

Summary
Glutamate is the main excitatory neurotransmitter in the central nervous system, and plays an excitatory role in generation of hypothalamic-pituitary-adrenocortical (HPA) axis responses to stress. The current study assesses the role of kainate-preferring receptors in glutamatergic excitation of the HPA axis. In situ hybridization and immunohistochemical analyses confirmed the existence of the GluR5 kainate subunit in the paraventricular nucleus of the hypothalamus (PVN). Importantly, GluR5 immunoreactivity was enriched in the external lamina of the median eminence, where it is co-localized with corticotropin releasing hormone (CRH). Intra-PVN infusion of LY382884 increased plasma adrenocorticotropin (ACTH), corticosterone and PVN Fos immunoreactivity. Infusions of LY382884 into the median eminence region, on the other hand, reduced restraint induced ACTH release without altering c-Fos expression. Together, these findings provide evidence for glutamate-mediated signaling in control of CRH release at the PVN and median eminence, mediated by way of kainate-preferring receptor complexes.
Keywords: HPA axis, GluR5, CRH, rats, presynaptic, median eminence
Introduction
The HPA axis is the main neuroendocrine arm of the stress response, and is activated in response to stimuli that threaten the homeostasis of an animal. Central drive of the HPA axis is mediated by the paraventricular nucleus of the hypothalamus (PVN). When activated, medial parvocellular PVN neurons release ACTH secretagogues, including CRH and vasopressin (AVP), into the hypophysial portal vasculature in the external zone of the median eminence. These hormones travel through the portal system to the anterior pituitary gland and stimulate corticotrophs to release ACTH (Vale and River, 1977) (Vale et al., 1981). ACTH is then released into the circulation, and induces the adrenal cortex to synthesize and release glucocorticoid hormones (corticosterone is the main glucocorticoid in rats).
A significant portion of the excitatory control of HPA axis activity is thought to be mediated by glutamate signaling (Herman et al., 2004; Jezova, 2005). Glutamate signals through presynaptic and postsynaptic ionotropic and metabotropic glutamate receptors, thereby regulating neuron excitation and excitability(Brann, 1995). With respect to the HPA axis, glutamate infusion into the third ventricle increases ACTH release, an effect that is blocked by deafferentation of the medial basal hypothalamus(Makara and Stark, 1975). This suggests that glutamate may regulate the HPA axis at the PVN or above. Because injection of glutamate directly into the PVN also induces ACTH release (Darlington et al., 1989), depletion of median eminence CRH content(Feldman and Weidenfeld, 1997), and release of CRH from hypothalamic slices(Joanny et al., 1997), it is likely that at least some of glutamate's effects on the HPA axis are mediated through the PVN itself. This idea is supported by evidence that blockade of ionotropic glutamate receptors by injection of the glutamate receptor antagonist kynurenate into the PVN decreases corticosterone responses to restraint stress(Ziegler and Herman, 2000).
These HPA axis actions of glutamate are most likely mediated solely by actions on the central nervous system. Although some glutamate receptor subtypes are expressed in both the adrenal cortex (Kristensen, 1993) and the pituitary gland (Petralia et al., 1994; Mahesh et al., 1999), there is little evidence that glutamate acting on these tissues increases secretion of ACTH or glucocorticoids (Hinoi et al., 2004). Some studies have shown activation of the HPA axis in response to peripherally-administered glutamate agonists(Gay and Plant, 1987; Carlson et al., 1989), but these effects are likely to have been caused by actions on the central nervous system, rather than on peripheral tissues(Zelena et al., 2005).
Although it is clear that glutamate acts in regulating the HPA axis at the PVN, it is not known which glutamate receptors are responsible for these regulatory actions. Anatomical evidence suggests that receptors from the n-methyl-d-aspartate (NMDA), alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainate receptor families, as well as metabotropic receptors, are localized in CRH-containing regions of the PVN (Herman et al., 2000; Scaccianoce et al., 2003; Eyigor et al., 2005; Ziegler et al., 2005). Antagonist studies have demonstrated functional roles for ionotropic (Ziegler and Herman, 2000) and metabotropic (Johnson et al., 2001; Scaccianoce et al., 2003) glutamate receptors in regulation of the HPA axis. It is not clear, however, which individual receptors from these classes are involved in regulating the HPA axis.
One possibility is suggested by the preferential expression of mRNA from the kainate family of receptors in hypophysiotrophic neurons in the PVN (Herman et al., 2000). In addition, kainate has an excitatory influence on the HPA axis when administered directly into the PVN (Zelena et al., 2005), suggesting a role for kainate receptors in HPA axis regulation. A possible role of kainate receptors in HPA axis regulation is further supported by increased expression of GluR5 and KA2 kainate receptors in response to hypoglycemia stress (Koenig and Cho, 2005). Because GluR5 mRNA and protein have both been reported to be expressed in the PVN (Herman et al., 2000; Eyigor et al., 2005), the current study investigates the role of GluR5 signaling in regulation of the HPA axis. These experiments were designed to test the hypothesis that GluR5 mediates stress-induced excitation of the PVN and activation of the HPA axis.
