

Summary
Hoiamide A, a novel bioactive cyclic depsipeptide, was isolated from an environmental assemblage of the marine cyanobacteria Lyngbya majuscula and Phormidium gracile collected in Papua New Guinea. This stereochemically complex metabolite possesses a highly unusual structure which likely derives from a mixed peptide-polyketide biogenetic origin, and includes a peptidic section featuring an acetate extended and S-adenosyl methionine modified isoleucine moiety, a triheterocyclic fragment bearing two a-methylated thiazolines and one thiazole, as well as a highly oxygenated and methylated C15-polyketide substructure. Pure hoiamide A potently inhibited [3H]batrachotoxin binding to voltage-gated sodium channels (IC50 = 92.8 nM) and activated sodium influx (EC50 = 1.73 μM) in mouse neocortical neurons, as well as exhibited modest cytotoxicity to cancer cells. Further investigation revealed that hoiamide A is a partial agonist of site 2 on the voltage gated sodium channel.
Introduction
For more than 30 years, cyanobacteria or blue-green algae have shown an exceptional capacity to produce structurally diverse and highly bioactive secondary metabolites (Tidgewell et al., 2009). Although chemical investigations of freshwater species occurred as early as the 1930’s (Fitch et al., 1934), it was the late Richard E. Moore who pioneered the chemical and biosynthetic exploration of marine cyanobacteria, beginning in 1977 with his report on majusculamides A and B (Marner et al., 1977). The continuing interest in the natural products of marine cyanobacteria is reflected in the rich literature which has reported more than 678 compounds from these organisms, many of which possess biomedically relevant properties (Tidgewell et al., 2009). Indeed, numerous reviews of the chemistry (Tan, 2007; Van Wagoner et al., 2007; Sivonen et al., 2008), biological properties (Grindberg et al., 2008; Ersmark et al., 2008; Paul et al., 2007; Sielaff et al., 2006), and biosynthetic pathways (Ramaswamy et al., 2006; Moore, 2005) of marine cyanobacteria have appeared in recent years.
Among the reported bioactivities of marine cyanobacterial metabolites, a growing number of these appear to target the mammalian voltage-gated sodium channels (VGSCs), either as blockers (Wu et al., 2000; Edwards et al., 2004) or activators (Li et al., 2001). VGSCs are responsible for the rapid influx of sodium that underlies the rising phase of the action potential in electrically excitable cells including neurons (Catterall et al., 2007). They are comprised of voltage-sensing and pore-forming elements in a single protein complex of one principal α subunit (220–260 kDa) and one or two auxiliary β 3 subunits (33–36 kDa) (Catterall et al., 2007; Denac et al., 2000; Taylor et al., 1995). Chemical substances with activity at the VGSCs may possess therapeutic value in the treatment of epilepsy, neuropathic pain, brain damage from ischemia and certain types of stroke, as well as heart failure (Denac et al., 2000). VGSCs serve as the molecular targets for toxins that act at six or more distinct receptor sites on the channel protein (Catterall et al., 2007). They include tetrodotoxin (TTX), saxitoxin, μ-conotoxin (neurotoxin site 1); batrachotoxin, veratridine, aconitine, grayanotoxin (neurotoxin site 2); α-scorpion toxins, sea anemone toxins (neurotoxin site 3); β-scorpion toxins (neurotoxin site 4); brevetoxins and ciguatoxin (neurotoxin site 5); δ-conotoxin (neurotoxin site 6); DDT and pyrethoids (neurotoxin site 7); and antillatoxin (undefined receptor site) (Catterall et al., 2007; Li et al., 2001; Denac et al., 2000; Taylor et al., 1995). Currently, therapeutic uses of drugs that modulate sodium channel function include local anesthetics, antiarrythmics, anticonvulsants, and neuroprotective agents (Taylor et al., 1995). Due to their high affinity and specificity, natural product sodium channel ligands have served as important tools to explore the structure and function of VGSCs (Catterall et al., 2007).
As part of our ongoing assay-based screening program for new neuroactive compounds from cyanobacteria, we found that the extract of a Papua New Guinea consortium of two cyanobacteria, Lyngbya majuscula and Phormidium gracile, exhibited potent inhibition of calcium oscillations and activation of sodium influx in mouse cerebrocortical neurons. Additionally, modest cytotoxicity against human lung adenocarcinoma (H460) and mouse neuroblastoma (Neuro2a) cell lines was recorded for this extract. A combination of bioassay and 1H NMR-guided fractionation led to the discovery of a highly unusual cyclic peptide, named hoiamide A (1), as the major bioactive component of the extract. Herein, we report on the isolation and structure elucidation of this novel natural product, including complete stereochemistry, as well as a detailed biological evaluation of its neuroactive properties and site of action at the mammalian VGSC.
Results and Discussion
Hoiamide A: Isolation and Planar Structure Elucidation
A dark purple composite collection of two intertwined cyanobacteria, Lyngbya majuscula and Phormidium gracile, was collected by SCUBA in Hoia Bay, Papua New Guinea. The cyanobacterial collection was extracted repeatedly with CH2Cl2/MeOH (2:1) and fractioned by silica gel vacuum column chromatography to produce nine fractions (A-I). Fraction F was found to be highly active in suppressing calcium oscillations in mouse neocortical neurons, and was thus subjected to a 1H NMR guided fractionation comprised of repetitive silica gel flash column chromatography followed by reversed-phased HPLC to afford the new natural product, hoiamide A (1) {[α]23D +5 (c 5.5, CHCl3)}. Pure hoiamide A showed exceptional potency in suppressing calcium oscillations in this neuron preparation (IC50 = 47.5 nM).
HRESIMS of 1 yielded an [M+H]+ peak at m/z 926.4446, with a complex isotopic pattern composed of m/z 926/927/928/929/930 (100:58:28:10:3 ratio), suggesting the presence of at least three sulfur atoms. Combining this information with NMR spectroscopic data (Table 1), it was possible to derive the molecular formula C44H70N5O10S3 (calcd for C44H71N5O10S3, 926.4441), exhibiting 12 degrees of unsaturation. Acetylation of 1 under standard conditions afforded a triacetate derivative (2) with an [M+H]+ ion at m/z 1052.4743 (calcd for C50H78N5O13S3, 1052.4758), suggesting the presence of three OH groups. A 1H NMR spectrum of 1 in DMSO-d6 (Table 1) displayed resonances characteristic of a peptide containing numerous aliphatic and oxygenated residues, including one aromatic singlet at δ 8.01, two exchangeable NH peaks at δ7.86 (d, J = 8.0 Hz) and δ6.82 (d, J = 9.8 Hz), eight methines adjacent to O/N at δ5.18 (d, J = 9.8 Hz), δ4.91 (d, J = 3.4 Hz), δ4.55 (dd, J = 7.7, 2.8 Hz), δ4.28 (m), δ3.80 (m), δ3.78 (d, J = 9.6 Hz), δ3.53 (dd, J = 11.4, 10.2 Hz) and δ3.18 (dd, J = 10.2, 1.4 Hz), one methoxy group at δ3.23, and two methyl singlets at δ1.63 and δ1.54 respectively. The readily interpretable 1H NMR data were completed by eight methyl doublets at δ1.15 (d, J = 6.6 Hz), δ0.95 (d, J = 6.8 Hz), δ0.90 (d, J = 6.9 Hz), δ0.85 (d, J = 6.0 Hz), δ0.838 (d, J = 6.3 Hz), δ0.82 (d, J = 6.8 Hz), δ0.76 (d, J = 6.9 Hz) and δ0.72 (d, J = 6.6 Hz), as well as two methyl triplets at δ0.838 (t, J = 6.3 Hz) and δ0.835 (t, J = 6.8 Hz). An extensive analysis by 2D NMR, including HSQC, HMBC, COSY, TOCSY, and NOESY, revealed three interconnected partial structures which constituted the entirety of hoiamide A (1) (Figure 1).
Table 1.
