorylation on Ser 370, which has been implicated in destabilizing the PTEN protein [11] by promoting subsequent phosphorylation at Thr 366 [12]. Moreover, we were unable to detect co-immunoprecipitation of PTEN with CKIε (Fig. 3B), suggesting that these proteins do not physically interact in mammary cells.
Fig. 3. CKIε does not interact with PTEN.

(A) MCF7 cells transiently transfected with WT CKIε were compared to DN or non-treated cells. PTEN function was measured by phosphorylation levels at S380/T382/383 and S370. No effect was seen at any of these sites, suggesting that CKIε does not phosphorylate PTEN. (B) Cell lysates expressing CKIε +/- IC261 treatment were subject to IP with anti-CKIε antibody, followed by WB analysis with either CKIε (IP positive control) or PTEN to determine co-IP. Lysate samples were included to help identify correct protein band. PTEN did not co-IP with CKIε, suggesting that they do no physically interact.
Physiological role of CKIε-mediated Akt up-regulation
Since MCF-7 cells do not naturally express CKIε, we tested whether the CKIε-mediated Akt signaling that we identified in MCF7 cells also occurs naturally in other breast cancer cell lines. The mammary carcinoma cell line Hs578T was chosen for this purpose because, like the MCF7 cell line, it has normal PTEN levels. In addition, it also has elevated phospho Akt levels [13], better mimicking the increase in phospho Akt that we saw with MCF7 cells transiently expressing CKIε. After establishing that Hs578T cells express endogenous CKIε, we activated the Akt pathway by Epidermal Growth Factor (EGF) stimulation after serum deprivation. Where indicated, cells were also treated with 20-40μM IC261, to test whether blocking endogenous CKIε could reduce Akt activation as measured by phospho-Akt 473 levels. Co-treatment with 40μM IC261 in the presence of insulin led to a drastic decrease in Akt phosphorylation levels, comparable to non insulin treated cells (Fig.4), suggesting that CKIε is required for full activation of the Akt pathway. The 20-50 μM dose range is commonly used for CKIε/δ -specific inhibition [13-14], while CK1α inhibition requires 100μM doses[15], suggesting that in our system the IC261 treatment is more likely to be affecting CKIε/δ than CKIα.
Figure 4. Inhibition of endogenous CKIε reduces Akt pathway in Hs578T cells.

Cells were serum starved overnight and then incubated in EGF containing media to induce PI3K pathway activation. Where indicated, cells were also incubated with IC261 to inhibit CKIε function. Even though very little effect was seen at 20 μM, at 40 μM IC261 led to a drastic decrease in pAkt S473, comparable to non-EGF treated cells.
Discussion
The Akt pathway has been shown to play a role in many different cellular processes, which, if misregulated, can lead to both cancer progression and resistance to chemotherapy. Our findings provide new insight into Akt regulation, identifying CKIε as a new positive regulator of the pathway.
In the presence of insulin, transient expression of CKIε in MCF7 cells was sufficient to induce substantial increase in Akt phosphorylation as well as phosphorylation of the direct target of Akt, GSK3β. Moreover, the reduction in phosphorylation levels caused by inhibition of CKIε function despite insulin treatment suggests that CKIε kinase activity is required for the increase in phosphorylation levels seen with Akt and GSK3β.
The mode of Akt activation that was recreated by introducing CKIε into MCF7 cells appears to also occur naturally in Hs578T cells. This breast cancer cell line, which expresses endogenous CKIε and CKIδ, was already known to have elevated Akt phosphorylation levels despite normal PTEN protein expression [13]. When the cells were treated with a CKIε/δ inhibitor, Akt phosphorylation was drastically inhibited despite growth factor stimulation, suggesting that CKIε is involved in the mechanism that leads to the increase in Akt phosphorylation in this model system.
Since inhibition of CKIε prevented both phosphorylation events on Akt, which have been shown to be carried out by different kinases, it is unlikely that CKIε acts directly on Akt. Therefore, the pathway activation seen above could be achieved in at least three major ways, given that CKIε is a Ser/Thr kinase: 1) by CKIε phosphorylating PTEN to inhibit its phosphatase activity on PI3K, 2) by CKIε regulating PDK1, which performs the priming Akt phosphorylation at Thr 308, which is required for subsequent phosphorylation at Ser 473, or 3) by CKIε inhibiting PP2A, which is responsible for dephosphorylating Akt and shutting down the pathway after stimulation [7].
In the breast cancer lines used here, CKIε did not phosphorylate PTEN at its most common inhibitory sites S380/T382/T383, nor at Ser 370, which is the priming phosphorylation to Thr 366. Moreover, while we were not able to test phosphorylation at Thr 366 itself, CKIδ, which shares 98% identity with CKIε in the catalytic domain, was not able to phosphorylate PTEN at either Ser 370 or Thr 366 (Leslie, N., personal communication). Therefore, it appears more likely that, rather than inhibiting PTEN, CKIε can increase Akt phosphorylation by either positively regulating PDK1 or by negatively regulating PP2A.
