

Abstract
Background
It has been proposed that plasma sphingomyelin (SM) plays a very important role in plasma lipoprotein metabolism and atherosclerosis. Sphingomyelin synthase (SMS) is the last enzyme for SM de novo biosynthesis. Two SMS genes, SMS1 and SMS2, have been cloned and characterized.
Methods and Results
To evaluate the in vivo role of SMS2 in SM metabolism, we prepared SMS2 knockout (KO) and SMS2 liver-specific transgenic (LTg) mice, and studied their plasma SM and lipoprotein metabolism. On a chow diet, SMS2 KO mice showed a significant decrease in plasma SM levels (25%, P<0.05), but no significant changes in total cholesterol, total phospholipids, or triglyceride, compared with wild-type (WT) littermates. On a high-fat diet, SMS2 KO mice showed a decrease in plasma SM levels (28%, P<0.01), whereas, SMS2LTg mice showed a significant increase in those levels (29%, P<0.05), but no significant changes in other lipids, compared with WT littermates. Atherogenic lipoproteins from SMS2LTg mice displayed a significantly stronger tendency toward aggregation after mammalian sphingomyelinase treatment, compared with controls. Moreover, SMS2 deficiency significantly increased plasma apoE levels (2.0-fold, P<0.001), while liver-specific SMS2 overexpression significantly decreased those levels (1.8-fold, P<0.01). Finally, SMS2 KO mouse plasma promoted cholesterol efflux from macrophages, while SMS2LTg mouse plasma prevented it.
Conclusions
We therefore believe that regulation of liver SMS2 activity could become a promising treatment for atherosclerosis.
Keywords: SMS2 knockout mice, SMS2 liver-specific transgenic mice, plasma sphingomyelin, plasma lipoproteins, plasma cholesterol
Sphingomyelin (SM), which is the second most abundant phospholipid in mammalian plasma, appears in all major lipoproteins, where it is part of the monolayer of polar lipids and cholesterol that surrounds a core of neutral lipids. Up to 18% of total plasma phospholipid exists as SM,1 with the ratio of phosphatidylcholine (PC)/SM varying widely among lipoprotein subclasses.2 Atherogenic lipoproteins such as VLDL remnants accumulating in cholesterol-fed rabbits tend to be SM-enriched.3 The SM content of atherosclerotic lesions is higher than that of normal arterial tissue, and SM accumulation is disproportionate to that of PC.4 However, the role of SM deposited or synthesized in atheromata still remains undefined.
It has been suggested that subendothelial retention and aggregation of atherogenic lipoproteins play a very important role in atherogenesis.5 SM-rich LDL retained in atherosclerotic lesions is acted on by an arterial wall sphingomyelinase, which appears to promote aggregation, initiating the early phase of atherosclerosis development.6 We have found that plasma SM levels in apoE KO mice are 4-fold higher than in WT mice,7 and this may partially explain the increased atherosclerosis found in these animals.8 Our laboratory and others have also discovered that inhibition of SM biosynthesis significantly decreases plasma SM and increases PC, thus lessening atherosclerotic lesions in apoE KO mice.9, 10 We have also reported that an SM-rich (1%) diet significantly increases plasma SM levels, LDL aggregation, and atherosclerotic lesions in LDL receptor KO mice.11 We have evidence that human plasma SM levels and SM/PC ratios are independent risk factors for coronary heart disease,12,13 and that plasma SM level are prognostic in patients with acute coronary syndrome.13 All of these data suggest that plasma SM plays a critical role in the development of atherosclerosis.
The biochemical synthesis of SM occurs through the action of serine palmitoyl-CoA transferase (SPT), 3-ketosphinganine reductase, ceramide synthase, dihydroceramide desaturase, and sphingomyelin synthase (SMS).14 SMS is the last enzyme for SM biosynthesis and it utilizes ceramide and PC as substrates to produce SM and diacylglycerol. Therefore, its activity should directly influence SM levels in cells and in the circulation. The liver and small intestine are the major contributions of plasma SM. The liver assembles lipids (SM, PC, cholesterol, and triglyceride) and apolipoproteins, and secretes the products, VLDL and HDL, into the circulation.15 Following hydrolysis of dietary SM in the lumen of small intestine,16 the backbone sphingoid bases and fatty acids, which are taken up by the enterocytes, and can be used to resynthesize SM. This SM can participate in chylomicron assembly, and then be secreted.17 Two SMS genes, SMS1 and SMS2, have been cloned and characterized for their cellular localizations.18 Both are expressed in the liver and small intestine.18 SMS1 is found in the trans-golgi apparatus, while SMS2 is predominantly located in the plasma membranes.18 Along with other investigators, we have shown that SMS1 and SMS2 expression positively correlates with levels of cellular SM and SM in membrane lipid rafts.19-21 However, the relationship between SMS activity and SM in the circulation is still not clear. For this study, we created both SMS2 gene knockout and SMS2 liver-specific transgenic mice, and investigated plasma SM levels in both models.