NMR data of hoiamide A (1) in DMSO-d6.
| Unit | Carbon | δCa | δH, multiplicity (Hz)b | COSY | HMBC (1H to 13C) | NOESY |
|---|---|---|---|---|---|---|
| Thr | 1 | 170.2 | ||||
| 2 | 58.0 | 4.55, dd (7.7, 2.8) | 2NH, 3 | 1, 3 | 4 | |
| NH 7.86, d (8.0) | 2 | 2, 3, 5 | 4, 6, 7 | |||
| 3 | 66.0 | 4.28, m | 2, 4 | |||
| 4 | 20.2 | 1.15, d (6.6) | 3 | 2, 3 | 2, 2NH | |
| Hiva | 5 | 169.4 | ||||
| 6 | 75.8 | 4.91, d (3.4) | 7 | 5, 7, 8, 9, 10 | 2NH, 8 | |
| 7 | 30.0 | 2.18, m | 6, 8, 9 | 8 | 2NH | |
| 8 | 18.9 | 0.90, d (6.9) | 7 | 6, 7, 9 | 6 | |
| 9 | 16.0 | 0.76, d (6.9) | 7 | 6, 7, 8 | ||
| Ahdhe | 10 | 174.2 | ||||
| 11 | 44.3 | 2.30, dq (9.0, 7.2) | 12, 18 | 10, 12, 18 | 13NH | |
| 12 | 71.1 | 3.78, d (9.6) | 11 | 10, 11, 18 | 17, 18 | |
| 13 | 52.6 | 3.53, dd (11.4, 10.2) | 13NH, 14 | 14, 17, 19 | 15a, 17, 18 | |
| NH 6.82, d (9.8) | 13 | 12, 13, 19 | 11, 14, 22 | |||
| 14 | 35.8 | 1.55, m | 13, 15b, 17 | 13NH | ||
| 15 | 25.1 | a1.04, ddq (13.8, 7.8, 7.2) | 15b, 16 | 13, 16, 17 | 13 | |
| b1.42, m | 14, 15a, 16 | 14 | ||||
| 16 | 11.1 | 0.838, t (6.3) | 15a, 15b | 15 | ||
| 17 | 15.6 | 0.85, d (6.0) | 14 | 13, 14, 15 | 12, 13 | |
| 18 | 13.8 | 0.95, d (6.8) | 11 | 10, 11, 12 | 12, 13 | |
| MoCys1 | 19 | 173.1 | ||||
| 20 | 84.6 | |||||
| 21 | 41.0 | a3.19, d (11.4) | 21b | 19, 20, 22, 23 | 22 | |
| b3.81, d (11.4) | 21a | 19, 20, 22, 23 | 22 | |||
| 22 | 25.4 | 1.54, s | 19, 20, 21 | 21a, 21b, 13NH | ||
| MoCys2 | 23 | 175.8 | ||||
| 24 | 83.3 | |||||
| 25 | 42.6 | a3.46, d (11.2) | 25b | 23, 24, 26, 27 | 26 | |
| b3.51, d (11.0) | 25a | 23, 24, 26 | ||||
| 26 | 24.1 | 1.63, s | 23, 24, 25 | 25a | ||
| MoCys3 | 27 | 162.1 | ||||
| 28 | 147.4 | |||||
| 29 | 122.4 | 8.01, s | 27, 28, 30 | |||
| Dmetua | 30 | 168.3 | ||||
| 31 | 33.1 | a2.92, dd (15.8, 2.0) | 31b, 32 | 30 | 34, 43 | |
| b2.99, dd (15.8, 9.2) | 31a, 32 | 30, 32 | 43 | |||
| 32 | 79.5 | 3.80, m | 31a, 31b, 33 | 30, 43, 44 | 43 | |
| 33 | 35.7 | 2.34, m | 32, 34, 43 | 32, 24 | 35, 44 | |
| 34 | 75.2 | 5.18, d (9.8) | 33, 35 | 1, 32, 33, 35, 36, 42 | 31a, 36, 43 | |
| 35 | 38.5 | 1.70, dq (7.8, 7.2) | 34, 36, 42 | 33, 36, 42 | 33, 41, 43 | |
| 36 | 72.1 | 3.18, dd (10.2, 1.4) | 35, 37 | 34, 38, 41 | 34, 38b, 42 | |
| 37 | 33.6 | 1.48, m | 36, 38a, 38b, 41 | 38, 39 | 42 | |
| 38 | 36.6 | a1.15, m | 37, 39 | 36, 37, 39, 40, 41 | ||
| b1.26, m | 37 | 37, 39, 40, 41 | 36 | |||
| 39 | 19.9 | 1.25, m | 38a, 40 | 38 | ||
| 40 | 14.3 | 0.835, t (6.8) | 39 | 39 | ||
| 41 | 11.9 | 0.72, d (6.6) | 37 | 36, 37, 38 | 35 | |
| 42 | 10.0 | 0.82, d (6.8) | 35 | 35, 36 | 36, 37 | |
| 43 | 10.8 | 0.838, d (6.3) | 33 | 32, 33, 34 | 31a, 31b, 32, 34, 35 | |
| 44 | 56.7 | 3.23, s | 32 | 33 | ||
Recorded at 125 MHz.
Recorded at 500 MHz.
Figure 1.
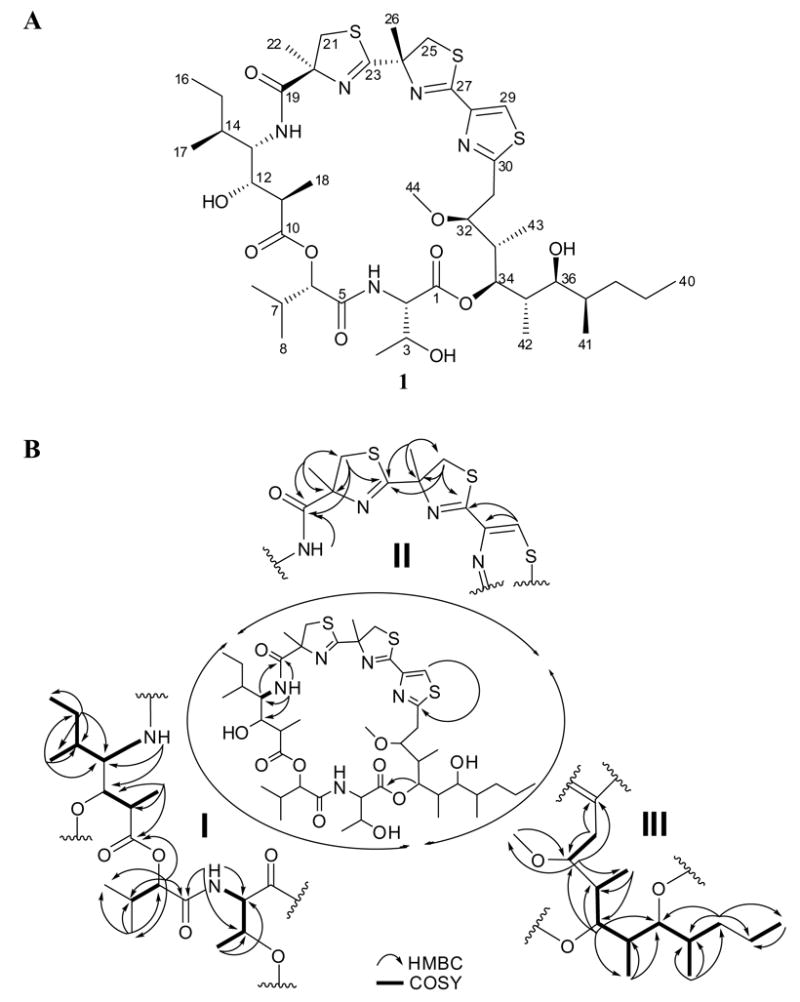
(A) Structure of hoiamide A (1) and (B) partial structures derived from NMR data and their assembly by key HMBC correlations.
The peptidic fragment I (Figure 1) was found to be comprised of threonine (C1-C4), 2-hydroxyisovaleric acid (Hiva, C5-C9), and a new C3-extended isoleucine residue (4-amino-3-hydroxy-2,5-dimethylheptanoic acid, Ahdhe, C10-C18). COSY correlations between a proton at δ3.53 (Ahdhe-H13) and an exchangeable proton at δ6.82 (Ahdhe-C13-NH) and a methine proton at δ1.55 (Ahdhe-H14), suggested an additional aliphatic amino acid subunit. The high field methine proton was further coupled to protons defining a sec-butyl side chain according to HMBC and TOCSY data, thus suggesting the amino acid isoleucine. However, the H13 ‘alpha’ proton showed additional coupling to an oxygenated methine at δ3.78 (d, J = 9.6 Hz, Ahdhe-H12) with an associated carbon at δ71.1 (Ahdhe-C12). This latter proton signal was coupled to an upfield methine at δ2.30 (dq, J = 9.0, 7.2 Hz, Ahdhe-H11) which in turn showed COSY correlations to a methyl doublet at δ0.95 (J = 6.8 Hz, Ahdhe-H18). Finally, the H11 methine was shown by HMBC to be adjacent to the carbonyl at δ174.2 (Ahdhe-C10), and hence, this residue was defined as a three carbon extended isoleucine residue. Long distance HMBC correlations between the exchangeable proton at δ7.86 (Thr-C2-NH) and carbon resonances at δ58.0 (Thr-C2), δ66.0 (Thr-C3) and δ169.4 (Hiva-C5), as well as between the α-proton at δ4.91 (Hiva-H6) and carbons at δ169.4 (Hiva-C5), δ30.0 (Hiva-C7), and δ174.2 (Ahdhe-C10), firmly established the connectivity among these three residues. Similar modifications of Ile to produce Ahdhe-like residues have been observed in several other marine natural products such as didemnins A-E (Rinehart et al., 1988) and various dolastatins (Pettit et al., 1987; Luesch et al., 2001). In the case of hoiamide A (1), the ketide-extended isoleucine is methylated at the α-carbon, likely from S-adenosyl methionine (SAM). This contrasts with the pattern of methylation seen in dolastatin 10 (Supplemental Data) wherein methylation occurs on the resulting secondary alcohol at the β position (e.g. the alcohol deriving from reduction of the former Ile carbonyl group). Notably, a consequence of this alternate site of methylation in hoiamide A is the creation of an additional stereocenter in the molecule.