PDK1, which is the kinase responsible for priming phosphorylation of Akt on Thr 308, is thought to be constitutively active and its regulation appears to involve recruitment to the plasma membrane by phosphatidylinositol-3,4,5-triphosphate (PIP3) upon activation of PI3K, which catalyses the conversion of PIP2 to PIP3 [16]. Even though PDK1 was identified as a substrate for CK1 phosphorylation with low stringency using the SCANSITE program from MIT (Table 1), PDK1 regulation appears to be fairly well understood [16], and phosphorylation does not seem to be involved. Therefore it seems an unlikely target for CKIε regulation of Akt, leaving PP2A as a potentially more interesting candidate. What makes PP2A even more appealing as the target for CKIε-mediated Akt phosphorylation is the observation that PP2A activity is higher in MCF7 cells than in Hs578T cells [17], consistent with the endogenous CKIε expression levels. PP2A is composed of several regulatory B subunits, each of which binds to the scaffolding A subunit and the catalytic C subunit to form different ABC heterotrimeric complexes, making for a complex mode of regulation [18]. Even though the exact regulation of PP2A is not fully understood, another point that makes PP2A a better target for CKIε than PDK1 is that, unlike for PDK1, phosphorylation has been proposed as a major mechanism for PP2A regulation [7]. In addition to phosphorylation at Tyr 307, extensive phosphorylation of Ser/Thr sites on PP2A has also been reported to have an inhibitory function on phosphatase activity [18]. We therefore performed a preliminary screen for PP2A subunits and found that several of them also appeared to be possible phosphorylation substrates for CK1, as summarized in Table 1. Based on our findings, as well as current knowledge on both PDK1 and PP2A regulation, we propose a tentative model in which, upon initial growth factor stimulation, CKIε is able to strengthen Akt signaling by negatively regulating PP2A. While further experiments are required to confirm that CKIε regulates Akt by inhibiting PP2A, our findings add to the evidence for a novel mechanism in which CKIε positively regulates the Akt pathway without requiring interaction with PTEN.
Table 1. Predicted binding motifs for PDK1 and PP2A.
| Name | ID | Site | Score | % |
|---|---|---|---|---|
| PDK1 | 015530 | S396 (*) | 0.4284 | 0.751 |
| PP2A | ||||
| Subunit A, PR65 α | P30153 | T97 | 0.5153 | 4.114 |
| T144 | 0.4527 | 1.323 | ||
| Subunit A, PR65 β | P30154 | T157 | 0.4527 | 1.323 |
| S215 | 0.4946 | 2.886 | ||
| Subunit B, PR55 α | P63151 | S194 | 0.4767 | 2.131 |
| T250 | 0.5180 | 4.267 | ||
| S276 | 0.5223 | 4.569 | ||
| Subunit B, PR55 β | Q00005 | S190 | 0.4767 | 2.131 |
| T246 | 0.5180 | 4.267 | ||
| S272 | 0.5223 | 4.569 | ||
| S408 | 0.5181 | 4.273 | ||
| Subunit B, PR55 γ | Q9Y2T4 | S246 | 0.5055 | 3.505 |
| S272 | 0.5223 | 4.569 | ||
| Subunit B, PR61 α | Q15172 | T151 | 0.5251 | 4.798 |
| Subunit B, PR61 β | Q15173 | T440 | 0.4752 | 2.063 |
| Subunit B, PR61 ε | Q16537 | T143 | 0.5251 | 4.798 |
| Subunit B, PR72/PR130 | Q06190 | T227 (*) | 0.4098 | 0.461 |
| S799 | 0.4473 | 1.182 | ||
| S263 | 0.4670 | 1.762 | ||
| T276 | 0.4974 | 3.030 | ||
| S266 | 0.5017 | 3.269 | ||
| S261 | 0.5045 | 3.440 | ||
| S1121 | 0.4376 | 0.941 | ||
| Subunit C, α and β | P67775, P62714 | S201 (#) | 0.6060 | 4.622 |
PDK1 and PP2A were identified by Swiss-Prot ID number and scanned for CK1 motif or the less stringent Acidophilic Ser/Thr motif (#).(*) marks sites also found at medium stringency. Each score represents how well the given sequence scored among the top XX percentile (%). The lower the %, the better the match. The most confident site was identified in PP2A, subunit B, PR72/130 at T227.
Acknowledgments
We thank Dr D. Virshup for the CKI constructs, Dr W. H. Lee for the MCF7 and Hs-578T cell lines, as well as Dr A. Edinger, Dr X. Dai, Dr T. Dolezal, Dr O. Marcu and Dr L. J. Marsh for their expert advice and critical revisions of the data and manuscript.