Methods
SMS2 KO mouse preparation
The overall strategy for gene targeting was to replace 90% of exon 2 with a neomycin-resistant gene (Supplement Fig. IA). Because exon 2 contains the translation initiation codon ATG, deletion of exon 2 would be expected to create an SMS2 null mouse allele. The homologues recombination was screened by PCR. Sense primer N1 (5′-tgcgaggccagaggccacttgtgtagc-3′) and antisense primer A1 (5′-tgtagccctggctgttctgtactc-3′) can amplify a 970 bp fragment (knockout, KO), while, sense primer S1 (5′-cgactccaccaacacttacacaag-3′) and antisense primer A1 (5′-tgtagccctggctgttctgtactc-3′) can amplify a 760 bp fragment (wild type, WT) (Supplement Fig.IB). The SMS2 KO mice originally on a 129 mouse genetic background have backcrossed with C57BL/6 mice for three generations. The animals (WT and KO) used in this study were 10-to-12-week-old littermates. The establishment of this mouse model has been reported by us previously.22
Liver-specific SMS2 transgenic mouse preparation
A 2.5-kilobase cDNA fragment of human SMS2 was cloned into the SalI and XhoI sites of the pLIV-7 plasmid,23 kindly provided by Dr. John M. Taylor (Gladstone Institute of Cardiovascular Disease, University of California, San Francisco) (Supplement Fig. II). A linearized fragment of the construct containing the promoter, first exon, first intron, and part of the second exon of the human apoE gene, the human SMS2 cDNA, and the polyadenylation sequence, and hepatic control region of the apoE/C-I gene locus were used to generate transgenic mice by standard procedures. Founder animals with a C57BL/6/CBA mixed background were backcrossed with C57BL/6J mice for two generations. The animals (WT and LTg) used in this study were 10-to-12-week-old littermates.
Atherogenic lipoprotein aggregation assay
Lipoprotein aggregation was assessed as previously described.7
Cholesterol efflux
WT macrophages (105 cells/well) were grown in DMEM with 10% FBS for 24 hours to reach a confluency of about 80%. Cell labeling was done by growing them overnight in DMEM with 10% FBS containing 50μg/ml acetylated LDL and 1 μCi/ml of [3H]cholesterol. Cells were washed three times with PBS to remove excess label, and equilibrated for 2 hours with DMEM without FBS. The medium was replaced with DMEM containing 10% plasma from SMS2 KO and WT mice, or SMS2LTg and WT mice. At 8 hours we measured [3H]cholesterol radioactivity in the medium, as well as radioactivity inside the cells. The percentage of cholesterol efflux was calculated by dividing the radioactive amounts in the extracellular media by the total of those amounts in the medium and cell fractions.
Statistical analysis
Data were typically expressed as mean ± S.D. Data between two groups were analyzed by the Student t-test. A P value of less than 0.05 was considered significant.
See the online-only Data Supplement (available online at http://atvb.ahajournals.org) for expanded Methods for: Lipid and lipoprotein assays, 24, 25 Apolipoprotein measurement.26, Lipid analyses by LC/MS/MS,22, mRNA analyses, SMS activity assay,22 Lysenin treatment and cell mortality measurement, Ceramidase assay, Ceramide synthase assay, and Sphingosine kinase assay.
Results
SMS2-deficient mouse
We have established SMS2 KO mice.22 (Supplement Fig.IB). As expected, SMS2 KO liver had no SMS2 mRNA (Supplement Fig. IC). Measurement of total liver SMS activity revealed that SMS2 deficiency causes a 60% reduction in that activity (P<0.001) (Supplement Fig. ID). However, SMS2 deficiency in the liver does not influence SMS1 mRNA levels (Supplement Fig. IE).
Creation of an SMS2 liver-specific transgenic mouse
One line of SMS2 transgenic mice was established. These animals demonstrated liver-specific expression of human SMS2 mRNA (Fig. 1A); there was a low expression in the brain, but none in other tissues (Fig. 1A). Measurement of total liver SMS activity revealed that human SMS2 expression causes a 2.5-fold increase in that activity (P<0.001) (Fig. 1B). However, SMS2 transgene expression in the liver does not influence SMS1 mRNA levels (Fig. 1C). We have thus established SMS2 liver-specific transgenic (LTg) mice.
Figure 1. The relationship between SMS2 overexpression and liver SMS activity.
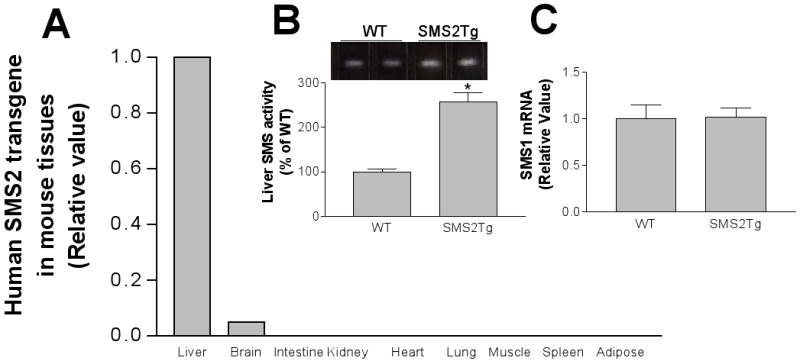
Panel A, Real-time PCR for tissue SMS2 mRNA levels; Panel B, liver SMS activity measurement in SMS2LTg mice; Panel C, Real-time PCR for liver SMS1 mRNA. Details, see “Methods.”