A second substructure in 1 (Figure 1) was initiated by HMBC correlations between a methyl singlet at δ1.54 (MoCys1-H22) and quaternary carbons at δ173.1 (MoCys1-C19), δ84.6 (MoCys1-C20), as well as a methylene at δ41.0 (MoCys1-C21), and suggested an a-methyl substituted cysteine-type residue (MoCys: modified cysteine). The diastereotopic methylene protons at δ3.81 (MoCys1-H21b) and δ3.19 (MoCys1-H21a), in combination with a key HMBC correlation between both H21 protons and the C23 deshielded carbon signal at d175.8, defined this as a cyclized thiazoline. Additional HMBC correlations between the various protons of this system and all neighboring carbons two or three bonds away helped confirm these assignments (Table 1). The deshielded signal at δ175.8 (MoCys2-C23) showed HMBC correlations to a second uniquely deshielded singlet methyl group at δ1.63 (MoCys2-C26), and thus provided linkage to an additional residue. A series of 1H and 13C NMR signals highly comparable to those in MoCys1 were identified from HMBC data, establishing this second methyl substituted thiazoline residue (Table 1). MoCys2 was linked to a thiazole ring via tandem HMBC correlations between a deshielded quaternary carbon at δ162.1 (MoCys3-C27) and both the C25 protons and a deshielded singlet proton (δ8.01, MoCys3-H29). The two other carbon atoms of this latter residue were also identified by HMBC couplings from the lone thiazole proton and from their distinctive 13C NMR shifts. A comparable arrangement of two methylated thiazolines followed by a thiazole heterocycle was first reported in the tantazole (Carmeli et al., 1990, 1993) and mirabazole (Carmeli et al., 1991) series of highly bioactive cyanobacterial metabolites, and the reported chemical shift data closely match that reported here for hoiamide A (1) at similar positions. Because substructure II was comprised of two thiazolines and one thiazole, all three of which are cysteine-derived, the sulfur component of the molecular formula was fully satisfied. Finally, HMBC correlations between the protons at δ6.82 (Ahdhe-C13-NH) and δ3.53 (Ahdhe-H13) with the carbonyl resonance at δ173.1 (MoCys1-C19) provided linkage between fragments I and II in hoiamide A.
The final fragment (III) comprising hoiamide A (1) was determined to be a C15-polyketide (C30-C44) possessing high levels of methylation and oxygenation (Figure 1). The thiazole carbon resonance in fragment II at δ168.3 (Dmetua-C30) showed HMBC correlations to diastereotopic methylene protons at δ2.99 (dd, J = 15.8, 9.2 Hz, Dmetua-H31b) and δ2.92 (dd, J = 15.8, 2.0 Hz, Dmetua-H31a) (δC 33.1). These methylene protons showed COSY correlations to a deshielded methine proton at δ3.80 (Dmetua-H32) which in turn was connected by HMBC to the methyl group of a methoxy resonance as well as to another deshielded methine (δ5.18, Dmetua-H34). By 1H and 13C NMR chemical shift arguments, this signal was proposed to be attached to an ester, a hypothesis later verified by HMBC (see below). However, because this ester methine signal was strongly coupled to two different high field methine resonances according to COSY data, it was placed beta to the δ3.80 methine. The δ2.34 methine (Dmetua-H33) predicted to intervene between these two deshielded methines was also proton coupled to a doublet methyl, and HMBC between the δ3.80 signal and the carbon atom of this methyl group (δ10.8, Dmetua-C43) confirmed these atom relationships. The methine to the other side of the δ5.18 signal was part of a connected spin system involving sequential methylmethine, oxymethine, methylmethine and methylene groups (Table 1). By chemical shift arguments (δ3.18, Dmetua-H36), and consideration of the molecular formula, the oxymethine at C36 was deduced to be a free secondary hydroxyl group. The latter methylene of this spin system was connected to a terminating ethyl group by HMBC, completing partial fragment III as a 5,7-dihydroxy-3-methoxy-4,6,8-trimethylundecanoyl derived residue (Dmetua). With fragments I-III accounting for all atoms of the molecular formula of 1 but only 11 of 12 degrees of unsaturation, it was concluded that the overall molecule formed a macrocycle. The deshielded methine at δ5.18 (Dmetua-H34) exhibited numerous HMBC cross peaks (Figure 1) including a highly prominent 3-bond correlation with an ester carbon at δ170.2 (Thr-C1), and thus established the macrocyclic planar structure of hoiamide A (1).
Hoiamide A: Relative and Absolute Stereochemistry
Next, we turned our attention to the absolute stereochemistry of hoiamide A (1). With 15 stereocenters, our initial strategy was to attempt crystallization to perform an X-ray analysis. Unfortunately, numerous efforts aimed at forming crystals from the natural product 1, its triacetate (2), or the freshly prepared mono p-bromobenzoic ester derivative (3), failed to produce X-ray quality crystals. Thus, hoiamide A (1) was hydrolyzed with 6 N HCl (110 °C, 22 h) and the hydrolyzate subjected to chiral HPLC, revealing the presence of (S)-Hiva in comparison with commercial standards. The same chromatographic technique was applied to another sample of 1 which was first subjected to ozonolysis and oxidative workup prior to hydrolysis, and led to the detection of 2-methylcysteic acid (MCya). Comparison of retention times with freshly synthesized MCya standards (Pattenden et al., 1993; Calmes et al., 1997) confirmed the occurrence of only (S)-MCya. Marfey’s analysis (Marfey, 1984) was performed using aliquots of the acid hydrolyzate as well as amino acid standards, employing N-(3-fluoro-4,6-dinitrophenyl)-L-alaninamide (L-FDAA) as the derivatizing agent, and revealed the presence of (2S,3R)-Thr in hoiamide A (1).
The relative stereochemistry of the C10-C18 (C3-extended Ile) and C30-C44 (Dmetua) sections was elucidated via J-based configuration analysis (Figure 2) (Matsumori et al., 1999). This procedure makes use of 3JH-H and 2–3JC-H values combined with NOE and ROE data to determine the conformation of adjacent stereocenters, and has been successfully applied to define the relative stereochemistry of substituted polyketides in other cyanobacterial metabolites such as phormidolide (Williamson et al., 2002), apratoxin A (Luesch et al., 2001), and largamide H (Plaza and Bewley, 2006). Homonuclear coupling constants were obtained from 1H-NMR, E.COSY (Griesinger et al., 1987) and 1D TOCSY (Uhrin and Barlow, 1997) spectra whereas heteronuclear coupling values were accurately measured from HETLOC (Uhrin et al., 1998) and HSQMBC (Williamson et al., 2000) experiments (Marquez et al., 2001). Starting with the C10-C18 section, the relative stereochemistry along C11-C12 could not be determined using J values alone because these did not distinguish between the two possible rotamers (threo A3, erythro B3) with H11/H12 in an anti relationship (3JH11-H12 = 9.0 Hz). Fortunately, NOESY data revealed the spatial proximity of H18 and H12, H18 and H13, as well as H11 and C13-NH, in agreement only with the erythro rotamer B3. Small homonuclear and heteronuclear J values were observed between C12 and C13, a result consistent only with the threo rotamer A1. The H13-H14 protons were also in an anti configuration (3JH13-H14 = 9.6 Hz) and thus, NOE correlations between H13/H17, H12/H17, and H14/C13-NH, confidently assigned it as rotamer B3.
Figure 2.

Homonuclear and heteronuclear coupling constant values used to assign the relative configuration of Ahdhe (C10-C18) and Dmetua (C30-C44). The magnitude of the coupling constants, (S) for small and (L) for large, allowed identification of gauche and anti orientations between the indicated atoms. For 1,2-methine systems in which the threo rotamer A3 can not be distinguished from the erythro rotamer B3, NOE data (↔) provided an unambiguous assignment. aRelative configuration and rotamer designation according to Matsumori et al. (1999). bWeak couplings with 3JH,H < 0.5 Hz were considered as 0 Hz. cNo correlation observed in HSQMBC as expected for a coupling of 0 Hz.
The C30-C44 section (Dmetua) exhibited anti configurations between adjacent methines C32-C33 (3JH32-H33 = 7.9 Hz), C33-C34 (3JH33-H34 = 9.6 Hz) and C35-C36 (3JH35-H36 = 10.2 Hz), which upon analysis of key NOE correlations (H31/H43, H43/H35 and H42/H37, respectively), were assigned as erythro rotamers B3. The remaining 1,2-methine systems C34-C35 (3JH34-H35 = ~0 Hz) and C36-C37 (3JH36-H37 = ~0 Hz) were deduced to possess gauche conformations between their protons, and were found to be threo rotamers A1 in accordance with their heteronuclear J values. This proposed configuration was in agreement with all of the NOEs observed within each of the substructures.