Materials and Methods
Animals
Adult male Sprague-Dawley rats weighing 250-300 g (Harlan, Indianapolis, IN) were used in these experiments. All experimental protocols were approved by the University of Cincinnati institutional animal care and use committee, and conform to the guidelines published in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Care was taken to minimize the number and suffering of animals used in these experiments. Animals were kept singly housed, on a 12h:12h light:dark cycle (light phase from 6:00 AM to 6:00 PM) and given ad libitum access to water and chow. Animals were allowed to adapt to the new environment for at least one week prior to undergoing surgery. Rats were anesthetized using a mix of ketamine and xylazine (87-90 mg/kg ketamine and 10-13 mg/kg xylazine) and given butorphanol as analgesic. They were implanted with 26-gauge unilateral chronic guide cannulae (inner diameter = 0.24 mm, outer diameter = 0.46 mm, Plastics One, Roanoke, VA) targeting the median eminence, or with bilateral guide cannulae targeting the right and left PVN. Stereotactic coordinates used for the guide cannulae were PVN: 1.9 mm posterior to bregma, 7.3 mm ventral (from the dura at the site of implant), centered over the sagittal sinus; and median eminence: 2.2 mm posterior to bregma, 0.3 mm lateral, and 9 mm ventral (from the skull at bregma). For the PVN injections, internal cannulae projected 0.5 mm past the guide cannula. Internal cannulae for the median eminence injections were 1 mm longer than the guide cannula. Cannula placement was verified by Nissl stain.
Microinjections and restraint stress challenge
Starting two days after surgery, the animals were handled daily to accustom them to having their cannulae handled, removed and re-inserted. On the day of the experiment, five to seven days after surgery, the obturators were removed from the animal and an internal guide cannula (33 gauge, inner diameter = 0.10 mm, outer diameter = 0.20 mm) was inserted in the guide cannula. The animals were given a 100 nl injection of either drug or vehicle (sterile 0.9% saline with 1% Chicago sky blue dye), using a PHD 2000 motorized syringe pump (Harvard Apparatus, Holliston, MA). Animals were given either 0.35 μg/μl (35 ng/injection for the PVN and high treatment group in the median eminence injections) or 0.035 μg/μl (low treatment group in the median eminence, 3.5 ng/injection) of LY382884, a GluR5-selective glutamate antagonist. Fifteen minutes post-injection, the animals were placed in plastic restraining tubes, and were restrained for 45 minutes. Blood samples were taken by tail clips at 15, 30, 60, and 120 minutes post-injection for measurement of plasma hormones. Chicago Sky Blue dye was purchased from Sigma (St. Louis, MO). LY382884 was generously provided by Eli Lilly. Blood samples were kept on ice until plasma isolation. Plasma was isolated from blood by centrifugation, and samples were stored at −20°C until being assayed.
Radioimmunoassay
Plasma corticosteronelevels were measured using a commercial 125I radioimmunoassay kit (ICN, Costa Mesa CA). ACTH radioimmunoassay was performed as previously described (Engeland et al., 1989), using 125I-labeled ACTH as tracer. The ACTH antiserum was a generous gift of W. Engeland (University of Minnesota).
Immunohistochemistry
After the final blood sample was taken from the rats (120 minutes post-injection), they were killed by barbiturate overdose. Animals were perfused intracardially with 0.9% saline solution, then with 4% paraformaldehyde in 50 mM potassium phosphate buffer. Brains were removed and placed in the paraformaldehyde solution and post-fixed overnight at 4°C. After post-fixation, the paraformaldehyde solution was removed and the brains were kept in 30% sucrose until sectioning. Perfused brains were frozen on dry ice, then sectioned at 25 μm using a sliding microtome (Leica, Bannockburn, IL), then stored in cryoprotectant (30 % sucrose, w/v; 1% polyvinylpyrrolidone (PVP-40), w/v; 30% ethylene glycol, v/v; in 50mM sodium phosphate buffer, pH 7.4) at -20°C until staining.
For sections to be stained using GluR5 antibody, antigen retrieval was performed as described (Eyigor et al., 2005). Briefly, after being washed in 50 mM potassium phosphate buffered saline (KPBS), sections were placed in 100 ml plastic beakers containing 50 ml of 50 mM citrate buffer (pH 6.0), then microwaved at high power until the temperature of the buffer reached 80-90°C (about two minutes for 4 beakers). Sections were allowed to sit in the heated buffer for 15 minutes, then immunohistochemistry was continued as outlined below. For sections not stained for GluR5, antigen retrieval was not used. Specificity for this antibody and protocol for GluR5 was demonstrated previously (Eyigor et al., 2005). In addition, we performed immunohistochemical controls, including omission of primary antibody and omission of antigen retrieval (see Figure 2 below).
Figure 2. Antigen Retrieval.

Antigen retrieval was necessary in order for GluR5-like immunoreactivity to be detected. A) Section of the hypothalamus at the level of the PVN stained by DAB immunohistochemistry for GluR5, without antigen retrieval. B) Immunostained section at the same level without primary antibody, for comparison. C) Section immunostained for GluR5, after antigen retrieval. D) Section immunostained for GluR5, at the level of the median eminence. 3V- third ventricle. Scale bar indicates 100 μm.
Sections were washed in KPBS, then native peroxide activity was blocked with 1.5% H2O2 in KPBS for 10 minutes. After washing again, tissue sections were incubated in a blocking solution (KPBS with 0.1% bovine serum albumin and 0.2% triton X-100). Primary antibodies were added and sections were incubated overnight at 4°C (48 hours at room temperature for GluR5). The sections were washed, then incubated in biotinylated goat anti-rabbit immunoglobulin (Vector Laboratories, Burlingame, CA) at 1:500 dilution for one hour at room temperature, then washed again. For sections visualized using 3,3′-diaminobenzidine (DAB) or amplified using biotinylated tyramide, the tissue was then treated with ABC (Vector Laboratories) at 1:1000 in KPBS for one hour, according to the manufacturer's instructions. Sections immunostained for CRH were further amplified using biotinylated tyramide at 1:250 in KPBS, according to the manufacturer's instructions. The sections were then developed in DAB (0.4 mg/ml in KPBS) with hydrogen peroxide (0.167 μl of 30%/ml KPBS) for 10 minutes, washed, then mounted on glass slides. Mounted sections were dehydrated in graded ethanol, cleared in xylene, and coverslipped using DPX mountant (Sigma, St. Louis, MO).