Plasma SM analysis: chow diet
As indicated in Table 1, SMS2 KO mice showed a significant decrease in plasma SM levels (25%, P<0.01), whereas SMS2LTg mice showed no significant changes, compared with WT littermates. Moreover, an SMS2 deficiency significantly increased plasma ceramide, sphingosine, and sphingosine-1-phosphate (43, 235, and 30%, respectively), while SMS2 liver overexpression had no effect on these lipids (Table 1). We also measured ceramidase (Supplement Fig. III, ceramide synthase (Supplement Fig. IV), and sphingosine kinase (Supplement Fig. V) activities in the livers from WT and SMS2 KO mice, respectively, we did not find any significant changes between WT and SMS2 KO mice, indicating these enzymes might not be rate limiting ones which determine the concentration of ceramide, sphingosine and S1P levels. SMS2 deficiency or overexpression had no significant impact on total cholesterol, total phospholipids, or triglyceride (Supplement Table I). The distribution of lipids was determined by FPLC of pooled plasma samples. HDL-SM was decreased in SMS2 KO mice (Fig. 2A), while no changes were observed in SMS2LTg ones (Fig. 2C), compared with WT animals. There were no changes of cholesterol levels or distribution in either SMS2 KO or SMS2LTg mice (Figs. 2B and D).
Table 1.
Plasma and liver lipid measurement in SMS2 KO, SMS2LTg, and WT mice
| Mice | SM (μM) | PC (μM) | Cer (ng/ml) | Sph (ng/ml) | S-1-P (ng/ml) | DHS-1-P (ng/ml) |
|---|---|---|---|---|---|---|
| Chow | ||||||
| WT | 71±9 | 1232±60 | 1344±164 | 17±2 | 314±10 | 83±5 |
| SMS2 KO | 53±5* | 1125±122 | 1925±185* | 57±7* | 407±9* | 94±14 |
| WT | 75±8 | 1281±151 | 1080±125 | 16±2 | 268±16 | 80±7 |
| SMS2LTg | 76±5 | 1334±92 | 877±81 | 19±3 | 272±30 | 73±2 |
| High-fat | ||||||
| WT | 115±6 | 1708±119 | 1096±92 | 20±8 | 485±55 | 156±21 |
| SMS2 KO | 83±7* | 1836±181 | 1779±215* | 27±3 | 516±60 | 158±33 |
| WT | 102±8 | 1868±150 | 1297±77 | 23±2 | 394±21 | 152±20 |
| SMS2LTg | 132±10* | 1970±75 | 906±97* | 12±2 | 304±23* | 120±13* |
Value: mean±SD; n=4-5.
SM, sphingomyelin; Cer, Ceramide; Sph, Sphingosine; S-1-P, Sphingosine-1-phosphate; DHS-1-P, Dihydroxyl-sphingosine-1-phosphate.
P<0.05.
Figure 2. Plasma lipoprotein analysis by FPLC in mice on chow and on a high fat high cholesterol diet.

A 200-μl aliquot of pooled plasma (from five animals) was loaded into a Sepharose 6B column and eluted with 50 mM Tris, 0.15 M NaCl (pH 7.5). An aliquot of each fraction was used for the determination of cholesterol and sphingomyelin. Panel A-D, SM and cholesterol distribution in SMS2 KO, SMS2Tg, WT mice on chow diet, respectively. Panel E-H, SM and cholesterol distribution in SMS2 KO, SMS2Tg, WT mice on a high fat diet, respectively.
Liver lipid analysis in SMS2 KO and SMS2LTg mice
As indicated in Supplement Table II, SMS2 deficient liver contained significantly less SM than controls (25%, P<0.05), while SMS2 overexpressed liver contained significantly more (21%, P<0.05), compared with controls. We also observed that liver S-1-P levels were significantly decreased in SMS2 KO (31%, P<0.01) and significantly increased in SMS2LTg mice (50%, P<0.001). Ceramide levels were significantly decreased in SMS2LTg mice (20%, P<0.05), compared with controls. There were no significant changes in PC, sphingosine, or dihydroxyl-sphingosine-1-phosphate (Supplement Table II), nor any in total cholesterol, phospholipids, or triglyceride levels (data not shown).