Connection of the relative stereochemistry of the Ahdhe (C10-C18) portion with the MoCys1 residue was made possible through NOE interactions between the methine protons at δ2.30 (Ahdhe-H11) and δ1.55 (Ahdhe-H14) with the NH proton at δ6.82 (Ahdhe-C13-NH) which in turn was correlated with the methyl singlet at δ1.54 (MoCys1-H22) (Figure 2). Moreover, because we had established the absolute configuration of the MoCys1 residue, these observed NOEs allowed assignment of the absolute stereochemistry of Ahdhe as 11R, 12S, 13S and 14S. Molecular modeling studies of compound 1 (Supplemental Data) confirmed that this NOE interaction was only possible with the S configuration at Ahdhe-C13. Thus, as with dolastatin 10, the natural configuration of Ile [e.g. L-(2S, 3S)-Ile] appears to have been one of the precursors to this portion of hoiamide A (1).
Our initial plan for determining the absolute stereochemistry of the Dmetua substructure (C30-C44) was to form the C36 Mosher’s ester derivative of compound 1. However, this position in hoiamide A (1) proved to be quite resistant to acylation using Mosher’s acid chloride. Instead, loss of H2O from the Thr residue yielded the C2-C3 olefinic derivative 4 (Scheme 1). Likely, steric effects disfavor reaction with the bulky MTPA acid chloride. As an alternative approach, 26 mg of the acid hydrolyzate of 1 were dissolved in MeOH/HCl and stirred at room temperature over an extended period so as to produce methyl esters of the various units comprising hoiamide A (1). Upon solvent removal under vacuum, the residue was partitioned between EtOAc and H2O, and both layers were subjected to extensive reversed-phase HPLC, 1H-NMR and LCMS analyses, in search for the C10-C18 and C30-C44 sections. One such fragment, 5 {[α]23D +18 (c 0.015, MeCN)} (Scheme 1), was a major component of the EtOAc fraction, and displayed an M+ at m/z 355.1806 consistent with the molecular formula C18H29NO4S (calcd 355.1812, 5 degrees of unsaturation).
Scheme 1a.

aReagents and conditions: (a) 6M HCl, 110 °C, 22 h; (b) MeOH, HCl(cat), 25 °C, 3 weeks; (c) solvent partitioning H2O/EtOAc; (d) MTPA, pyridine, DMAP, 25 °C, 18 h.
The 1H NMR spectrum of 5 (Table 2) showed diagnostic resonances for portions of the Dmetua substructure of 1: a thiazole aromatic singlet at δ8.09, a methoxy singlet at δ3.95, three oxygenated methines at δ3.70 (d, J = 9.6 Hz), δ3.43 (dd, J = 10.2, 4.8 Hz) and δ2.88 (dd, J = 10.2, 1.8 Hz), three methyl doublets at δ0.96 (J = 6.6 Hz), δ0.88 (J = 6.0 Hz) and δ0.87 (J = 7.2 Hz), as well as one methyl triplet at δ0.79 (J = 7.2 Hz). Surprisingly, the singlet at δ3.95 (H18) exhibited HMBC correlations with quaternary carbon resonances at δ161.6 (C1) and δ145.6.6 (C2), indicating that this was a methyl ester at the C-terminus of the Dmetua residue. The diastereotopic methylene protons at δ3.22 (dd, J = 14.4, 10.2 Hz, H5b) and δ3.12 (dd, J = 14.4, 1.8 Hz, H5a) showed HMBC cross peaks with carbons at δ169.2 (C4) in the thiazole ring, a methyl-bearing methine at δ39.2 (C7), and the oxygenated methine at δ77.5 (C6), which in turn was connected to another methine at δ83.2 (C10) through an ether linkage. This latter linkage was established on the basis of reciprocal HMBC correlations between their attached protons (H6/C10 and H10/C6). Moreover, this ether bridge formed a new six-membered heterocycle in derivative 5, thereby satisfying the required 5 degrees of unsaturation. Additional 2D NMR data placed a 2-pentyl side chain on the carbon at δ83.2 (C10) and the final oxygenated methine at δ3.43 (H8) flanked by methyl-bearing methines at δ1.95 (H7) and δ1.68 (H9) to either side; each of the latter were connected to the bridged system described above. This analysis allowed assignment of the planar structure of derivative 5 (Scheme 1) which likely results from the Dmetua residue via displacement of the methoxy group at C32 by the C36 hydroxy group with inversion of configuration (the absence of an epimer at C6 in 5 supports an SN2 mechanism).*
Table 2.
NMR data for derivative (5) in CDCl3.
| Carbon | δCa | δH, multiplicity (Hz)b | COSY | HMBC (1H to 13C) | NOESY |
|---|---|---|---|---|---|
| 1 | 161.6 | ||||
| 2 | 145.6 | ||||
| 3 | 127.8 | 8.09, s | 1, 2, 4 | ||
| 4 | 169.2 | ||||
| 5 | 37.7 | a3.12, dd (14.4, 1.8) | 6, 5b | 4, 6, 7 | 17 |
| b3.22, dd (14.4, 10.2) | 6, 5a | 4, 6, 7 | 17 | ||
| 6 | 77.5 | 3.70, d (9.6) | 5a, 5b, 7 | 4, 5, 8, 10, 17 | 8, 10 |
| 7 | 39.2 | 1.95, m | 6, 8, 17 | 8, 9, 17 | |
| 8 | 76.7 | 3.43, dd (10.2, 4.8) | 7, 9 | 9, 16, 17 | 6, 10, 16 |
| 9 | 33.9 | 1.68, m | 8, 10, 16 | 8, 10 | 15, 17 |
| 10 | 83.2 | 2.88 dd (10.2, 1.8) | 9, 11 | 6, 8, 11, 12, 15, 16 | 6, 8, 16 |
| 11 | 33.2 | 1.66, m | 10, 15, 12a, 12b | 12, 13, 15 | 14 |
| 12 | 37.1 | a1.24, m | 11, 12b | 10, 11, 13, 14, 15 | 15 |
| b1.33, m | 11, 12a | 10, 11, 13, 14, 15 | 15 | ||
| 13 | 20.4 | 1.16, m | 14 | 11, 12, 14 | |
| 14 | 14.4 | 0.79, t (7.2) | 13 | 12, 13 | 11 |
| 15 | 13.1 | 0.87, d (7.2) | 11 | 10, 11, 12 | 9, 12a, 12b |
| 16 | 13.0 | 0.88, d (6.0) | 9 | 8, 9, 10 | 8, 10 |
| 17 | 5.6 | 0.96, d (6.6) | 7 | 6, 7, 8 | 5a, 5b, 9 |
| 18 | 52.5 | 3.95, s | 1, 2 |
Recorded at 125 MHz.
Recorded at 500 MHz.
Further NMR analysis of compound (5) provided insightful information on the relative configuration of Dmetua which confirmed the assignments made by J-based configurational analysis. The highly shielded methyl doublet at δ0.96 (H17) was situated in an axial position, together with protons at δ3.70 (H6), δ3.43 (H8), δ1.68 (H9) and δ2.88 (H10), based on coupling constant information. Such an arrangement was consistent with NOESY data (Scheme 1), which additionally defined the stereocenter at C11 outside the tetrahydropyran ring. The absolute stereochemistry at C8 was determined as S by the modified Mosher method (Ohtani et al., 1991), and with the relative configuration of 1 established via J-based analysis and of 5 by 1H-1H coupling constants and NOE data, the absolute stereochemistry of the Dmetua fragment could be assigned as 32S, 33S, 34R, 35S, 36S and 37R. This completed the assignment of structure and stereochemistry of hoiamide A (1).
Hoiamide A (1) represents a particularly interesting example of a natural product deriving from a mixture of the polyketide synthase (PKS) and nonribosomal peptide synthetase (NRPS) pathways. The initial polyketide section, the Dmetua fragment, is unique to 1 and shows a typical remnant oxidation pattern at several sites, as well as methylation at the C-2 positions of predicted acetate units which entirely derive from SAM methylation events in cyanobacteria. Excluding the branching carbons of the Dmetua moiety, the carbon chain is eleven carbons long, and suggests either a propionate starter unit, the SAM methylation of an acetate starter, or a truncation of a hexaketide. Of these alternatives, we favor the SAM methylation possibility because a) propionate is unknown as a starter unit in cyanobacterial polyketides (Grindberg et al., 2008) and b) the remaining oxidations and methylations are positionally inconsistent with a terminal decarboxylation. Next, an intriguing tetrapeptide is created, with the three cysteine residues subject to heterocyclization reactions followed by dehydration. The first of these is further modified by oxidation to the thiazole ring whereas the latter two are stereospecifically methylated at the alpha carbon, again, likely involving SAM as the methyl source. Cysteine-derived linear arrangements of 2,4-disubstituted thiazolines/thiazole rings possess a diverse range of biological activities (Carmeli et al., 1990, 1991, 1993; Naegeli and Zahner, 1980; Jansen et al., 1992) and are believed to play an important role in their mechanism of action (Wipf et al., 1998; Kwan et al., 2008). The fourth residue, isoleucine, is modified by a PKS extension, reduction of the ketone to an alcohol, and methylation of the acetate C-2 carbon. Due to these tailoring reactions, this homologated Ile residue is chirally dense with four contiguous asymmetric centers. Next, a hydroxy acid common to many cyanobacterial natural products, hydroxyisovaleric acid (Hiva), is incorporated into hoiamide A (1). In the one other case where the gene structure for Hiva incorporation has been studied in a cyanobacterium [e.g. the natural product hectochlorin (Ramaswamy et al., 2007)], this occurs through initial activation of the a-keto acid and subsequent reduction to the hydroxy-acid once tethered to the NRPS condensation domain. Finally, the only amino acid in hoiamide A (1) not modified by a secondary tailoring reaction, L-threonine, is attached as the final residue. This terminating amino acid sequence, Thr-Hiva, has not been described in any other known cyanobacterial natural product. Finally, the C34 hydroxyl group in the Dmetua group is presumably involved in both hydrolysis from the NRPS as well as ester formation to the final 26-membered cyclic depsipeptide structure.