For sections visualized using immunofluorescence, after the incubation with biotinylated secondary antibody, sections were washed, then incubated in streptavidin-conjugated Cy3 (1:500), washed, and mounted on glass slides. For double-stained sections, we used Alexa-488 conjugated-donkey anti-rabbit immunoglobulin (Vector Laboratories). The slides were coverslipped using gelvatol. Primary antibodies used were rabbit anti-GFAP (DAKO, Carpinteria, CA) at 1:20,000 dilution, rabbit anti-fos (Calbiochem, La Jolla, CA) at 1:20,000, goat anti-GluR5 (Santa Cruz Biotechnology, Santa Cruz, CA) at 1:1000 (DAB) or 1:100 (immunofluorescence), and rabbit anti-CRH (courtesy Dr. Wylie Vale) at 1:100000. The CRH immunostain was amplified using biotinylated tyramide.
In situ hybridization
In situ hybridization was performed as described previously (Ziegler et al., 2005)(Ziegler et al., 2002). Briefly, 35S-labeled antisense RNA probes were made against a cDNA for GluR5. Tissue sections mounted on glass slides were hybridized using standard hybridization protocols as described. Following hybridization and washing, slides were coated with Kodak photographic emulsion NTB2, developed, and counterstained with cresyl violet Nissl stain. These slides were then coverslipped using DPX mountant
Imaging
DAB-stained slides were photographed using an Axioplan 2 microscope (Zeiss, Thornwood, NY). c-Fos activation was measured using immunostaining visualized with DAB. The number of c-Fos positive cells in the PVN was determined by counting particles, using the ImageJ software (Rasband, 1997-2007). Two sections were counted for each animal. Confocal pictures of immunofluorescence-stained slides were taken using an LSM 510 microscopy system (Zeiss, Thornwood, NY), with an image size of 1024 × 1024 pixels, and 0.48 μm optical slice depth. Digital images were cropped, and brightness and contrast were adjusted to improve clarity. No other alterations were made.
Statistics
Plasma hormone concentrations were analyzed using two-way or three-way analysis of variance (ANOVA) with repeated measures. c-Fos data were analyzed using one-way ANOVA or an unpaired t test, as appropriate. Because there were no differences in c-Fos expression in the right PVN vs. the left PVN, both sides were pooled in each animal for these analyses (except where otherwise indicated). Outliers were identified using Grubb's test (Barnett and Lewis, 1994). Significant main effects shown on ANOVA were further analyzed using the Fisher least significant difference post-hoc test. Statistical analyses were performed using GB Stat (Dynamic Microsystems, Silver Spring, MD) and the Prism software program (GraphPad, San Diego, CA). Significance was set at p < 0.05.
Results
GluR5 expression
In situ hybridization for GluR5 mRNA in the PVN agreed with previous studies that document expression of GluR5 mRNA in the parvocellular PVN (Figure 1). We performed immunohistochemistry to confirm that GluR5 protein is also expressed in the PVN. Similar to the experience of Eyigor et al. (Eyigor et al., 2005), we found that GluR5 immunoreactivity could not be detected without using an antigen retrieval technique (Figure 2A). We also did not see any immunoreactivity after antigen retrieval in the absence of the anti-GluR5 primary antibody (Figure 2B). However, when the tissues were subjected to antigen retrieval prior to immunostaining, immunoreactivity was revealed. We observed GluR5-like immunoreactivity in both the PVN (Figure 2C) and median eminence, especially in the external lamina (Figure 2D). Overall, the pattern of expression for GluR5 immunoreactivity was similar to that seen for GluR5 mRNA expression using in situ hybridization. For example, large numbers of GluR5 immunoreactive cells were detected in regions known to express high levels of GluR5 mRNA, including the nucleus reuniens of the thalamus and hippocampus (not shown).
Figure 1. GluR5 expression in PVN by in situ hybridization.

A) Bright field image of the PVN on an emulsion-dipped slide after in-situ hybridization. The PVN is outlined. Scale bar represents 100 μm. B) Higher magnification of the boxed area in A above. Cells associated with silver grains representing GluR5 mRNA expression are indicated with arrowheads. The third ventricle is indicated as 3V. Scale bar represents 50 μm.
The presence of GluR5-like immunoreactivity in both the PVN and the external lamina of the median eminence suggested the possibility of co-localization with the ACTH secretagogue CRH. Therefore, we performed dual-label confocal microscopy to determine whether GluR5 immunoreactivity was co-localized with CRH in median eminence terminals. Nearly all of the nerve terminals in the external lamina of the median eminence that contained CRH also contained GluR5-like immunoreactivity, although not all GluR5-containing terminals contained CRH (Figure 3).
Figure 3. GluR5/CRH co-localization.

Tissue was double-labeled for GluR5 and CRH, and imaged using confocal microscopy. Pictures shown are photomicrographs of the median eminence taken at low magnification, to show the distribution of CRH and GluR5 in the median eminence (A-C), or high magnification, showing co-localization of CRH and GluR5 immunoreactivity in axon terminals of the external lamina of the median eminence (D-F). GluR5 is pictured as green (A, D) and CRH as red (B, E). Areas co-expressing GluR5 and CRH are depicted in yellow (C, F). Terminals co-expressing CRH and GluR5 are indicated with arrowheads. Scale bars represent 20 μm.
