

Abstract
Fanconi anemia (FA) is an autosomal recessive disorder characterized by multiple congenital anomalies, progressive bone marrow failure, and high cancer risk. Cells from FA patients exhibit spontaneous chromosomal instability and hypersensitivity to DNA interstrand cross-linking (ICL) agents. Although the precise mechanistic details of the FA/BRCA pathway of ICL-repair are not well understood, progress has been made in the identification of the FA proteins that are required for the pathway. Among the 13 FA complementation groups from which all the FA genes have been cloned, only a few of the FA proteins are predicted to have direct roles in DNA metabolism. One of the more recently identified FA proteins, shown to be responsible for complementation of the FA complementation group J, is the BRCA1 Associated C-terminal Helicase (BACH1, designated FANCJ), originally identified as a protein associated with breast cancer. FANCJ has been proposed to function downstream of FANCD2 monoubiquitination, a critical event in the FA pathway. Evidence supports a role for FANCJ in a homologous recombination (HR) pathway of double strand break (DSB) repair. In this review, we will summarize the current knowledge in terms of FANCJ functions through its enzymatic activities and protein interactions. The molecular roles of FANCJ in DNA repair and the response to replicational stress will be discussed.
Keywords: FANCJ, Fanconi anemia, breast cancer, cancer, DNA damage, DNA repair, helicase
THE FANCONI ANEMIA PATHWAY
Fanconi anemia (FA) is a rare hereditary disorder characterized by skeletal abnormalities, bone marrow failure, and an increased incidence of cancer [1–3]. The basic cellular abnormality in FA has been postulated to lie in the DNA repair mechanisms because cells from FA patients display chromosomal abnormalities, and are hypersensitive to DNA interstrand crosslinks (ICLs) agents such as mitomycin C (MMC) and cisplatin. FA is a genetically heterogeneous disease with 13 complementation groups currently recognized. It is possible that more FA-associated genes may be identified since some FA patients have not been assigned a complementation group.
Thirteen causative genes (FANCA/B/C/D1/D2/E/F/G/I/J/L/M/N) have been identified to date. Increasing evidence suggests that these genes encode proteins that function as signal transducers [4–7] or DNA processing molecules [8–10] in a DNA-damage response pathway. The eight FA gene products (FANCA/B/C/E/F/G/L/M) assemble to form the nuclear FA core complex, and co-immunoprecipitation studies revealed that any known mutation in components of the core complex found in FA patients appears to disrupt integrity of the complex [11–14]. Two new proteins, FAAP24 and FAAP100, were recently found in this core complex and are essential for the stability of this core complex [15, 16]. Certain FA proteins have been identified to be the same as proteins in the BRCA pathway or physically associated with the BRCA pathway. For example, FANCD1 (mutated in FA-D1) is identical to the breast/ovarian cancer susceptibility factor BRCA2 [17], and the most recently identified FA gene, FANCN, encodes PALB2 (partner and localizer of BRCA2) [14, 18], a crucial regulator of the BRCA2 protein [19]. Additionally, FANCJ is identical to BACH1/BRIP1 [20–22], a DNA helicase that interacts directly with BRCA1 [8]. Accumulating evidence has suggested that the proteins in the FA pathway constitute a novel DNA-damage response network that involves breast cancer proteins BRCA1 and BRCA2, which is more generally referred to as the FA/BRCA pathway.
Monoubiquitination of FANCD2 and its paralog, FANCI, play critical roles in the FA/BRCA pathway. Mutational inactivation of any one of the FA core complex results in loss of FANCD2 monoubiquitination and as a result, the loss of function of the FA repair pathway. FANCD2 monoubiquitylation and its nuclear foci formation occur in response to DNA damaging agents and during S phase of the cell cycle even in the absence of exogenous DNA damage [23]. A DNA damage-activated signaling kinase, ATR (ATM and rad3 related), and a single-strand DNA binding protein heterotrimeric complex Replication Protein A (RPA) are required for DNA damage-inducible monoubiquitylation and foci formation of FANCD2, indicating an upstream role for these factors in the activation of the FA pathway [4, 11, 12]. The exact signal and activation cascade required for FANCD2 monoubiquitylation, however, remain elusive. The newly discovered FANCI protein was shown to be monobiquitylated in response to DNA damage and replication, and its monoubiquitylation also depends on the other FA core complex proteins [24–26]. FANCD1, FANCJ and FANCN are thought to act downstream or in a pathway parallel to that of the FA pathway because cells lacking these proteins have normal levels of monoubiquitylated FANCD2 in response to DNA damage.