Hoiamide A: Pharmacological Studies
Hoiamide A Produces Sodium Influx in Neocortical Neurons
Systematic screening of natural product extracts, using a combination of high throughput Ca2+ and Na+ influx assays in neocortical neurons, led to the identification of the novel bioactive cyclic peptide hoiamide A (1). Further characterization demonstrated that hoiamide A produced a rapid and concentration-dependent elevation of neuronal [Na+]i in neocortical neurons as detected with the sodium sensitive fluorescent dye, SBFI (Figure 3a). The concentration-response curve for hoiamide-induced sodium influx was best fit by a three-parameter logistic equation yielding an EC50 value of 2.31 μM (0.82 – 6.53, 95% Confidence Intervals, [95% CI]) (Figure 3c). We and others have demonstrated that batrachotoxin is a full agonist at neurotoxin site 2 on the sodium channel α subunit (Cao et al., 2008; Catterall, 1977). We therefore compared the relative efficacies of hoiamide A and batrachotoxin as stimulators of neuronal [Na+]i. As shown in Figure 3b, batrachotoxin produced a rapid and concentration-dependent elevation of neuronal [Na+]i with an EC50 value of 11.4 nM (6.92–18.8, 95% CI). The EC50 value for batrachotoxin-induced sodium influx is consistent with that reported by Cao et al (2008). The maximum response for batrachotoxin-induced elevation of neuronal [Na+]i was greater than 60 mM, whereas the maximal response for hoiamide A-induced increase in [Na+]i was less than 20 mM. Nonlinear regression analysis of the hoiamide A concentration-response data revealed that the relative efficacy of hoiamide A for elevation of neuronal [Na+]i was 0.38 (Figure 3c). These data are consistent with hoiamide A (1) acting as a partial agonist at neurotoxin site 2.
Figure 3.

(A) Time-response relationships for hoiamide A-induced elevation of neuronal [Na+]i in neocortical neurons; (B) Time-response relationships for batrachotoxin-induced elevation of neuronal [Na+]i in neocortical neurons; (C) Nonlinear regression analysis of the peak SBFI (340/380) response versus hoiamide A and batrachotoxin concentration; (D) TTX inhibition of hoiamide A-induced elevation of neuronal [Na+]i. These data were obtained from at least three different cultures performed in duplicate.
TTX Antagonizes Hoiamide A-induced Elevation of Neuronal [Na+]i in Neocortical Neurons
VGSCs, and the N-methyl-D-aspartic acid (NMDA) and a-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) subtypes of glutamate receptors represent the main routes for sodium influx in neocortical neurons. To further explore the mechanism of hoiamide A-induced sodium influx, we evaluated the effects of TTX (VGSC inhibitor), MK-801 (NMDA receptor antagonist) and NBQX (AMPA receptor antagonist) on hoiamide A-induced elevation of neuronal [Na+]i. Pretreatment with TTX (1 μM) eliminated the hoiamide A-induced elevation of neuronal [Na+]i demonstrating the requirement for activation of VGSCs for hoiamide A (1) induced sodium influx (Figure 3d). Pretreatment with MK-801 (1 μM) produced a partial, but statistically significant, decrease in hoiamide A-induced elevation of neuronal [Na+]i (Figure S24a, Supplemental Data). This effect may be due to the engagement of NMDA receptors as a result of membrane depolarization; a consequence of activation of VGSCs. The AMPA receptor antagonist NBQX (5 μM) had no effect on hoiamide A-induced Na+ influx (Figure S24b). Considered together these data demonstrate that hoiamide A (1) is a VGSC activator.
Deltamethrin and PbTx-3 Enhancement of Hoiamide A-induced Elevation of Neuronal [Na+]i in Neocortical Neurons
Although the multiple neurotoxin sites on VGSCs are topologically distinct, there are strong allosteric interactions between them. Pyrethroids are synthetic insecticidal compounds that resemble natural pyrethrins (Lombet et al., 1988). Pyrethroids specifically bind to a site on the sodium channel α subunit that is distinct from neurotoxin site 1–6 and is allosterically coupled to sites 2, 3 and 5 (Trainer et al., 1997, 1993). Therefore, to further explore the molecular mechanism of action for hoiamide A (1), we assessed the effect of a subthreshold concentration of deltamethrin (0.1 μM) on the hoiamide A-induced elevation of neuronal [Na+]i in neocortical neurons. A concentration of deltamethrin (0.1 μM), which was inactive alone (Figure S25b), significantly increased the maximum response of hoiamide A-induced sodium influx from an AUC (fluorescence x min) value of 89.9 (81.5–98.2, 95%CI) to 141.4 (119.7–162.5, 95% CI) without altering the hoiamide A EC50 value (Figure 4a, 4c, 4e). These data provide direct evidence that hoiamide A (1) binds to a site on the sodium channel that is allosterically coupled to the pyrethroid insecticide binding site. Brevetoxins are potent lipid-soluble polyether neurotoxins produced by the marine dinoflagellate, Karenia brevis (Baden, 1989). Brevetoxins interact specifically with neurotoxin site 5 on the α subunit of VGSC. Evidence for the allosteric coupling of neurotoxin site 2 and site 5 has been provided by demonstrating that the binding of [3H]batrachotoxin to neurotoxin site 2 is enhanced by brevetoxins (Li et al., 2001; Sharkey et al., 1987). We therefore determined whether an allosteric interaction existed between site 5 and the hoiamide A (1) binding site. A concentration of brevetoxin 3 (PbTx-3, 5 nM), which was inactive alone (Figure S25a), also produced a significant increase in the maximum response of hoiamide A-induced sodium influx from an AUC (fluorescence x min) value of 89.9 (81.5–98.2, 95% CI) to 143.4 (129.7–157.4, 95% CI) with no corresponding effect on the hoiamide A EC50 value (Figure 4a, 4b, 4d). These data suggest that a positive allosteric interaction exists between the hoiamide A (1) binding site and neurotoxin site 5 on VGSCs. These positive allosteric interactions between hoiamide A, and PbTx-3 and deltamethrin are therefore consistent with hoiamide A interacting with neurotoxin site 2.
Figure 4.

Deltamethrin (0.1 μM) and PbTx-3 (5 nM) enhance hoiamide A-induced elevation of neuronal [Na+]i. (A) Time-response relationships for hoiamide A-induced elevation of neuronal [Na+]i ; (B) Time-response relationships for hoiamide A-induced elevation of neuronal [Na+]i in the presence of 5 nM PbTx-3; (C) Time-response relationships for hoiamide A-induced elevation of neuronal [Na+]i in the presence of 0.1 μM deltamethrin; (D) Nonlinear regression analysis of the SBFI response [area under the curve (AUC)] versus the concentration of hoiamide A in the presence and absence of 5 nM PbTx-3; (E) Nonlinear regression analysis of the SBFI response [area under the curve (AUC)] versus the concentration of hoiamide A in the presence and absence of 0.1 μM deltamethrin. Each data point represents the Mean ± S.E.M. from two experiments performed in triplicate.
Hoiamide A Inhibits the Specific Binding of [3H]batrachotoxin A 20-α-benzoate ([3H]BTX) to Neurotoxin Site 2 on VGSCs in Neocortical Neurons
[3H]BTX is a radioligand probe that binds to neurotoxin site 2 on the VGSC α subunit3 (Catterall et al., 1981). This ligand interacts preferentially to the active or open conformation of the channel, and the specific binding is sensitive to conformational changes induced by the binding of neurotoxins at other sites on the sodium channel α subunit. We therefore used [3H]BTX as a probe to provide further evidence for hoiamide A (1) interaction with neurotoxin site 2 on VGSCs. Inasmuch as the specific binding of [3H]BTX is quite low in the absence of other sodium channel gating modifiers and is dramatically increased by the allosteric interaction of deltamethrin and brevetoxin, we assessed the influence of hoiamide A on [3H]BTX binding in the presence of these allosteric modulators. As depicted in Figure 5, hoiamide A (1) produced a concentration-dependent inhibition of [3H]BTX specific binding in the presence of PbTx-3 (300 nM) and deltamethrin (10 μM). The hoiamide A IC50 value was 92.8 nM (56.6–152.1, 95% CI). The prototypic neurotoxin site 2 ligand veratridine also inhibited [3H]BTX specific binding to VGSCs with an IC50 value of 33.0 μM (24.2–45.1, 95% CI) indicating that it was much less potent than hoiamide A (Figure 5). Considered together these data directly demonstrate that hoiamide A (1) interacts with neurotoxin site 2 on the VGSC α subunit.