Injection site confirmation
We confirmed the sites of injection in the PVN and median eminence cannulation experiments by two methods. First, we injected Chicago Sky Blue dye with the drug or vehicle, which allowed us to visualize the location of the injection grossly while cutting the brain. Although the dye was not visible when the tissue was immunostained and mounted on slides, the dye allowed us to confirm the presence and approximate location of an injection. We used Nissl stain (Figure 4) to visualize the cannula tracks and injection sites. Injections were counted as a hit if the center of the injection site was within 0.1 mm of the targeted structure (PVN or median eminence). Missed injections were omitted from the analyses of plasma hormones and c-Fos expression.
Figure 4. PVN Cannula Placement.
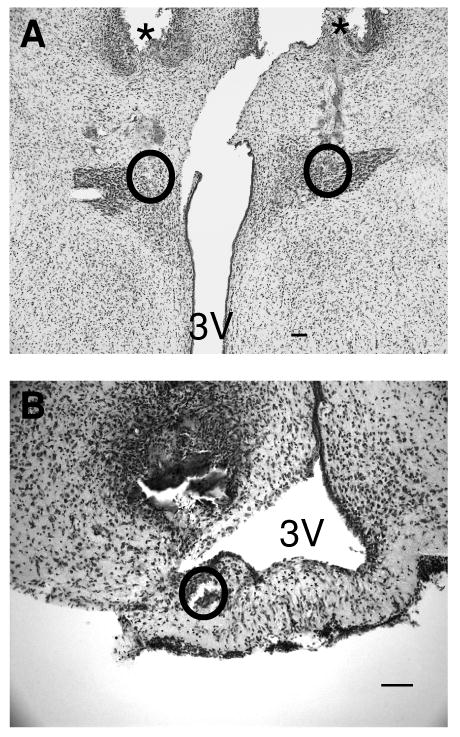
Cannula placement was verified by microscopic examination of Nissl stained slices. A) Photomicrograph of Nissl stained section, showing the injection sites for a PVN injection. The tracks made by the guide cannula are indicated with asterisks, and the injection sites are outlined. B) Photomicrograph of Nissl stained section, showing the injection site for a median eminence injection. The injection site is outlined. 3V- 3rd ventricle. The scale bar represents 100 μm.
GluR5 Antagonist Infusion: PVN
Previous data from our group indicates that the corticosterone response to restraint is blunted by intracerebroventricular injection of LY382884 (van Hooren and Herman, unpublished observations), suggesting that there is a role for GluR5 in excitatory regulation of the HPA axis. Since GluR5 mRNA and protein are expressed in the PVN, we hypothesized that paraventricular GluR5 is responsible for a major part of this excitatory input. To test this hypothesis, we injected vehicle (n=10) or LY382884 (35 ng per side, n=8) into the PVN via cannula 15 minutes before subjecting the rats to restraint. We then measured plasma ACTH and corticosterone, and c-Fos expression in the PVN. In contrast to the results of the third ventricle infusion, intra-PVN infusion of LY382884 led to a significant increase in ACTH (F1,71 = 5.38, p < 0.05). Post-hoc analysis revealed that this increase was significant at the 60 min time point. There was also a significant main effect of LY382884 treatment on corticosterone (F1,71 = 6.00, p < 0.05)(see figure 5A,B), consistent with an overall increase in the integrated corticosterone response. However, post-hoc tests did not reveal a significant difference at any given point. Fos protein expression in the PVN was significantly elevated by LY382884 treatment (p < 0.05, see figure 5C).
Figure 5. PVN Injection of LY382884.

Animals were injected bilaterally into the PVN with LY382884, and subjected to restraint stress. A) Plasma ACTH levels. There was a significant main effect of LY382884 treatment on plasma ACTH. B) Plasma corticosterone levels. There was a significant main effect of LY382884 treatment on corticosterone, although post-hoc tests did not reveal significant differences at any individual time points. C) c-Fos immunoreactive cells in the PVN. Results are expressed as mean ± SEM. Restraint time is indicated by a bar on the time course graphs. * denotes p < 0.05 vs vehicle.
GluR5 Antagonist Infusion: Median Eminence
The intense staining of the external lamina of the hypothalamic median eminence with the GluR5 antibody suggested that there is also GluR5 protein present on nerve terminals there. Thus, we hypothesized that GluR5 may also influence release of CRH into the portal circulation, and thus act to modulate activation of the HPA axis at the median eminence. To test this hypothesis, we implanted rats with cannulae aimed at the median eminence. Rats injected with either 35 ng (high, n=6) or 3.5 ng (low, n=7) of LY382884, or vehicle (n=7) were subjected to restraint stress challenge after the injection. LY382884 injection significantly reduced the ACTH responses to restraint stress (treatment × time interaction F6,51 = 2.356, p < 0.05) at 60 minutes after the injection (the end of the restraint challenge, Figure 6A). There was also a trend toward a decreased corticosterone response to restraint in LY382884 treated animals, but this failed to reach statistical significance (Figure 6B). There was no effect of the LY382884 treatment on c-Fos protein expression in the PVN, either between the drug and vehicle treated animals (Figure 6C), or between the right and left PVN (not shown). There also did not appear to be a dose-response effect of LY382884 in the median eminence, on either ACTH or corticosterone responses to restraint, at the doses tested.
Figure 6. Median Eminence Injection of LY382884.