Recent evidence suggests that in addition to FANCM’s requirement for FANCD2 and FANCI monoubiquitination, ATP-dependent activities of FANCM are required for MMC resistance but not FANCD2/FANCI monoubiquitination [27], suggesting that FANCM and its associated FA core complex plays a role in downstream events of the DNA repair pathway. Consistent with a direct role of FANCM in DNA repair, the protein has intrinsic DNA translocase [10] and branch-point translocase activities [9, 27].
The FA proteins interact with a cast of DNA repair factors, and it is becoming increasingly evident that these FA-interacting proteins have a role in genome stability and their interactions are critical for a proper DNA damage response. For example, the Bloom’s syndrome helicase (BLM) is associated with the FA core complex [12] and BLM mutations lead to a disease characterized by elevated sister chromatid exchange and cancer [28]. The FA/BRCA pathway and its cross-talk with other DNA repair proteins has been extensively reviewed elsewhere [1–3, 13, 29–32]. In this review, we will summarize the current knowledge in terms of FANCJ functions through its enzymatic activities and protein interactions. The molecular roles of FANCJ in DNA repair and the response to replicational stress will be discussed in terms of potential protein partners that operate in the FA pathway of DNA ICL repair and those that serve to enable smooth progression of the replication fork.
IDENTIFICATION AND CLASSIFICATION OF FANCJ
FANCJ was found to have a role in DSB repair by its interaction with the tumor suppressor BRCA1 [8, 22] and be genetically linked to FA [20–22]. We will summarize the identification of FANCJ as a tumor suppressor and the classification of FANCJ in a unique family of DNA helicases that are important for genomic stability.
Discovery of FANCJ as a Helicase Associated with Breast Cancer and Linked to Fanconi Anemia
FANCJ was first discovered by Cantor et al. as a protein that binds to the breast cancer C-terminal (BRCT) repeats of BRCA1 [8]. Because a transcription factor was also named BACH1 [33], the BRCA1-interacting helicase was later called BRIP1 (BRCA1 interacting protein 1). BACH1/BRIP1 was later identified as the gene mutated in the J complementation group of FA by three independent groups [20–22]. Now, BACH1 (BRIP1) is widely referred to as FANCJ because of its genetic linkage to FA-J complementation group. A genetic interaction between FANCJ and BRCA1 in DSB repair was established [8], suggesting a tumor suppressor role of FANCJ. In support of a clinical importance of FANCJ as a tumor suppressor, two females among a cohort of 65 women with early-onset breast cancer were identified carrying 2 independent germ line sequence changes (P47A or M299I) in the FANCJ coding region and normal genotypes for BRCA1 and BRCA2 [8]. More recently, Seal et al. reported that truncating mutations in FANCJ are low-penetrance breast cancer susceptibility alleles [34]. A very recent study provided evidence that transcription of FANCJ was controlled by the E2F/retinoblastoma pathway through a conserved E2F-responsive site, and its elevated expression is detected in primary invasive breast carcinomas with unfavorable characteristics [35]. Since FANCD2 monoubquitination is intact in FA-J cells, FANCJ appears to act downstream in the FA pathway akin to BRCA2/FANCD1 or in a parallel pathway to FANCD2 monoubiquitination. However, a clear understanding of the molecular and cellular roles of FANCJ in relation to the FA pathway is lacking.