Figure 5.

Hoiamide A and veratridine inhibit the specific binding of [3H]BTX to VGSCs. Each data point represents the mean ± S.E.M. from three experiments performed in triplicate.
Inhibition of BTX-induced Increase in Neuronal [Na+]i by Hoiamide A
We have previously demonstrated that saturating concentrations of neurotoxin site 2 ligands produce different maximal responses in sodium influx (Cao et al., 2008). We have demonstrated herein that hoiamide A (1) produced a maximum sodium influx that was less than that of the full agonist, batrachotoxin (Figure 3c). This suggests that hoiamide A acts as a partial agonist at neurotoxin site 2 on VGSCs. Catterall (1995) first demonstrated partial agonism at neurotoxin site 2 with a combination of a full activator (batrachotoxin) and a weak activator (aconitine). This interaction resulted in an inhibition of the response to the full activator by the weak activator. We have previously demonstrated that gambierol, a low efficacy partial agonist at neurotoxin site 5, inhibited the full agonist PbTx-1 induced elevation of neuronal [Na+]i (Cao et al., 2008). Thus, a low-efficacy agonist may act as a functional antagonist of a high-efficacy agonist at either neurotoxin site 2 or 5 on VGSCs (Cao et al., 2008). Given the modest efficacy of hoiamide A (1) as a stimulator of neuronal [Na+]i, we performed a titration with hoiamide A in the presence of a fixed concentration of the full agonist, batrachotoxin (30 nM). As depicted in Figure 6, the titration with hoiamide A produced a concentration-dependent reduction in batrachotoxin-induced sodium influx (SBFI AUC). These data demonstrate that hoiamide A (1) and batrachotoxin interact in a mutually exclusive manner with a common recognition site on VGSCs. The partial agonist activity of hoiamide A is therefore demonstrated by the ability of hoiamide A to produce a concentration-dependent inhibition of batrachotoxin-induced elevation of [Na+]i in neocortical neurons.
Figure 6.

(A) Time-response relationships for hoiamide A reversal of batrachotoxin (30 nM)-induced elevation of neuronal [Na+]i; (B) Nonlinear regression analysis of the hoiamide-induced reversal of the integrated SBFI response [area under the curve (AUC)] to batrachotoxin (30 nM) in neocortical neurons. Histogram represents the responses to batrachotoxin (30 nM) and hoiamide A (30 μM) alone. These experiments were performed twice in triplicate with similar results.
Significance
Marine cyanobacteria represent a particularly rich source of structurally diverse neurotoxic secondary metabolites, such as antillatoxin, kalkitoxin and jamaicamide A (Grindberg et al., 2008). In the present study, we describe the isolation and structure elucidation of hoiamide A (1), a novel cyclic depsipeptide discovered in an environmental assemblage of Lyngbya majuscula and Phormidium gracile. Featuring three heavily modified regions (peptidic, triheterocyclic and polyketide), hoiamide A (1) illustrates the intricate biosynthetic machinery responsible for the production of cyanobacterial compounds. Using a combination of neurochemical and pharmacological approaches, we have demonstrated that hoiamide A (1) represents a new chemotype to exhibit partial agonism at the neurotoxin site 2 on VGSCs. VGSCs are vital for central nervous system function. We and others have shown that an array of sodium channel gating modifiers produce [Na+]i increments that are of sufficient magnitude to increase NMDA receptor channel activity (Cao et al., 2008; Yu and Salter, 1998, 1999). Sodium channel activators therefore appear capable of mimicking activity-dependent control of neuronal development by upregulating NMDA receptor signaling pathways that influence neuronal growth and plasticity (George et al., 2009). Voltage-gated sodium channel activators may accordingly represent a novel pharmacologic strategy to regulate neuronal plasticity. Hoiamide A (1) therefore represents a structurally novel lead compound for drugs capable of promoting neuronal growth and plasticity.
Experimental Procedures
[See Supporting Information for General Experimental Procedures and Analytical Data for Derivatives of Hoiamide A (1)]
Collection of Biological Material
A composite collection of the marine cyanobacteria Lyngbya majuscula and Phormidium gracile (voucher specimen available from WHG as collection number PNG5-28-02-12) was obtained at a depth of 5–10 m from Hoia Bay, Papua New Guinea, on May 2002 (10° 15.990 S, 150° 46.169 E). Samples were stored in 70% EtOH at −20°C prior to extraction. Taxonomy was assigned by microscopic comparison with the description given by Komarek and Anagnostidis (2005), as well as Geitler (1932) (Supplemental Data).
Extraction and Isolation
Approximately 186 g (dry wt) of the cyanobacteria were extracted repeatedly with CH2Cl2/MeOH (2:1) to afford 4.377 g of crude extract. A portion of this material (3.5474 g) was fractionated by silica gel vacuum liquid chromatography using a stepwise gradient solvent system of increasing polarity starting from 10% EtOAc in hexanes to 100% MeOH, to produce nine fractions (A-I). The bioactive fraction F (0.7890 g) was subjected to a 1H NMR-guided fractionation comprised of two silica gel columns (60 and 50% EtOAc in hexanes, respectively) and reversed-phase HPLC (Phenomenex Jupiter 10 μ C18, 250 x 10 mm, 65% MeCN/H2O with 0.1% TFA at 3 mL/min, detection at 210, 254 and 280 nm) to yield 150.0 mg of compound (1).
Hoiamide A (1): colorless amorphous solid; [α]23D +5 (c 5.5, CHCl3); UV (MeCN) λmax 250 nm (log ε 3.99); IR (neat) 3379, 2967, 2932, 2879, 1743, 1607, 1519, 1154, 1085, 735 cm−1; 1H and 13C NMR data, see Table 1; HR ESIMS m/z [M+H]+ 926.4446 (calcd for C44H71N5O10S3, 926.4441).
Neocortical Neuron Culture
Primary cultures of neocortical neurons were obtained from embryonic day 16 Swiss-Webster mice as described previously (Cao et al., 2007). Briefly, pregnant mice were euthanized by CO2 asphyxiation, and embryos were removed under sterile conditions. Neocortices were collected, stripped of meninges, minced by trituration with a Pasteur pipette and treated with trypsin for 25 min at 37 °C. The cells were then dissociated by two successive trituration and sedimentation steps in soybean trypsin inhibitor and DNase containing isolation buffer, centrifuged and resuspended in Eagle’s minimal essential medium with Earle’s salt (MEM) and supplemented with 1 mM L-glutamine, 10% fetal bovine serum, 10% horse serum, 100 IU/ml penicillin and 0.10 mg/ml streptomycin, pH 7.4. Cells were plated onto poly-L-lysine-coated 96-well (9 mm) clear-bottomed black-well culture plates (Costar) at a density of 1.5 x 105 cells/well. Cells were then incubated at 37 °C in a 5% CO2 and 95% humidity atmosphere. Cytosine arabinoside (10 μM) was added to the culture medium on day 2 after plating to prevent proliferation of nonneuronal cells. The culture media was changed both on days 5 and 7 using a serum-free growth medium containing Neurobasal Medium supplemented with B-27, 100 I.U./mL penicillin, 0.10 mg/mL streptomycin, and 0.2 mM L-glutamine. Neocortical cultures were used in experiments between 8–13 days in vitro (DIV). All animal use protocols were approved by the Institutional Animal Care and Use Committee (IACUC).
Intracellular Sodium Concentration ([Na+]i) Measurement
The neocortical neurons cultured in 96-well plate (DIV 8–13) were washed four times with Lock’s buffer (in mM: 8.6 Hepes, 5.6 KCl, 154 NaCl, 5.6 Glucose, 1.0 MgCl2, 2.3 CaCl2, 0.0001 glycine, pH 7.4) using an automated cell washer (Bioteck instrument Inc., VT, USA). The background fluorescence of each well was measured and averaged prior to dye loading. Cells were then incubated for 1 h at 37 ºC with dye loading buffer (50 μL/well) containing 10 μM SBFI-AM and 0.02% Pluronic F-127. After 1 h incubation in dye loading medium, cells were washed five times with Locke’s buffer, leaving a final volume of 150 μL in each well. The plate was then transferred to the plate chamber of a FLEXstation™ II (Molecular Devices, Sunnyvale, CA, USA). Cells were excited at 340 nm and 380 nm and Na+-bound SBFI emission was detected at 505 nm. Fluorescence readings were taken once every 5 s for 60 s to establish the baseline and then 50 μL of neurotoxin containing solution (4x) was added to each well from the compound plate at the rate of 52 μL/s, yielding a final volume of 200 μL/well.