Animals were injected with LY382884 into the median eminence, and subjected to restraint stress. A) Plasma ACTH levels. LY382884 treatment significantly blunted the ACTH response to restraint stress at 60 minutes after the injection, vs. vehicle, in both low and high treatment groups. B) Plasma corticosterone levels. There was no significant main effect of LY382884 treatment on the corticosterone response. C) Particle counts from the PVN of c-Fos-immunostained slices. The open bar represents the vehicle treated group, the grey bar the low treatment group, and the black bar the high treatment group. There were no significant differences between treatment groups. Restraint time is indicated by a horizontal bar on the time course graphs for ACTH and corticosterone. Low treatment group rats received 3.5 ng of LY382884, while high treatment group rats received 35 ng. There was no significant difference between these two doses. Results are expressed as mean ± SEM. * denotes p < 0.05.
Discussion
Previous studies suggested a role for paraventricular glutamate signaling in regulation of the HPA axis (Darlington et al., 1989; Feldman and Weidenfeld, 1997; Ziegler and Herman, 2000; Zelena et al., 2005). The current studies tested the hypothesis that GluR5 mediates the effects of glutamate on the HPA axis. Our anatomical data confirm that GluR5 protein is present in both the PVN and median eminence, and co-localizes with CRH immunoreactivity in median eminence terminals, suggesting the capacity for GluR5 signaling in PVN CRH neurons. Functional tests revealed that injection of a GluR5 antagonist into the PVN increased ACTH and PVN Fos induction after restraint stress, consistent with an inhibitory role for GluR5 in modulating central activation of the HPA axis. In contrast, injection of LY382884 directly into the median eminence reduced HPA axis stress responses, consistent with an excitatory role for glutamate in ACTH secretagogue release. Together, the results suggest that GluR5 receptors play a role in both activation and inhibition of HPA axis stress responses.
Glutamate is an excitatory amino acid neurotransmitter, so it is interesting that blocking its actions in the PVN led to excitation of the HPA axis response to restraint. Reduced HPA axis activation may be mediated by presynaptic inhibition via GluR5-containing receptors, as has been seen in other brain regions. In the hippocampus, for example, GluR5 is found presynaptically (Pinheiro et al., 2005), and presynaptic GluR5-mediated signaling leads to decreased excitatory activity (Clarke et al., 1997). One possible explanation for this phenomenon is that GluR5 mediates an autoreceptor function, where GluR5-mediated signaling leads to decreased glutamate release from the synapse. Presynaptic regulation of neurotransmitter release has been reported in the amygdala and neocortex, where high concentrations of GluR5 agonists suppress GABA release from interneurons (Braga et al., 2003; Campbell et al., 2007). If this is the case in the PVN, presynaptic GluR5 may decrease excitatory neurotransmission at PVN synapses. Inhibiting this GluR5-mediated signaling would result in the release of presynaptic inhibition, leading to enhanced HPA axis activity and enhanced c-Fos activation in the PVN, consistent with our observed data.
A second explanation is that activation of presynaptic GluR5-containing receptors increases GABA release onto parvocellular neurons in the PVN. GluR5 agonists are known to facilitate the release of neurotransmitters from presynaptic terminals (Xu et al., 2006; Aroniadou-Anderjaska et al., 2007; Campbell et al., 2007; Wu et al., 2007). Therefore, inhibition of presynaptic GluR5 could lead to enhanced HPA axis activation and PVN c-Fos expression by removal of inhibition, consistent with the results that we observed. In support of this hypothesis, previous studies demonstrate that GluR5 mediates increased presynaptic GABA release in hippocampal neurons (Christensen et al., 2004)
A third explanation for our results is that the inhibitory signaling seen is mediated by post-synaptic GluR5 on GABAergic interneurons close to the PVN. In this case, GluR5-mediated excitatory signaling at these interneurons would cause increased GABA release onto CRH-containing parvocellular neurons in the PVN. Activation of GluR5 would thus indirectly inhibit HPA axis activity by increasing inhibition of CRH-containing cells in the PVN. These possible mechanisms for GluR5 action in the PVN proper are summarized in Figure 7A-C.
Figure 7. Model of GluR5-mediated effects on the HPA axis.

GluR5 activation in the PVN and at the median eminence have opposite actions on activation of the HPA axis. Based on what is known about GluR5 actions in the central nervous system, there are several ways to explain this. A) Presynaptic GluR5-mediated signaling at a GABAergic synapse causes enhanced glutamate release onto the parvocellular neuron, thus decreasing HPA axis activity. B) Presynaptic GluR5-mediated signaling at a glutamatergic synapse causes decreased glutamate release onto the parvocellular neuron, thus decreasing HPA axis activity. C) Post-synaptic GluR5 in an inhibitory interneuron leads to increased GABA release onto the parvocellular neuron, leading to HPA axis activity. D) At the median eminence, presynaptic GluR5-mediated signaling leads to enhanced CRH release, thus increasing activity of the HPA axis.
Inhibition of GluR5 in the median eminence reduces ACTH release, indicative of an excitatory effect of local glutamate on secretagogue release. Since there are very few neuronal cell somata in the median eminence, it is possible that GluR5 excitation of the HPA axis is mediated by presynaptic enhancement of CRH release, consistent with its known role in presynaptic facilitation of neurotransmitter release elsewhere (Christensen et al., 2004)(Campbell et al., 2007)(Xu et al., 2006)(Wu et al., 2007). This mechanism for GluR5 signaling is outlined in Figure 7D.