Classification of FANCJ in a Unique Family of DNA Helicases
FANCJ protein is 1249 amino acids in length, with a predicted molecular mass of ~130 kDa. The N-terminal 888 amino acid sequence of FANCJ shares homology with proteins that belong to a DEAH subfamily of superfamily 2 helicases [8]. The BRCA1 interaction domain resides in the C-terminal region of FANCJ after the helicase core domain (Fig. (1)). In addition to a core helicase domain comprised of the seven conserved ATPase/helicase motifs, the N-terminal region of FANCJ has a predicted nuclear localization sequence (NLS) (residues 159–174) and a conserved Fe-S cluster (residues 270–363) (Fig. (1) and Fig. (2)). Three recently solved crystal structures of a protein that is mutated in Xeroderma pigmentosum group D (XPD) and related to FANCJ by sequence homology in the helicase core domain confirmed the existence of a novel Fe-S domain [36–38], first identified biochemically in 2006 by the White lab [39]. Structural and biochemical evidence indicate that the conserved Fe-S domain is essential for XPD helicase activity and suggests that it serves as a recognition domain for the ssDNA-dsDNA junction, acting as the wedge for separation of complementary strands [36–39]. Biochemical characterization of a FANCJ-M299I variant (Fig. (2)) suggests that the conserved Fe-S domain in FANCJ plays a role in unwinding damaged DNA substrates [40].
Fig. (1). Map of FANCJ protein.

The conserved helicase motifs are indicated by yellow boxes and the positions of the metal binding domain (red), nuclear localization sequence (NLS in green), and BRCA1 interaction domain (blue). MLH1 binding domain and four conserved cysteines in iron-sulfur domain are indicated above and below the protein schematic in pink.
Fig. (2). Sequence alignment of the Fe-S domain.

Human DNA helicases FANCJ and XPD, mouse RTel, yeast Chl1, and Sulfolobus acidocaldarius (Sac) XPD and Ferroplasma acidarmanus (Fac) Rad3 were shown. The four conserved cysteine residues of the metal binding domain are highlighted in yellow. The FANCJ amino acid substitution (M299I) adjacent to a conserved cysteine residue and representing a FANCJ polymorphic variant is highlighted. Another substitution (A349P) arising from a mutation genetically linked to Fanconi Anemia (Complementation Group J) and adjacent to a conserved cysteine residue is also highlighted. Some conserved residues (L, PYY and A) are highlighted in grey.
The Fe-S domain contains four conserved cysteines found in a number of helicases sharing sequence homology in the helicase core domain (Fig. (2)). The presence of Fe-S clusters in helicases engaged in the DNA damage response is interesting because a number of proteins involved in damaged DNA recognition and repair such as base excision repair glycosylases also contain Fe-S domains [41–43]. Biochemical and structural evidence indicates that Fe-S clusters in DNA repair glycolsylases are involved in DNA binding, which ultimately impacts base removal catalysis [44]. DNA substrate binding by a glycoslyase can facilitate oxidation of the [4Fe-4S]2+ cluster to the [4Fe-4S]3+ form, which has been proposed to help in locating damaged bases [44]. One model suggests that DNA-mediated charge transfer between DNA repair enzymes provides a mechanism to facilitate scanning of the DNA for damage searching [45]. Most recently, an Fe-S cluster domain was discovered in human DNA primase that contributes to enzyme activity, raising the possibility that the DNA replication machinery has redox-sensitive activities [46]. It is of immediate interest to ascertain if DNA helicases involved in repair or replicational stress like FANCJ utilize their Fe-S clusters to scan for DNA damage or alternate DNA structures during an early step of DNA damage processing.
The interaction domain of FANCJ with MutLα, discussed later, was also mapped to a portion of the N-terminal helicase domain (residues 128–158) [47] (Fig. (1)). In the C-terminal region of FANCJ (residues 888–1063) is located the BRCA1-interacting domain that directly binds to the two BRCT repeats of BRCA1. Since FANCJ has been shown to exist in protein complexes [47–49], its interaction with other DNA repair factors such as the single-stranded DNA binding protein replication protein A (RPA) [50] may be mediated by the helicase domain or flanking regions.