Whole Cell Binding Assay
At 8–10 days in culture, neocortical neurons cultured in 12-well plates were used for measurement of [3H]BTX binding. Cells were first rinsed three times each with 1 mL of buffer A containing 140 mM choline chloride, 5 mM KCl, 1.8 mM CaCl2, 0.8 mM MgSO4, and 10 mM Hepes (pH 7.4 with 1M Tris). Cells were then incubated with 0.5 mL of buffer B (buffer A plus 2 mg/mL BSA) containing 2 nM [3H]BTX, 300 nM PbTx-3, 10 μM deltamethrin and various concentrations of hoiamide for 3–4 h. After incubation, cells were rinsed three times with 1 mL of buffer A before lysis with 500 μL of 1% Triton X-100 under a condition of constant shaking overnight. A 400-μL aliquot of the resulting lysate was collected, and [3H]BTX bound was measured by liquid scintillation counter. Nonspecific binding of [3H]BTX was determined as [3H]BTX bound in the presence of 200 μM veratridine. Our preliminary results indicated that whole cell assays conducted at room temperature (22 °C) provided an optimum signal-to noise ratio; all binding experiments were conducted at 22 °C.
Data Analysis
The raw emission data at each excitation wavelength were exported to an Excel work sheet and corrected for background fluorescence. The SBFI fluorescence ratios (340/380) versus time were then analyzed, and time-response and concentration-response graphs generated using Graphpad Prism software (Graphpad Software Inc., San Diego, CA). The EC50 and maximum response values were determined by non-linear regression analysis using a logistic equation.
Supplemental Data
General experimental procedures and analytical data for derivatives of compound 1, 1D and 2D NMR spectra for 1 and 5, as well as 1H NMR spectra for 2, 3, 4 and the MTPA ester derivatives of 5; MM2 energy minimizations of 1, comparison of Ile-extended residues in 1 and 6, details in the taxonomic identification of PNG5-28-02-12, and a procedure for the isolation of 4, are available as Supplemental Data at http://www.cell.com/chemistry-biology/supplemental/XXXXXXXXX
Supplementary Material
Acknowledgments
We thank D. Edwards and L. T. Simmons for collection of the assemblage of L. majuscula and P. gracile from Papua New Guinea, as well as A. Jansma and X. Huang for assistance with the Bruker 600 MHz TCI cryoprobe and Bruker 800 MHz NMR spectrometers, respectively. We appreciate N. Engene input in the morphological characterization and taxonomic identification of the source organisms. Support of chemical and pharmacological aspects of the work was provided by NIH (NS053398).
Footnotes
Neighboring group participation (anchimeric assistance) by the thiazole-ring sulfur lone pair might stabilize the protonated and thus positively charged oxygen atom of the leaving group (MeOH) via a five-membered ring intermediate, thus favoring an SN2 nucleophilic substitution.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baden DG. Brevetoxins: unique polyether dinoflagellate toxins. Faseb J. 1989;3:1807–1817. doi: 10.1096/fasebj.3.7.2565840. [DOI] [PubMed] [Google Scholar]
- Calmes M, Escale F, Paolini F. Viable use of 2-substituted thiazolidine-4-methanol diastereoisomeric mixtures during asymmetric borane reduction of aromatic ketones. Tetrahedron: Asymm. 1997;8:3691–3697. [Google Scholar]
- Cao Z, George J, Gerwick WH, Baden DG, Rainier JD, Murray TF. Influence of lipid-soluble gating modifier toxins on sodium influx in neocortical neurons. J Pharmacol Exp Ther. 2008;326:604–613. doi: 10.1124/jpet.108.138230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z, George J, Baden DG, Murray TF. Brevetoxin-induced phosphorylation of Pyk2 and Src in murine neocortical neurons involves distinct signaling pathways. Brain Res. 2007;1184:17–27. doi: 10.1016/j.brainres.2007.09.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeli S, Moore RE, Patterson GML. Tantazoles: unusual cytotoxic alkaloids from the blue-green alga Scytonema mirabile. J Am Chem Soc. 1990;112:8195–8197. [Google Scholar]
- Carmeli S, Paik S, Moore RE, Patterson GML, Yoshida WY. Revised structures and biosynthetic studies of tantazoles A and B. Tetrahedron Lett. 1993;34:6681–6684. [Google Scholar]
- Carmeli S, Moore RE, Patterson GML. Mirabazoles, minor tantazole-related cytotoxins from the terrestrial blue-green alga Scytonema mirabile. Tetrahedron Lett. 1991;32:2593–2596. [Google Scholar]
- Catterall WA, Cestele S, Yarov-Yarovoy V, Yu FH, Konoki K, Scheuer T. Voltage-gated ion channels and gating modifier toxins. Toxicon. 2007;49:124–141. doi: 10.1016/j.toxicon.2006.09.022. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Activation of the action potential Na+ ionophore by neurotoxins. An allosteric model J Biol Chem. 1977;252:8669–8676. [PubMed] [Google Scholar]
- Catterall WA, Morrow CS, Daly JW, Brown GB. Binding of batrachotoxinin A 20-alpha-benzoate to a receptor site associated with sodium channels in synaptic nerve ending particles. J Biol Chem. 1981;256:8922–8927. [PubMed] [Google Scholar]
- Denac H, Mevissen M, Scholtysik G. Structure, function and pharmacology of voltage-gated sodium channels. Naunyn-Schmiedeberg’s Arch Pharmacol. 2000;362:453–479. doi: 10.1007/s002100000319. [DOI] [PubMed] [Google Scholar]
- Edwards DJ, Marquez BL, Nogle LM, McPhail K, Goeger DE, Roberts MA, Gerwick WH. Structure and biosynthesis of the jamaicamides, new mixed polyketide-peptide neurotoxins from the marine cyanobacterium Lyngbya majuscula. Chem Biol. 2004;11:817–833. doi: 10.1016/j.chembiol.2004.03.030. [DOI] [PubMed] [Google Scholar]
- Ersmark K, Del Valle JR, Hanessian S. Chemistry and biology of the Aeruginosin family of serine protease inhibitors. Angew Chem, Intl Ed. 2008;47:1202–1223. doi: 10.1002/anie.200605219. [DOI] [PubMed] [Google Scholar]
- Fitch CP, Bishop LM, Boyd WL, Gortner RA, Rogers CF, Tilden JE. “Water bloom” as a cause of poisoning in domestic animals. Cornell Veterinarian. 1934;24:30–39. [Google Scholar]
- Geitler L. Kryptogamen-flora von Deutschland, Osterreich und der Schweiz. In: Rabenhorst L, editor. Cyanophyceae. Leipzig, Germany: Akademische Verlag; 1932. pp. 1060–1061. 1985 reprint: Konigstein: Koeltz Scientific Books) [Google Scholar]
- George J, Dravid SM, Prakash A, Xie J, Peterson JH, Jabba SV, Baden D, Murray TF. Sodium channel activation augments NMDA receptor function and promotes neurite outgrowth in immature cerebrocortical neurons. J Neuroscience. 2009 doi: 10.1523/JNEUROSCI.6104-08.2009. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesinger C, Sorensen OW, Ernst RR. Practical aspects of the E.COSY technique. Measurement of scalar spin-spin coupling constants in peptides. J Magn Res. 1987;75:474–492. [Google Scholar]
- Grindberg RV, Shuman CF, Sorrels CM, Wingerd J, Gerwick WH. Neurotoxic alkaloids from cyanobacteria. In: Fattorusso E, Taglialatela-Scafati O, editors. Modern Alkaloids, Structure, Isolation, Synthesis and Biology. Weinheim, Germany: Wiley-VCH Verlang GmbH; 2008. pp. 139–170. [Google Scholar]
- Jansen R, Kunze B, Reichenbach H, Jurkiewicz E, Hunsmann G, Hofle G. Antibiotics from gliding bacteria XLVII. Thiangazole: a novel inhibitor of HIV-1 from Polyangium sp. Liebigs Ann Chem. 1992:357–359. [Google Scholar]
- Komarek J, Anagnostidis K. Cyanoprokaryota. In: Budel B, Krienitz L, Gartner G, Schagerl M, editors. Susswasserflora von mitteleuropa. 19/2. Munchen, Germany: Elsevier GmbH; 2005. pp. 441–625. [Google Scholar]
- Kwan JC, Rocca JR, Abboud KA, Paul VJ, Luesch H. Total structure determination of grassypeptolide, a new marine cyanobacterial cytotoxin. Org Lett. 2008;10:789–792. doi: 10.1021/ol702946d. [DOI] [PubMed] [Google Scholar]