The GluR5-like immunoreactivity in the median eminence suggests a mechanism by which glutamate can excite the HPA axis at the level of the neurosecretory terminal. CRH is the primary hypothalamic secretagogue for ACTH release from the anterior lobe of the pituitary (Vale et al., 1981; Rivier and Vale, 1983), and is secreted into the hypophyseal portal vasculature in the external lamina of the median eminence. The co-localization of GluR5 with CRH in the external lamina suggests that presynaptic GluR5 may enhance CRH release, such that glutamate signaling through GluR5 in the median eminence enhances activity of the HPA axis. The fact that GluR5 antagonist injection into the median eminence reduced HPA axis stress responses without affecting PVN c-Fos induction further supports a local action of GluR5 signaling at the nerve terminal.
In the studies described, animals were given LY382884 infusions 15 minutes prior to the onset of restraint stress. Pilot studies indicated that this time period was sufficient to reveal HPA axis-modifying effects of GluR5 inhibition after intraventricular injection (data not shown). The time interval between drug administration and stress application was designed to allow sufficient time for the drug to diffuse into the cellular area of the injection and affect ACTH and corticosterone secretion.
The lack of a dose-response to LY382884 in the animals injected into the median eminence suggests that the dose of LY382884 given in these experiments may be higher than the maximally efficacious concentration. The incomplete inhibition of HPA axis activation by local LY382884 treatment suggests that glutamate effects mediated by GluR5 modulate stress excitation of the HPA axis, but are not required for the initiation of the response.
Our studies could not determine whether drugs injected into the median eminence were able to diffuse out of the median eminence and reach the pituitary gland. This could potentially present a problem, as there are glutamate receptors expressed in the anterior pituitary (Hinoi et al., 2004), and glutamate binds to the anterior lobe of the pituitary, albeit weakly (Meeker et al., 1994). However, kainate does not compete with glutamate for binding to pituitary membranes in binding assays (Yoneda and Ogita, 1989), suggesting that functional kainate receptors, including GluR5, are not found in the anterior pituitary. Additionally, peripherally administered glutamatergic drugs that affect the HPA axis are thought to exert these effects via brain areas deficient in blood-brain barrier, rather than through the pituitary gland (Jezova, 2005). Therefore, even if some of the LY382884 did diffuse to the pituitary gland, it does not seem likely that it affected HPA axis signaling in the pituitary gland. We can not, however, conclusively rule out effects on the pituitary, nor on structures near the median eminence, such as the arcuate nucleus.
The external lamina of the median eminence contains axon terminals from neurons located almost exclusively in the PVN and other parts of the hypothalamus (Wiegand and Price, 1980), although there may be afferent fibers from the medial septum and diagonal band of Broca (Silverman et al., 1987). There is evidence that CRH-expressing neurons in the PVN also express VGlut2 protein in the median eminence (Hrabovszky et al., 2005), suggesting that glutamate acting on GluR5 in the median eminence could be released by CRH nerve terminals. Indeed, neuropeptidergic neurons almost uniformly co-release classical neurotransmitters, including glutamate (Hokfelt et al., 2000). This suggests that presynaptic glutamate signaling through GluR5 in the median eminence could be acting as a feed-forward signal in activation of the HPA axis response to stress.
In conclusion, the current report indicates novel, GluR5-mediated presynaptic mechanisms for glutamatergic regulation of the HPA axis. Our data support a role for kainate-preferring receptors in gating of CRH release at median eminence terminals, as well as in local inhibition of PVN activation. Presynaptic modulation of HPA activation provides a mechanism whereby release of CRH can be adjusted at the level of the nerve terminal via glutamatergic projections to the median eminence. Importantly, the localized blood-brain barrier deficiency in the median eminence provides a potential route for specific pharmacological modulation of HPA stress reactivity in disorders characterized by glucocorticoid hypersecretion.
Acknowledgments
We thank Mark Dolgas, Ben Packard, Kenny Jones, Dennis Choi, Amy Furay, Miyuki Tauchi, Michelle Ostrander, Yve Ulrich-Lai, Rong Zhang, Ingrid Thomas, Amanda Jones, and Nancy Mueller for help with blood and brain collections in these experiments. We also thank Eli Lilly, Inc. for the generous gift of LY382884.