So far, it is uncertain whether SMS2 deficiency or an SMS2 liver overexpression changes the SM levels on the hepatocyte plasma membranes, where the lipid transporters are located and signal transduction is initiated. Lysenin is a recently discovered SM-specific cytotoxin. Lysenin recognizes SM only when it forms aggregates or microdomains.27 Based on our results above, we expected that an SMS2 deficiency would reduce, while an SMS2 liver overexpression would induce hepatocyte plasma membrane SM levels, thus influencing the formation of aggregates or microdomains recognizable by lysenin. To investigate the effect of liver SMS2 deficiency or liver SMS2 overexpression on the formation of these microdomains, we isolated primary hepatocytes from SMS2 KO, SMS2LTg, and control mice, and tested the sensitivity of the cells to lysenin-mediated cytolysis. As indicated in Figure 3, hepatocytes lacking SMS2 showed significantly less sensitivity to lysenin-mediated cytolysis than controls. This was also true for red blood cells, embryo fibroblast cells, and macrophages (Fig. 3A). Hepatocytes overexpressing SMS2 showed significantly more sensitivity to lysenin-mediated cytolysis than did controls (Fig. 3B). These results suggest that liver SMS2 deficiency or SMS2 overexpression not only significantly influence SM levels in the liver, but also significantly alter SM-rich microdomains (probably lipid rafts) in the hepatocyte plasma membranes.
Figure 3. SMS2 deficiency decreased lysenin-mediated cell mortality, while SMS2 overexpression increased it.

Panel A, Primary hepatocytes, red blood cells, embryo fibroblasts, and peritoneal macrophages were isolated from SMS2 KO and WT mice and treated with lysenin (200 ng/ml) for 2 hours. Panel B, Primary hepatocytes were isolated from SMS2LTg and WT mice and treated with lysenin (200 ng/ml) for 2 hours. Cell mortality was monitored by WST-1 Cell Proliferation Reagent (Roche). Values are mean ± S.D., n= 5, P<0.001.
Plasma SM analysis: high-fat high-cholesterol diet
We next sought to investigate the effect of a high-fat high-cholesterol diet on the SMS2 KO, SMS2LTg, and control mice. As indicated in Table 1, SMS2 KO animals showed a significant decrease in plasma SM levels (28%, P<0.01), whereas SMS2 LTg mice demonstrated a significant increase (20%, P<0.01), compared with WT littermates. SMS2 deficiency or overexpression had no significant impact on total cholesterol, total phospholipids, or triglyceride (Supplement Table I). FPLC of pooled plasma revealed that both HDL-SM and non-HDL-SM are markedly decreased in SMS2 KO mice (Fig. 2E), whereas both HDL-SM and non-HDL-SM are increased in SMS2LTg mice (Fig. 2G), compared with WT animals. There were no changes in cholesterol levels or distribution in either SMS2 KO or SMS2LTg mice, compared with controls (Figs. 2F and H).
SMS2 deficiency increases plasma apoE levels, while SMS2 overexpression decreases them
Assessment of plasma apolipoprotein levels in mice on a chow diet by reducing SDS-PAGE revealed a significant decrease of apoE (71%, P<0.001) in SMS2LTg mice (Supplement Fig. VIC), compared with controls. However, no significant difference was observed between SMS2 KO and WT mice (Supplement Fig. VIA). On a high-fat high-cholesterol diet, we found that SMS2 KO mice had significantly more plasma apoE (65%, P<0.01), while SMS2LTg mice had significantly less (51%, P<0.001), compared with controls (Supplement Figs. VIB and D). Western blot analysis of FPLC fractions revealed that an SMS2 deficiency elicited more apoE in plasma HDL, while an SMS2 overexpression elicited less, compared with controls (Supplement Fig. VII). There were no significant changes in plasma apoA-I or apoB levels in either SMS2 KO or SMS2LTg mice, compared with controls under tested conditions (Supplement Figs. VIA-D). We also measured liver mRNA levels of apoE, apoB, and apoA-I in SMS2 KO, SMS2LTg, and control mice by real-time PCT, finding no significant changes (data not shown).
SM-rich non-HDL particles from SMS2LTg mice are significantly aggregated after sphingomyelinase treatment
We then set out to determine whether enrichment of SM in non-HDL from SMS2LTg mice would contribute to the atherogenicity of these particles. As mentioned above, there is evidence to suggest that hydrolysis of lipoprotein SM by an arterial wall sphingomyelinase may lead to lipoprotein aggregation.7 We therefore reasoned that enrichment of lipoproteins with SM might increase their susceptibility to aggregation induced by sphingomyelinase, which is secreted by J774 macrophages. This might occur through increasing substrate availability to the enzyme.7 As shown in Figure 4A, non-HDL particles from SMS2LTg mice on a high-fat high-cholesterol diet were indeed significantly aggregated after treatment with sphingomyelinase, compared with controls (P<0.01).
Figure 4. SMS2 activity has proatherogenic properties.

Panel A, In vitro aggregation of atherogenic lipoproteins induced by macrophage-derived secreted sphingomyelinase. The Non-HDL particles aggregation was assessed as previously described.7 Panel B and C, macrophage cholesterol efflux was evaluated in SMS2 KO and WT mice, and in SMS2LTg and WT mice, respectively. The procedure is same as that described in “Methods”. Values are mean ± S.D., n= 4, P<0.01.