A close inspection of the FANCJ helicase domain reveals that this region of the protein shares sequence homology with a number of putative or bonafide DNA helicases that are implicated in the maintenance of genomic stability in a variety of organisms (Table (1)). The bacterial DNA damage-inducible G (DinG) helicase is regulated by the SOS circuit, and it has been suggested that DinG serves a backup role in DNA repair or replication restart after DNA damage [51]. Mutations in FANCJ-related genes in yeast (Chl1) and human (ChlR1) result in sister chromatid cohesion defects [52–54]. Moreover, loss of ChlR1 in mouse causes placental malformation and embryonic lethality due to the accumulation of aneuploid cells characterized by cohesion defects [55]. Nematodes mutated in the FANCJ homolog dog-1 (deletions of guanine-rich DNA) show germline as well as somatic deletions in genes containing guanine-rich DNA, suggesting a possible role in the metabolism of G-quadruplex structures [56]. Recent genetic evidence indicate that DOG-1 is the C. elegans FANCJ homologue based on the sensitivity of dog-1 mutants to ICL agents, and its epistatic relationship to fcd-1 (C. elegans FANCD2) [57]. A mouse gene sharing sequence homology in the helicase domain with dog-1 and FANCJ is designated Rtel (Regulator of telomere length). Rtel knockout mice die within the first two weeks, and embryonic stem cells from these knockout mice show telomere loss and display many chromosome breaks and fusions, suggesting a role of the FANCJ homolog in the maintenance of telomere length [58]. FANCJ also shares sequence homology with the human XPD and yeast RAD3 helicases [8], which are subunits of the transcription factor IIH (TFIIH)/DNA repair complex required for nucleotide excision repair [59, 60]. Mutation in XPD results in the skin cancer-prone disease Xeroderma pigmentosum [61]. Clearly, the FANCJ-like family of DNA helicases plays critical roles in DNA repair and genome integrity.
Table 1.
FANCJ-like DNA Helicases
| Organism | Helicase | Disease and Organic or Cellular Phenotype |
Proposed Function | References |
|---|---|---|---|---|
| Human | FANCJ | Fanconi anemia and ICL sensitivity |
ICL repair/HR repair, G4 resolution |
[8, 62, 76] |
| ChlR1 | Sister chromatid cohesion defects |
Sister-chromatid cohesion |
[52] | |
| XPD | Xeroderma pigmentosum and UV sensitivity |
NER | [61] | |
| Mouse | Rtel1 | Telomere loss and chromosome instability |
Telomere maintance | [58] |
| ChlR1 | Sister chromatid cohesion defects |
Sister chromatid cohesion |
[55] | |
| Nematode | DOG-1 | Deletion of G-rich DNA | G4 resolution, ICL repair | [56, 57, 101] |
| Yeast | Rad3 | UV sensitivity | NER | [59] |
| CHL1 | Unusual mating and cell cycle delay at G2/M, sister chromatid cohesion defects |
Sister chromatid cohesion |
[54, 105, 106] | |
| Bacteria | DinG | Recombinational repair | [51] |
Pathogenic Mutations and Polymorphic Variations of FANCJ
FANCJ missense and truncating mutations resulting in FA are found in the helicase core domain (see LOVD FA Database (http://www.rockefeller.edu/fanconi/mutate/). Truncating FANCJ mutations that cause FA in biallelic carriers confer susceptibility to breast cancer in monoallelic carriers [34]. Clinically relevant mutations exist in the conserved Fe-S cluster of the FANCJ helicase domain [8, 21, 39, 62] (Fig. (2)). A number of naturally occurring polymorphisms in FANCJ exist whose pathological significance is not yet known. Characterization of the biochemical and cellular defects of pathogenic FANCJ mutations and analysis of FANCJ variants not yet linked to a human disease or cancer should be of wide interest to the field. In the following section, we will summarize what is known about the catalytic properties and protein interactions of FANCJ to envision how FANCJ acts in the FA pathway and other pathways.
DNA UNWINDING PROPERTIES OF THE FANCJ HELICASE
Biochemical studies have begun to address the molecular functions of FANCJ helicase, including its DNA substrate specificity and tracking mechanism. These studies suggest that FANCJ has certain unique properties that are distinct from helicases of other families, and these functions are likely to be important for its mechanism of action and roles in cellular DNA metabolism.