Abbreviations
- ACTH
adrenocorticotropic hormone
- AMPA
alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- ANOVA
analysis of variance
- ATPA
(RS)-2-Amino-3-(3-hydroxy-5-tert-butylisoxazol-4-yl)propanoic acid
- CRH
corticotropin releasing hormone
- DAB
diaminobenzidine
- GFAP
glial fibrillary acidic protein
- HPA
hypothalamic-pituitary-adrenocortical
- KPBS
potassium phosphate buffered saline
- NMDA
n-methyl-d-aspartate
- PVN
paraventricular nucleus of the hypothalamus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aroniadou-Anderjaska V, Qashu F, Braga MF. Mechanisms regulating GABAergic inhibitory transmission in the basolateral amygdala: implications for epilepsy and anxiety disorders. Amino Acids. 2007;32:305–315. doi: 10.1007/s00726-006-0415-x. [DOI] [PubMed] [Google Scholar]
- Barnett V, Lewis T. Outliers in statistical data. 3rd. New York: John Wiley & Sons; 1994. [Google Scholar]
- Braga MF, Aroniadou-Anderjaska V, Xie J, Li H. Bidirectional modulation of GABA release by presynaptic glutamate receptor 5 kainate receptors in the basolateral amygdala. J Neurosci. 2003;23:442–452. doi: 10.1523/JNEUROSCI.23-02-00442.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brann DW. Glutamate: a major excitatory transmitter in neuroendocrine regulation. Neuroendocrinology. 1995;61:213–225. doi: 10.1159/000126843. [DOI] [PubMed] [Google Scholar]
- Campbell SL, Mathew SS, Hablitz JJ. Pre- and postsynaptic effects of kainate on layer II/III pyramidal cells in rat neocortex. Neuropharmacology. 2007;53:37–47. doi: 10.1016/j.neuropharm.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson HE, Miglietta JT, Roginsky MS, Stegink LD. Stimulation of pituitary hormone secretion by neurotransmitter amino acids in humans. Metabolism. 1989;38:1179–1182. doi: 10.1016/0026-0495(89)90156-x. [DOI] [PubMed] [Google Scholar]
- Christensen JK, Paternain AV, Selak S, Ahring PK, Lerma J. A mosaic of functional kainate receptors in hippocampal interneurons. J Neurosci. 2004;24:8986–8993. doi: 10.1523/JNEUROSCI.2156-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke VR, Ballyk BA, Hoo KH, Mandelzys A, Pellizzari A, Bath CP, Thomas J, Sharpe EF, Davies CH, Ornstein PL, Schoepp DD, Kamboj RK, Collingridge GL, Lodge D, Bleakman D. A hippocampal GluR5 kainate receptor regulating inhibitory synaptic transmission. Nature. 1997;389:599–603. doi: 10.1038/39315. [DOI] [PubMed] [Google Scholar]
- Darlington DN, Miyamoto M, Keil LC, Dallman MF. Paraventricular stimulation with glutamate elicits bradycardia and pituitary responses. Am J Physiol. 1989;256:R112–119. doi: 10.1152/ajpregu.1989.256.1.R112. [DOI] [PubMed] [Google Scholar]
- Engeland WC, Miller P, Gann DS. Dissociation between changes in plasma bioactive and immunoreactive adrenocorticotropin after hemorrhage in awake dogs. Endocrinology. 1989;124:2978–2985. doi: 10.1210/endo-124-6-2978. [DOI] [PubMed] [Google Scholar]
- Eyigor O, Minbay Z, Cavusoglu I, Jennes L. Localization of kainate receptor subunit GluR5-immunoreactive cells in the rat hypothalamus. Brain Res Mol Brain Res. 2005;136:38–44. doi: 10.1016/j.molbrainres.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Feldman S, Weidenfeld J. Hypothalamic mechanisms mediating glutamate effects on the hypothalamo-pituitary-adrenocortical axis. J Neural Transm. 1997;104:633–642. doi: 10.1007/BF01291881. [DOI] [PubMed] [Google Scholar]
- Gay VL, Plant TM. N-methyl-D,L-aspartate elicits hypothalamic gonadotropin-releasing hormone release in prepubertal male rhesus monkeys (Macaca mulatta) Endocrinology. 1987;120:2289–2296. doi: 10.1210/endo-120-6-2289. [DOI] [PubMed] [Google Scholar]
- Herman JP, Mueller NK, Figueiredo H. Role of GABA and glutamate circuitry in hypothalamo-pituitary-adrenocortical stress integration. Ann N Y Acad Sci. 2004;1018:35–45. doi: 10.1196/annals.1296.004. [DOI] [PubMed] [Google Scholar]
- Herman JP, Eyigor O, Ziegler DR, Jennes L. Expression of ionotropic glutamate receptor subunit mRNAs in the hypothalamic paraventricular nucleus of the rat. J Comp Neurol. 2000;422:352–362. [PubMed] [Google Scholar]
- Hinoi E, Takarada T, Ueshima T, Tsuchihashi Y, Yoneda Y. Glutamate signaling in peripheral tissues. Eur J Biochem. 2004;271:1–13. doi: 10.1046/j.1432-1033.2003.03907.x. [DOI] [PubMed] [Google Scholar]
- Hokfelt T, Broberger C, Xu ZQ, Sergeyev V, Ubink R, Diez M. Neuropeptides--an overview. Neuropharmacology. 2000;39:1337–1356. doi: 10.1016/s0028-3908(00)00010-1. [DOI] [PubMed] [Google Scholar]
- Hrabovszky E, Wittmann G, Turi GF, Liposits Z, Fekete C. Hypophysiotropic thyrotropin-releasing hormone and corticotropin-releasing hormone neurons of the rat contain vesicular glutamate transporter-2. Endocrinology. 2005;146:341–347. doi: 10.1210/en.2004-0856. [DOI] [PubMed] [Google Scholar]
- Jezova D. Control of ACTH secretion by excitatory amino acids: functional significance and clinical implications. Endocrine. 2005;28:287–294. doi: 10.1385/ENDO:28:3:287. [DOI] [PubMed] [Google Scholar]
- Joanny P, Steinberg J, Oliver C, Grino M. Glutamate and N-methyl-D-aspartate stimulate rat hypothalamic corticotropin-releasing factor secretion in vitro. J Neuroendocrinol. 1997;9:93–97. doi: 10.1046/j.1365-2826.1997.00548.x. [DOI] [PubMed] [Google Scholar]