Plasma from SMS2 deficient mice promotes cholesterol efflux from macrophages, while that from SMS2LTg animals prevents it
Macrophage cholesterol efflux plays an important role in reverse cholesterol transport, an antiatherogenic process. We found that apoE levels were significantly increased in SMS2 KO mouse plasma, and decreased in SMS2LTg plasma following a high-fat high-cholesterol feeding. Since apoE is a well-known agent mediating macrophage cholesterol efflux,28 we speculated that SMS2 KO plasma might promote cholesterol efflux, while SMS2LTg plasma could prevent it. WT mouse macrophages derived from bone marrow were labeled with [3H]cholesterol, and cholesterol efflux was monitored with the application of 10% plasma from either WT or SMS2 KO mice, and from either WT or SMS2LTg mice. As shown in Figure 4B and C, SMS2 KO plasma significantly increased cholesterol efflux (22%, P<0.01), while SM S2LTg plasma significantly decreased it (26%, P<0.01) from the macrophages, compared with plasma from WT mice. We also measured cholesterol mass effluxed from macrophages with 10% WT, SMS2LTg, and SMS2 KO mouse plasma in the culture medium and found that SMS2LTg plasma significantly decreased cholesterol mass efflux (42%, P<0.01) from the macrophages, compared with plasma from WT mice (Supplement Fig.VIIA), while SMS2 KO plasma increased it (46%, P<0.001) (Supplement Fig.VIIB). This indicates an antiatherogenic result of SMS2 liver deficiency and a proatherogenic result of SMS2 overexpression.
Discussion
In this study, we have demonstrated for the first time that disruption of the SMS2 gene caused: 1) a 60% reduction of SMS activity in the liver, although SMS1 mRNA levels were not influenced; 2) a significant decrease of plasma SM and increase of ceramide levels; 3) a significant reduction of liver and hepatocyte plasma membrane SM levels; 4) a significant increase in plasma apoE levels, but not in those of apoA-I or apoB; and 5) a significant induction of cholesterol efflux from macrophages toward the plasma. Moreover, we have shown that overexpression of SMS2 in the liver had the opposite effect from a deficiency of it.
SM, an amphathic phospholipid located in the surface monolayer of all classes of plasma lipoproteins (LDL/VLDL, 70-75%; HDL, 25–30%),29 has significant effects on lipoprotein metabolism. But there is even now no clear answer to the one fundamental question: what factors determine the levels of SM in the circulation? In this study, we have partially answered that question: SMS2 is one of those factors, and it influences tissue and also plasma SM levels.
We found that human plasma SM level is an independent risk factor for coronary heart disease.12, 13 We believe that SM on LDL retained in atherosclerotic lesions is hydrolyzed by an arterial wall sphingomyelinase, which promotes aggregation by converting SM to ceramide.4, 6 There are two ways of preventing this atherogenic event, the first being to reduce sphingomyelinase levels. Indeed, it has recently been reported that apoE KO mice lacking sphingomyelinase have decreased development of early atherosclerotic lesions and, more important, decreased retention of atherogenic lipoproteins, compared with apoE KO matched for similar lipoprotein levels.30 The second way of preventing atherogenicity is by reducing SM levels in the atherogenic lipoproteins, through inhibition of the SM biosynthesis pathway in the lipoprotein-producing tissues, such as the liver and small intestine. We have demonstrated that SMS2 deficiency causes lower plasma SM levels, while liver-specific SMS2 overexpression causes higher ones, compared with controls. We also found that SM-enriched non-HDL particles from SMS2LTg mice have a stronger potential for aggregation after mammalian sphingomyelinase treatment, compared with controls (Fig. 4A), indicating a proatherogenic property in these particles. The non-HDL aggregation study also confirmed our previous study, that SM-enriched non-HDL particles from adenovirus-mediated SMS2 overexpressed mice have a stronger potential for aggregation after sphingomyelinase treatment.31
It is interesting that there were no effects on cholesterol levels in our animals. It is known that cholesterol levels in membranes paralleled SM levels. It is expected that decreasing SM levels would influence cholesterol levels, but this phenominon was not observed in this study. Previously, we also found that inhibition of SM de novo synthesis does not influence plasma cholesterol levels9 and that decreasing membrane SM levels does not always accompany with decreasing cholesterol levels.11 There are other mechanisms other than cholesterol-binding govern SM levels on the membrane and in the circulation.