- Li WI, Berman FW, Okino T, Yokokawa F, Shioiri T, Gerwick WH, Murray TF. Antillatoxin is a marine cyanobacterial toxin that potently activates voltage-gated sodium channels. Proc Natl Acad Sci USA. 2001;98:7599–7604. doi: 10.1073/pnas.121085898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombet A, Mourre C, Lazdunski M. Interaction of insecticides of the pyrethroid family with specific binding sites on the voltage-dependent sodium channel from mammalian brain. Brain Res. 1988;459:44–53. doi: 10.1016/0006-8993(88)90284-3. [DOI] [PubMed] [Google Scholar]
- Luesch H, Moore RE, Paul VJ, Mooberry SL, Corbett TH. Isolation of Dolastatin 10 from the Marine Cyanobacterium Symploca Species VP642 and Total Stereochemistry and Biological Evaluation of Its Analogue Symplostatin 1. J Nat Prod. 2001;64:907–910. doi: 10.1021/np010049y. [DOI] [PubMed] [Google Scholar]
- Luesch H, Yoshida WY, Moore RE, Paul VJ, Corbett TH. Total structure determination of apratoxin A, a potent novel cytotoxin from the marine cyanobacterium Lyngbya majuscula. J Am Chem Soc. 2001;123:5418–5423. doi: 10.1021/ja010453j. [DOI] [PubMed] [Google Scholar]
- Marfey P. Determination of D-amino acids. II Use of a bifunctional reagent 1,5-difluoro-2,4-dinitrobenzene. Carlsberg Res Commun. 1984;49:591–596. [Google Scholar]
- Marner FJ, Moore RE, Hirotsu K, Clardy J. Majusculamides A and B, two epimeric lipodipeptides from Lyngbya majuscula Gomont. J Org Chem. 1977;42:2815–2819. [Google Scholar]
- Marquez BL, Gerwick WH, Williamson RT. Survey of NMR experiments for the determination of nJ(C,H) heteronuclear coupling constants in small molecules. Magn Reson Chem. 2001;39:499–530. [Google Scholar]
- Matsumori N, Kaneno D, Murata M, Nakamura H, Tachibana K. Stereochemical determinations of acyclic structures based on carbon-proton spin-coupling constants. A method of configuration analysis for natural products. J Org Chem. 1999;64:866–876. doi: 10.1021/jo981810k. [DOI] [PubMed] [Google Scholar]
- Moore BS. Biosynthesis of marine natural products: Microorganisms (Part A) Nat Prod Rep. 2005;22:580–593. doi: 10.1039/b404737k. [DOI] [PubMed] [Google Scholar]
- Naegeli HU, Zahner H. Metabolites of microorganisms. Part 193 Ferrithiocin. Helv Chim Acta. 1980;63:1400–1406. [Google Scholar]
- Ohtani I, Kusumi T, Kashman J, Kakisawa H. High-field FT NMR application of Mosher’s method. The absolute configurations of marine terpenoids. J Am Chem Soc. 1991;113:4092–4096. [Google Scholar]
- Paul VJ, Arthur KE, Ritson-Williams R, Ross C, Sharp K. Chemical defenses: from compounds to communities. Biol Bull. 2007;213:226–251. doi: 10.2307/25066642. [DOI] [PubMed] [Google Scholar]
- Pattenden G, Thom SM, Jones MF. Enantioselective synthesis of 2-alkyl substituted cysteines. Tetrahedron. 1993;49:2131–2138. [Google Scholar]
- Pettit GR, Kamano Y, Herald CL, Tuinman AA, Boettner FE, Kizu H, Schmidt JM, Baczynskyj L, Tomer KB, Bontems RJ. The Isolation and Structure of a Remarkable Marine Animal Antineoplastic Constituent: Dolastatin 10. J Am Chem Soc. 1987;109:6883–6885. [Google Scholar]
- Plaza A, Bewley CA. Largamides A-H, unusual cyclic peptides from the marine cyanobacterium Oscillatoria sp. J Org Chem. 2006;71:6898–6907. doi: 10.1021/jo061044e. [DOI] [PubMed] [Google Scholar]
- Ramaswamy AV, Flatt PM, Edwards DJ, Simmons TL, Han B, Gerwick WH. The secondary metabolites and biosynthetic gene clusters of marine cyanobacteria. Applications in biotechnology. Front Mar Biotech. 2006;175:175–224. [Google Scholar]
- Ramaswamy AV, Sorrels CS, Gerwick WH. Cloning and biochemical characterization of the hectochlorin biosynthetic gene cluster from the marine cyanobacterium Lyngbya majuscula. J Nat Prod. 2007;70:1977–1986. doi: 10.1021/np0704250. [DOI] [PubMed] [Google Scholar]
- Rinehart KL, Kishore V, Bible KC, Sakai R, Sullins DW, Li K. Didemnins and tunichlorin: novel natural products from the marine tunicate Trididemnum solidum. J Nat Prod. 1988;51:1–21. doi: 10.1021/np50055a001. [DOI] [PubMed] [Google Scholar]
- Sharkey RG, Jover E, Couraud F, Baden DG, Catterall WA. Allosteric modulation of neurotoxin binding to voltage-sensitive sodium channels by Ptychodiscus brevis toxin 2. Mol Pharmacol. 1987;31:273–278. [PubMed] [Google Scholar]
- Sielaff H, Christiansen G, Schwecke R. Natural products from cyanobacteria: Exploiting a new source for drug discovery. IDrugs. 2006;9:119–127. [PubMed] [Google Scholar]
- Sivonen K, Borner T. Bioactive compounds produced by cyanobacteria. In: Herrero A, Flores E, editors. Cyanobacteria. Norwich, UK: Caister Acad. Press; 2008. pp. 159–197. [Google Scholar]
- Tan LT. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry. 2007;68:954–979. doi: 10.1016/j.phytochem.2007.01.012. [DOI] [PubMed] [Google Scholar]
- Taylor CP, Meldrum BS. Na+ channels as targets for neuroprotective drugs. Trends Pharmacol Sci. 1995;16:309–316. doi: 10.1016/s0165-6147(00)89060-4. [DOI] [PubMed] [Google Scholar]
- Tidgewell K, Clark BT, Gerwick WH. The Natural Products Chemistry of Cyanobacteria. In: Moore B, Crews P, editors. Comprehensive natural products chemistry. 2. Oxford, UK: Elsevier Limited; 2009. in press. [Google Scholar]
- Trainer VL, Moreau E, Guedin D, Baden DG, Catterall WA. Neurotoxin binding and allosteric modulation at receptor sites 2 and 5 on purified and reconstituted rat brain sodium channels. J Biol Chem. 1993;268:17114–17119. [PubMed] [Google Scholar]
- Trainer VL, McPhee JC, Boutelet-Bochan H, Baker C, Scheuer T, Babin D, Demoute JP, Guedin D, Catterall WA. High affinity binding of pyrethroids to the alpha subunit of brain sodium channels. Mol Pharmacol. 1997;51:651–657. doi: 10.1124/mol.51.4.651. [DOI] [PubMed] [Google Scholar]
- Uhrin D, Barlow PN. Gradient-enhanced one-dimensional proton chemical-shift correlation with full sensitivity. J Magn Res. 1997;126:248–255. [Google Scholar]
- Uhrin D, Batta G, Hruby VJ, Barlow PN, Kover KE. Sensitivity and gradient-enhanced hetero (ω1) half-filtered TOCSY experiment for measuring long-range heteronuclear coupling constants. J Magn Res. 1998;130:155–161. doi: 10.1006/jmre.1997.1308. [DOI] [PubMed] [Google Scholar]
- Van Wagoner RM, Drummond AK, Wright JLC. Biogenetic diversity of cyanobacterial metabolites. Adv Appl Microbiol. 2007;61:89–217. doi: 10.1016/S0065-2164(06)61004-6. [DOI] [PubMed] [Google Scholar]
- Williamson RT, Boulanger A, Vulpanovici A, Roberts MA, Gerwick WH. Structure and absolute stereochemistry of phormidolide, a new toxic metabolite from the cyanobacterium Phormidium sp. J Org Chem. 2002;67:7927–7936. doi: 10.1021/jo020240s. [DOI] [PubMed] [Google Scholar]
- Williamson RT, Marquez BL, Gerwick WH, Kover KE. One- and two-dimensional gradient-selected HSQMBC NMR experiments for the efficient analysis of long-range heteronuclear coupling constants. Magn Reson Chem. 2000;38:265–273. [Google Scholar]
- Wipf P, Fritch PC, Geib SJ, Sefler AM. Conformational Studies and Structure-Activity Analysis of Lissoclinamide 7 and Related Cyclopeptide Alkaloids. J Am Chem Soc. 1998;120:4105–4112. [Google Scholar]
- Wu M, Okino T, Nogle LM, Marquez BL, Williamson RT, Sitachitta N, Berman FW, Murray TF, McGough K, Jacobs R, et al. Structure, synthesis, and biological properties of kalkitoxin, a novel neurotoxin from the marine cyanobacterium Lyngbya majuscula. J Am Chem Soc. 2000;122:12041–12042. [Google Scholar]
- Yu XM, Salter MW. Gain control of NMDA-receptor currents by intracellular sodium. Nature. 1998;396:469–474. doi: 10.1038/24877. [DOI] [PubMed] [Google Scholar]
- Yu XM, Salter MW. Src, a molecular switch governing gain control of synaptic transmission mediated by N-methyl-D-aspartate receptors. Proc Natl Acad Sci. 1999;96:7697–7704. doi: 10.1073/pnas.96.14.7697. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.