DNA Substrate Specificity of the FANCJ Helicase
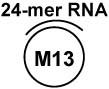
The first biochemical characterization of the FANCJ helicase showed it is a DNA-dependent ATPase that catalytically unwinds M13 partial duplex DNA substrates characterized by a covalently closed ssDNA circle annealed to a shorter oligonucleotide (Table (2)) in an ATP-dependent manner, and unwinds duplex DNA with a 5’ to 3’ directionality on a linearized M13 directionality substrate [62]. To determine the minimal loading requirements for optimal unwinding, FANCJ was tested on oligonucleotide-based substrates with increasing lengths of the 5’ ssDNA tail [63] (Table (2)). These studies demonstrated that FANCJ requires a 5’ tail of ~15 nucleotide (nt) to catalyze appreciable unwinding and that increasing the 5' ssDNA tail up to 35 nt increases the efficiency of FANCJ helicase activity. Further analysis revealed that the additional presence of a 3’ ssDNA tail, creating a forked duplex substrate (Table (2)), confers a significantly greater ability for FANCJ to both bind and unwind the DNA molecule compared to a simple 5’ tailed substrate [63]. Although neither the assembly state nor tertiary structure of FANCJ is known, it is possible that the 3' tail may stimulate FANCJ unwinding by sterically determining whether one or two DNA strands pass through a central channel of FANCJ, as proposed for DnaB [64]. In support of the notion that FANCJ may exist as a multimer, it was observed by gel exclusion chromatography that recombinant wild-type FLAG-tagged FANCJ (~130-kDa polypeptide) migrates as a ~500-kDa enzymatically active species [62]. Future studies will likely address the potential importance of FANCJ subunit interactions for its catalytic activity as a helicase in DNA repair pathways.
Table 2.
Substrate Specificity of FANCJ Helicase
| Substrate Name | Substrate Structure | Relative Helicase Activity |
|---|---|---|
| 3'-tailed duplex a | No | |
| 5'-tailed duplex a | + | |
| Forked duplex a | 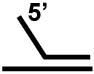 |
+++ |
| 5' flap substrate a | 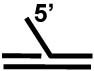 |
+++ |
| 3' flap substrate a | 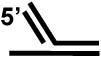 |
No |
| Replication fork a | 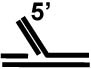 |
No |
| Holliday junction a |  |
No |
| D-loop with a 5' ssDNA tail a |
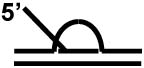 |
+++ |
| D-loop with a 3' ssDNA tail a |
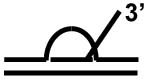 |
++ |
| Flush D-loop a |  |
+ |
| G-quadruplex DNA b | 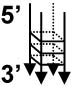 |
++++ |
| M13 partial duplex DNA c | 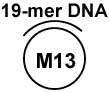 |
++ |
| M13 partial DNA-RNA hybrid c |
 |
+ |
| RNA double strand c | 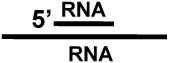 |
No |
The preference of FANCJ to unwind a forked duplex substrate with both 3’ and 5’ ssDNA arms raised the possibility that FANCJ, like certain RecQ helicases (e.g., Werner syndrome helicase or BLM [65], might have a relaxed DNA substrate specificity and unwind a variety of DNA replication and repair intermediates. FANCJ was able to efficiently unwind a 5’ flap DNA substrate (Table (2)), a strand displacement intermediate that arises during lagging strand synthesis or during DNA repair synthesis in a process such as base excision repair. However, FANCJ failed to unwind a 3’ flap substrate that might arise due to equilibrating strands during strand displacement. FANCJ also failed to unwind a synthetic replication fork (with duplex leading and lagging strand arms) that might arise when the replication fork is stalled (Table (2)). These studies demonstrated that FANCJ requires a pre-existing ssDNA tail of defined 5’ to 3’ polarity to unwind the adjacent B-form duplex DNA [63].