The relationship between SMS2 activity and apoE in the plasma is unexpected. In this study, we found that SMS2 deficiency increases plasma apoE (Supplement Figs. VIA and B), while SMS2 overexpression in the liver decreases it (Supplement Figs. VIC and D). It is very likely that an SM increase or decrease alters the structure of the cellular membranes. Lipid rafts and caveolae are a subset of membrane microdomains that are enriched with SM, cholesterol, and glycosphingolipids.32 The changes of SM levels in such microdomains might thereby influence the conformation of nascent apoE associated with these structures. It has been reported that apoE expression leads to increases in the secretion of SM, which is colocalized with apoE-enriched lipoproteins, suggesting the importance of the SM-containing membranes.33 The SM content of the Golgi is in rapid equilibrium with SM in the plasma membranes, and SM-enriched microdomains have been described in the Golgi membranes.34
Indeed, we found that SMS2 KO cells have lower SM levels in the plasma membranes, while SMS2LTg hepatocytes have higher ones (Figs. 3A and B), suggesting that SMS2 activity might be related to liver apoE secretion. Another possibility is that changes in levels of ceramide (a substrate of SMS) may also have an impact on plasma apoE, since ceramide is a known second messenger,35 and one of the components of large lipid rafts.36 Increased ceramide in the SMS2 KO liver (Table 1) might be related to increased apoE secretion, while decreased ceramide in SMS2LTg liver (Table 1) might be related to the opposite. However, this possibility may be quite illusory. It has been reported that inhibiting the degradation of cellular ceramide, or supplementing exogenous ceramide to the macrophages, decreases rather than increases apoE secretion from the macrophages.33 Of course, the mechanism of apoE secretion from the macrophages may be different from that of the hepatocytes. The details of this mechanism deserve further investigation.
Plasma from SMS2 deficient mice significantly enhanced cholesterol efflux from the macrophages, while that from SMS2LTg animals significantly reduced it, compared with their respective controls (Fig. 4B and C). This observation could have important implications for the vessel wall homeostatic response to atherogenic insult. There are two possibilities that might explain this phenomenon. ApoE is a mediator of cholesterol efflux28 which plays an important role in reverse cholesterol transport.37 Therefore, changes in plasma ApoE may account for the differences in cholesterol efflux. Alternatively, changes in plasma SM may be the explanation. SM-depletion in tissue culture medium can prevent apoA-1 mediated cholesterol efflux from CHO cells, while SM supplement promote the efflux from these cells.38 Although we observed opposite in macrophage cholesterol efflux (Fig.4B and C), one thing seems to be clear that SM levels in microenvironment can influence cholesterol efflux and different cells have different outcomes.
SMS2 deficiency also has impact on other sphingolipid levels, including three important second messengers, ceramide, sphingosine, and sphingosine-1-phosphate (Table 1). The metabolism of these lipids is tightly linked. While we are considering the effect of SM on atherosclerosis, we should not ignore the potential impact of these bioactive lipids. Indeed, pharmacological inhibition of SM de novo synthesis, not only decrease SM levels, but also decrease ceramide, sphingosine, and sphingosine-1-phosphate.9 These changes might contribute to the reduction of atherosclerosis observed in apoE KO mice. 9
In summary, SMS2 deficiency has antiatherogenic properties, while SMS2 liver overexpression has very much the opposite effect. We therefore believe that regulation of liver SMS2 activity could become a promising treatment for atherosclerosis.
Supplementary Material
Acknowledgments
The authors are grateful to Mr. Thomas Beyer for his support for lipid analysis and to Mr. Calvin Yeang for English Editing.
Sources of Funding: This work was supported by an American Heart Association Heritage Affiliate Grant-in-Aid (to Dr. Jiang) and National Institute of Health grants (HL-64735 and HL-69817 to Dr Jiang)
Footnotes
Disclosures: None
References
- 1.Phillips GB, Dodge JT. Composition of phospholipids and of phospholipid fatty acids of human plasma. J Lipid Res. 1967;8:676–681. [PubMed] [Google Scholar]
- 2.Subbaiah PV, Davidson MH, Ritter MC, Buchanan W, Bagdade JD. Effects of dietary supplementation with marine lipid concentrate on the plasma lipoprotein composition of hypercholesterolemic patients. Atherosclerosis. 1989;79:157–166. doi: 10.1016/0021-9150(89)90120-2. [DOI] [PubMed] [Google Scholar]