This work was supported by the National Institute of Health, Grant #CA91043.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Engelman J, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nature Rev Genet. 2006;7:606–618. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 2.Nahta R, Yu D, Hung M, Hortobagyi GN, Esteva FJ. Mechanisms of Disase: understanding resisance to HER2-targeted therapy in human breast cancer. Nature Clinical Practice. 2006;3:269–279. doi: 10.1038/ncponc0509. [DOI] [PubMed] [Google Scholar]
- 3.Kim D, Dan HC, Park S, Yang L, Liu Q, Kaneko S, Ning J, He L, Yang H, Sun M, Nicosia SV, Cheng JQ. AKT/PKB Signaling mechanisms in Cancer and Chemoresistance. Frontiers in Bioscience. 2005;10:975–987. doi: 10.2741/1592. [DOI] [PubMed] [Google Scholar]
- 4.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 5.Feng J, Park J, Cron P, Hess D, Hemmings BA. Identification of a PKB/Akt Hydrophobic Motif Ser-473 Kinase as DNA-dependent Protein Kinase. J Biol Chem. 2004;279:41189–41196. doi: 10.1074/jbc.M406731200. [DOI] [PubMed] [Google Scholar]
- 6.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 7.Millward TA, Zolnierowics S, Hemmings BA. Regulation of protein kinase cascades by protein phosphatase 2A. TiBS. 1999;24:186–191. doi: 10.1016/s0968-0004(99)01375-4. [DOI] [PubMed] [Google Scholar]
- 8.Okamura A, Iwata N, Yakushijin K, Nishikawa S, Hamaguchi M, Fukui C, Yamamoto K, Matsui T. Casein Kinase Iε down-regulates phospho-Akt via PTEN, following genotoxic stress-induced apoptosis in hematopoietic cells. Life Sciences. 2006;78:1624–1629. doi: 10.1016/j.lfs.2005.07.041. [DOI] [PubMed] [Google Scholar]
- 9.Fuja TJ, Lin F, Osan KE, Bryant PJ. Somatic Mutations and Altered Expression of the Candidate Tumor Supressors CSNK1ε, DLG1, and EDD/hHYD in Mammary Ductal Carcinoma. Cancer Res. 2004;64:942–951. doi: 10.1158/0008-5472.can-03-2100. [DOI] [PubMed] [Google Scholar]
- 10.Gericke A, Munson M, Ross AH. Regulation of the PTEN phosphatase. Gene. 2006;374:1–9. doi: 10.1016/j.gene.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 11.Macario H, Perera NM, Davidson L, Downes CP, Leslie NR. PTEN is destabilized by phoshorylation on Thr366. Biochem J. 2007;405:439–44. doi: 10.1042/BJ20061837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Al-Khouri AM, Ma Y, Togo SH, Williams S, Mustelin T. Cooperative Phosphorylation of the Tumor Suppressor Phosphatase and Tensin Homologue (PTEN) by Casein Kinases and Glycogen Synthase Kinase 3 β. J Biol Chem. 2005;280:35195–35202. doi: 10.1074/jbc.M503045200. [DOI] [PubMed] [Google Scholar]
- 13.Weng L, Smith WM, Dahia PL, Ziebold U, Gil E, Lees JA, Eng C. PTEN Suppresses Breast Cancer Cell Growth by Phosphatase Activity-dependent G1 Arrest followed by Cell Death. Cancer Res. 1999;59:5808–5814. [PubMed] [Google Scholar]
- 14.Flajolet M, He G, Heiman M, Lin A, Nairn AC, Greengard P. Regulation of Alzheimer's disease amyloid-β formation by casein kinase I. Proc Natl Acad Sci USA. 2007;104:4159–4164. doi: 10.1073/pnas.0611236104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kattapuram T, Yang S, Maki JL, Stone JR. Protein Kinase CK1α Regulates mRNA Binding by Heterogenous Nuclear Ribonucleoprotein C in Response to Physiologic Levels of Hydrogen Peroxide. J Biol Chem. 2005;280:15340–15347. doi: 10.1074/jbc.M500214200. [DOI] [PubMed] [Google Scholar]
- 16.Komander D, Fairservice A, Deak M, Kular GS, Prescott AR, Downes CP, Safrany ST, Alessi DR, van Aalten DMF. Structural insights into the regulation of PDK1 by phosphoinositides and inositol phosphates. EMBO J. 2004;23:3918–3928. doi: 10.1038/sj.emboj.7600379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gopalakrishna R, Gundimeda U, Fontana JA, Clarke R. Differential distribution of protein phosphatase 2A in human breast carcinoma cell lines and its relation to estrogen receptor status. Cancer Lett. 1999;136:143–151. doi: 10.1016/s0304-3835(98)00315-2. [DOI] [PubMed] [Google Scholar]
- 18.Lechward K, Awotunde OS, Swiatek W, Muszynska G. Protein phosphatase 2A: Variety of forms and diversity of functions. Acta Biochimica Polonica. 2001;48:921–933. [PubMed] [Google Scholar]