- Johnson MP, Kelly G, Chamberlain M. Changes in rat serum corticosterone after treatment with metabotropic glutamate receptor agonists or antagonists. J Neuroendocrinol. 2001;13:670–677. doi: 10.1046/j.1365-2826.2001.00678.x. [DOI] [PubMed] [Google Scholar]
- Koenig JI, Cho JY. Provocation of kainic acid receptor mRNA changes in the rat paraventricular nucleus by insulin-induced hypoglycaemia. J Neuroendocrinol. 2005;17:111–118. doi: 10.1111/j.1365-2826.2005.01285.x. [DOI] [PubMed] [Google Scholar]
- Kristensen P. Differential expression of AMPA glutamate receptor mRNAs in the rat adrenal gland. FEBS Lett. 1993;332:14–18. doi: 10.1016/0014-5793(93)80472-7. [DOI] [PubMed] [Google Scholar]
- Mahesh VB, Zamorano P, De Sevilla L, Lewis D, Brann DW. Characterization of ionotropic glutamate receptors in rat hypothalamus, pituitary and immortalized gonadotropin-releasing hormone (GnRH) neurons (GT1-7 cells) Neuroendocrinology. 1999;69:397–407. doi: 10.1159/000054442. [DOI] [PubMed] [Google Scholar]
- Makara GB, Stark E. Effect of intraventricular glutamate on ACTH release. Neuroendocrinology. 1975;18:213–216. doi: 10.1159/000122400. [DOI] [PubMed] [Google Scholar]
- Meeker RB, Greenwood RS, Hayward JN. Glutamate receptors in the rat hypothalamus and pituitary. Endocrinology. 1994;134:621–629. doi: 10.1210/endo.134.2.7905409. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Yokotani N, Wenthold RJ. Light and electron microscope distribution of the NMDA receptor subunit NMDAR1 in the rat nervous system using a selective anti-peptide antibody. J Neurosci. 1994;14:667–696. doi: 10.1523/JNEUROSCI.14-02-00667.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro PS, Rodrigues RJ, Rebola N, Xapelli S, Oliveira CR, Malva JO. Presynaptic kainate receptors are localized close to release sites in rat hippocampal synapses. Neurochem Int. 2005;47:309–316. doi: 10.1016/j.neuint.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Rasband W. ImageJ In. 1.38×. Bethesda, Maryland, USA: National Institutes of Health; 19972007. [Google Scholar]
- Rivier C, Vale W. Interaction of corticotropin-releasing factor and arginine vasopressin on adrenocorticotropin secretion in vivo. Endocrinology. 1983;113:939–942. doi: 10.1210/endo-113-3-939. [DOI] [PubMed] [Google Scholar]
- Scaccianoce S, Matrisciano F, Del Bianco P, Caricasole A, Di Giorgi Gerevini V, Cappuccio I, Melchiorri D, Battaglia G, Nicoletti F. Endogenous activation of group-II metabotropic glutamate receptors inhibits the hypothalamic-pituitary-adrenocortical axis. Neuropharmacology. 2003;44:555–561. doi: 10.1016/s0028-3908(03)00027-3. [DOI] [PubMed] [Google Scholar]
- Silverman AJ, Jhamandas J, Renaud LP. Localization of luteinizing hormone-releasing hormone (LHRH) neurons that project to the median eminence. J Neurosci. 1987;7:2312–2319. [PMC free article] [PubMed] [Google Scholar]
- Vale W, River C. Substances modulating the secretion of ACTH by cultured anterior pituitary cells. Fed Proc. 1977;36:2094–2099. [PubMed] [Google Scholar]
- Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science. 1981;213:1394–1397. doi: 10.1126/science.6267699. [DOI] [PubMed] [Google Scholar]
- Wiegand SJ, Price JL. Cells of origin of the afferent fibers to the median eminence in the rat. J Comp Neurol. 1980;192:1–19. doi: 10.1002/cne.901920102. [DOI] [PubMed] [Google Scholar]
- Wu LJ, Xu H, Ren M, Zhuo M. Genetic and pharmacological studies of GluR5 modulation of inhibitory synaptic transmission in the anterior cingulate cortex of adult mice. Dev Neurobiol. 2007;67:146–157. doi: 10.1002/dneu.20331. [DOI] [PubMed] [Google Scholar]
- Xu H, Wu LJ, Zhao MG, Toyoda H, Vadakkan KI, Jia Y, Pinaud R, Zhuo M. Presynaptic regulation of the inhibitory transmission by GluR5-containing kainate receptors in spinal substantia gelatinosa. Mol Pain. 2006;2:29. doi: 10.1186/1744-8069-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda Y, Ogita K. Solubilization of stereospecific and quisqualate-sensitive activity of [3H]glutamate binding in the pituitary of the rat. Neuropharmacology. 1989;28:611–616. doi: 10.1016/0028-3908(89)90140-8. [DOI] [PubMed] [Google Scholar]
- Zelena D, Mergl Z, Makara GB. Glutamate agonists activate the hypothalamic-pituitary-adrenal axis through hypothalamic paraventricular nucleus but not through vasopressinerg neurons. Brain Res. 2005;1031:185–193. doi: 10.1016/j.brainres.2004.10.034. [DOI] [PubMed] [Google Scholar]
- Ziegler DR, Herman JP. Local integration of glutamate signaling in the hypothalamic paraventricular region: regulation of glucocorticoid stress responses. Endocrinology. 2000;141:4801–4804. doi: 10.1210/endo.141.12.7949. [DOI] [PubMed] [Google Scholar]
- Ziegler DR, Cullinan WE, Herman JP. Distribution of vesicular glutamate transporter mRNA in rat hypothalamus. J Comp Neurol. 2002;448:217–229. doi: 10.1002/cne.10257. [DOI] [PubMed] [Google Scholar]
- Ziegler DR, Cullinan WE, Herman JP. Organization and regulation of paraventricular nucleus glutamate signaling systems: N-methyl-D-aspartate receptors. J Comp Neurol. 2005;484:43–56. doi: 10.1002/cne.20445. [DOI] [PubMed] [Google Scholar]