- 3.Rodriguez AL, Ghiselli GC, Torreggiani D, Sirtori CR. Very low density lipoproteins in normal and cholesterol-fed rabbits: lipid and protein composition and metabolism. Part 1: chemical composition of very low density lipoprotein in rabbits. Atherosclerosis. 1976;23:73–83. doi: 10.1016/0021-9150(76)90119-2. [DOI] [PubMed] [Google Scholar]
- 4.Schissel SL, Tweedie-Hardman J, Rapp JH, Graham G, Williams KJ, Tabas I. Rabbit aorta and human atherosclerotic lesions hydrolyze the sphingomyelin of retained low density lipoprotein. J Clin Invest. 1996;98:1455–1464. doi: 10.1172/JCI118934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams KJ, Tabas I. The response-to-retention hypothesis of early atherosclerosis. Arterioscler Thromb Vasc Biol. 1995;15:551–561. doi: 10.1161/01.atv.15.5.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schissel SL, Tweedie-Hardman J, Rapp JH, Graham G, Williams KJ, Tabas I. Rabbit aorta and human atherosclerotic lesions hydrolyze the sphingomyelin of retained low density lipoprotein. J Clin Invest. 1996;98:1455–1464. doi: 10.1172/JCI118934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeong Ts, Schissel SL, Tabas I, Pownall HJ, Tall AR, Jiang XC. Increased sphingomyelin content of plasma lipoproteins in apolipoprotein E knockout mice reflects combined production and catabolic defects and enhances reactivity with mammalian sphingomyelinase. J Clin Invest. 1998;101:905–912. doi: 10.1172/JCI870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Plump AS, Breslow JL. Apolipoprotein E and the apolipoprotein E-deficient mouse. Annu Rev Nutr. 1995;15:495–518. doi: 10.1146/annurev.nu.15.070195.002431. [DOI] [PubMed] [Google Scholar]
- 9.Hojjati MR, Li Z, Zhou H, Tang S, Huan C, Ooi E, Lu S, Jiang XC. Effect of myriocin on plasma sphingolipid metabolism and atherosclerosis in apoE-deficient mice. J Biol Chem. 2005;280:10284–10289. doi: 10.1074/jbc.M412348200. [DOI] [PubMed] [Google Scholar]
- 10.Park TS, Panek RL, Mueller SB, Hanselman JC, Rosebury WS, Robertson AW, Kindt EK, Homan R, Karathanasis SK, Rekhter MD. Inhibition of sphingomyelin synthesis reduces atherogenesis in apolipoprotein E-knockout mice. Circulation. 2004;110:3465–3471. doi: 10.1161/01.CIR.0000148370.60535.22. [DOI] [PubMed] [Google Scholar]
- 11.Li Z, Basterr MJ, Hailemariam TK, Hojjati MR, Lu S, Liu J, Liu R, Zhou H, Jiang XC. The effect of dietary sphingolipids on plasma sphingomyelin metabolism and atherosclerosis. Biochim Biophys Acta. 2005;1735:130–134. doi: 10.1016/j.bbalip.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 12.Jiang XC, Paultre F, Pearson TA, Reed RG, Francis CK, Lin M, Berglund L, Tall AR. Plasma sphingomyelin level as a risk factor for coronary artery disease. Arterioscler Thromb Vasc Biol. 2000;20:2614–2618. doi: 10.1161/01.atv.20.12.2614. [DOI] [PubMed] [Google Scholar]
- 13.Schlitt A, Blankenberg S, Yan D, von Gizycki H, Buerke M, Werdan K, Bickel C, Lackner KJ, Meyer J, Rupprecht HJ, Jiang XC. Further evaluation of plasma sphingomyelin levels as a risk factor for coronary artery disease. Nutr Metab. 2006;3:5–12. doi: 10.1186/1743-7075-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Merrill AH, Jones DD. An update of the enzymology and regulation sphingomyelin metabolism. Biochim Biophysi Acta. 1990;1044:1–12. doi: 10.1016/0005-2760(90)90211-f. [DOI] [PubMed] [Google Scholar]
- 15.Hamilton RL, Moorehouse A, Havel RJ. Isolation and properties of nascent lipoproteins from highly purified rat hepatocytic Golgi fractions. J Lipid Res. 1991;32:529–543. [PubMed] [Google Scholar]
- 16.Nisson A. The presence of sphingomyelin and ceramide-cleaving enzymes in the intestinal tract of the rat. Biochim Biophys Acta. 1969;176:339–347. doi: 10.1016/0005-2760(69)90192-1. [DOI] [PubMed] [Google Scholar]
- 17.Nilsson A, Duan RD. Absorption and lipoprotein transport of sphingomyelin. J Lipid Res. 2006;47:154–171. doi: 10.1194/jlr.M500357-JLR200. [DOI] [PubMed] [Google Scholar]
- 18.Huitema K, van den Dikkenberg J, Brouwers JF, Holthuis JC. Identification of a family of animal sphingomyelin synthases. EMBO J. 2004;23:33–44. doi: 10.1038/sj.emboj.7600034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Z, Hailemariam TK, Zhou H, Li Y, Duckworth DC, Peake DA, Zhang Y, Kuo MS, Cao G, Jiang XC. Inhibition of sphingomyelin synthase (SMS) affects intracellular sphingomyelin accumulation and plasma membrane lipid organization. Biochim Biophys Acta. 2007;1771:1186–1194. doi: 10.1016/j.bbalip.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miyaji M, Jin ZX, Yamaoka S, Amakawa R, Fukuhara S, Sato SB, Kobayashi T, Domae N, Mimori T, Bloom ET, Okazaki T, Umehara H. Role of membrane sphingomyelin and ceramide in platform formation for Fas-mediated apoptosis. J Exp Med. 2005;202:249–259. doi: 10.1084/jem.20041685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van der Luit AH, Budde M, Zerp S, Caan W, Klarenbeek JB, Verheij M, Van Blitterswijk WJ. Resistance to alkyl-lysophospholipid-induced apoptosis due to downregulated sphingomyelin synthase 1 expression with consequent sphingomyelin- and cholesterol-deficiency in lipid rafts. Biochem J. 2007;401:541–549. doi: 10.1042/BJ20061178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hailemariam TK, Huan C, Liu J, Li Z, Roman C, Kalbfeisch M, Bui HH, Peake DA, Kuo MS, Cao G, Wadgaonkar R, Jiang XC. Sphingomyelin synthase 2 deficiency attenuates NFkappaB activation. Arterioscler Thromb Vasc Biol. 2008;28:1519–1526. doi: 10.1161/ATVBAHA.108.168682. [DOI] [PubMed] [Google Scholar]
- 23.Fan J, Wang J, Bensadoun A, Lauer SJ, Dang Q, Mahley RW, Taylor JM. Overexpression of hepatic lipase in transgenic rabbits leads to a marked reduction of plasma high density lipoproteins and intermediate density lipoproteins. Proc Natl Acad Sci U S A. 1994;91:8724–8728. doi: 10.1073/pnas.91.18.8724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hojjati MR, Jiang XC. Rapid, specific, and sensitive measurements of plasma sphingomyelin and phosphatidylcholine. J Lipid Res. 2006;47:673–676. doi: 10.1194/jlr.D500040-JLR200. [DOI] [PubMed] [Google Scholar]
- 25.Jiang XC, Bruce C, Mar J, Lin M, Ji Y, Francone OL, Tall AR. Targeted mutation of plasma phospholipid transfer protein gene markedly reduces high-density lipoprotein levels. J Clin Invest. 1999;103:907–914. doi: 10.1172/JCI5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rohlmann A, Gotthardt M, Hammer RE, Herz J. Inducible inactivation of hepatic LRP gene by cre-mediated recombination confirms role of LRP in clearance of chylomicron remnants. J Clin Invest. 1998;101:689–695. doi: 10.1172/JCI1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishitsuka R, Yamaji-Hasegawa A, Makino A, Hirabayashi Y, Kobayashi T. A lipid-specific toxin reveals heterogeneity of sphingomyelin-containing membranes. Biophys J. 2004;86:296–307. doi: 10.1016/S0006-3495(04)74105-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Innerarity TL, Pitas RE, Mahley RW. Modulating effects of canine high density lipoproteins on cholesteryl ester synthesis induced by β-very low density lipoproteins in macrophages. Possible in vitro correlates with atherosclerosis. Arteriosclerosis. 1982;2:114–124. doi: 10.1161/01.atv.2.2.114. [DOI] [PubMed] [Google Scholar]
- 29.Nilsson A, Duan RD. Absorption and lipoprotein transport of sphingomyelin. J Lipid Res. 2006;47:154–171. doi: 10.1194/jlr.M500357-JLR200. [DOI] [PubMed] [Google Scholar]
- 30.Tabas I, Williams KJ, Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116:1832–1844. doi: 10.1161/CIRCULATIONAHA.106.676890. [DOI] [PubMed] [Google Scholar]
- 31.Dong J, Liu J, Lou B, Li Z, Ye X, Wu M, Jiang XC. Adenovirus-mediated overexpression of sphingomyelin synthases 1 and 2 increases the atherogenic potential in mice. J Lipid Res. 2006;47:1307–1314. doi: 10.1194/jlr.M600040-JLR200. [DOI] [PubMed] [Google Scholar]
- 32.Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–72. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 33.Lucic D, Huang ZH, Gu D, Subbaiah PV, Mazzone T. Cellular sphingolipids regulate macrophage apolipoprotein E secretion. Biochemistry. 2007;46:11196–11204. doi: 10.1021/bi701106v. [DOI] [PubMed] [Google Scholar]
- 34.Gkantiragas I, Brugger B, Stuven E, Kaloyanova D, Li XY, Lohr K, Lottspeich F, Wieland FT, Holms JB. Sphingomyelin-enriched microdomains at the Golgi complex. Mol Biol Cell. 2001;12:1819–1833. doi: 10.1091/mbc.12.6.1819. 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hannun YA, Luberto C, Argraves KM. Enzymes of sphingolipid metabolism: from modular to integrative signaling. Biochemistry. 2001;40:4893–4903. doi: 10.1021/bi002836k. [DOI] [PubMed] [Google Scholar]
- 36.Grassmé H, Jekle A, Riehle A, Schwa H, Berge J, Sandhoff K, Kolesnick R, Gulbins E. CD95 signaling via ceramide-rich membrane rafts. J Biol Chem. 2001;276:20589–20596. doi: 10.1074/jbc.M101207200. [DOI] [PubMed] [Google Scholar]
- 37.Mahley RW, Huang Y, Weisgraber KH. Putting cholesterol in its place: apoE and reverse cholesterol transport. J Clin Invest. 2006;116:1226–1229. doi: 10.1172/JCI28632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagao K, Takahashi K, Hanada K, Kioka N, Matsuo M, Ueda K. Enhanced apoA-I-dependent cholesterol efflux by ABCA1 from sphingomyelin-deficient Chinese hamster ovary cells. J Biol Chem. 2007;282:14868–14874. doi: 10.1074/jbc.M611230200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.