Since FANCJ is proposed to be involved in HR repair, its activity on key DNA intermediates of this pathway is relevant. An early intermediate of HR is the three-stranded D-loop which is produced by strand invasion into a recipient duplex (Table (2)). FANCJ was able to unwind D-loop structures by catalytically releasing the invading strand of the HR intermediate [63]. The four-stranded Holliday Junction (HJ) is a mobile DNA structure that occurs as an intermediate in homologous and site-specific DNA recombination and/or replication restart. Although FANCJ could bind model HJ structures, it failed to unwind a synthetic HJ. The specificity of FANCJ helicase for certain HR substrate intermediates suggests that FANCJ might operate on the D-loop structure which arises in an early stage of HR after stand invasion, but not in the branch migration or resolution of a HJ that might form later in the HR process. Furthermore, the 5’ to 3’ directionality of FANCJ helicase activity and its inability to unwind HJ structures suggests that FANCJ is likely to operate in a distinct manner from the RecQ helicases, which are also implicated in HR repair. This does not exclude, however, the possibility that a RecQ helicase may co-localize and operate with FANCJ in a DNA repair center or even a direct interaction between the two helicases.
FANCJ helicase activity was found to be profoundly sensitive to a polyglycol backbone modification in either the translocating or non-translocating strands of the duplex, suggesting that FANCJ interactions with the sugar phosphate backbone of both strands of the duplex are important as FANCJ tracks along the DNA molecule and separates the strands [63]. Importantly, these studies revealed that FANCJ does not display strand-specific inhibition with respect to the strand that it translocates on as observed for other helicases (vaccina virus NPH-II [66], E. coli Dda [67, 68], bacteriophage T7 gene 4 [69]). The ability of FANCJ to sense backbone discontinuity in both strands of the DNA duplex as it unwinds may be important for its physiological functions in DNA damage signaling or repair. The existence of FANCJ in a BRCA1-containing complex with other DNA repair factors involved in the recognition and repair of aberrant DNA structures suggests that the complex functions in the DNA damage response [49]. It is conceivable that a specialized helicase like FANCJ might have the ability to sense DNA damage in either strand of the duplex and facilitate DNA damage processing.
In addition to DNA damage in standard duplex DNA, non-B form DNA with alternate secondary structure may impede the replication fork, and be a source of genomic instability (Fig. (3)). Guanine-rich nucleic acids have the potential to form G-quadruplex or ‘G4 DNA’ stabilized by Hoogsteen hydrogen bonding between guanine residues, and G4 DNA influences gene expression and genomic stability (for review, see [70]). The number of distinct sites with potential to form G4 DNA in the human genome is estimated at more than 300,000 [71]. The unique metabolism of G-rich chromosomal regions that form quadruplexes may influence a number of biological processes including immunoglobulin gene rearrangements [72], promoter activation [73], and telomere maintenance [74, 75].
Fig. (3). Proposed role of FANCJ in resolving G4 quaduplex DNA during replication.

According to the model presented, the replication fork moves in from the left toward the poly(G) sequences (in red). Single-stranded poly(G) sequences used as a template for lagging-strand DNA synthesis are postulated to occasionally give rise to stable secondary structures such as G4 structure. FANCJ helicase is recruited to unwind the G4 quadstructure, permitting polymerization using the template strand (in blue). Failure to resolve G-quadruplex DNA in FA-J cells results in genomic instability (dashed line in red).
FANCJ was shown to efficiently unwind a variety of model G4 substrates in a reaction that required the presence of ATP and was dependent on intrinisic FANCJ ATP hydrolysis [76]. A 5’ ssDNA tail is required for FANCJ to load and unwind the adjacent G-tetraplex. The 5’ tail requirement distinguishes FANCJ from the human WRN or BLM syndrome helicases that require a 3’ ssDNA tail to unwind G4 substrates [77, 78]. Evidence that the FANCJ helicase activity on G-quadruplex DNA is biologically important is discussed below in the Replication section.
DNA REPAIR PROTEINS TEAM UP WITH FANCJ TO INSURE A PROPER DNA DAMAGE RESPONSE
Understanding the pathway(s) that FANCJ participates in to facilitate an appropriate DNA damage response will require a thorough analysis of its protein interactions and genetic functions. We will summarize our current knowledge of how FANCJ performs its cellular functions through its timely interactions with DNA repair factors.
Interaction of FANCJ with the Tumor Suppressor BRCA1 Is Required for Normal Double Strand Break Repair
Germ line mutations in BRCA1 lead to an increased lifetime risk of breast and/or ovarian cancer [79]. Moreover, the tumor suppressor gene BRCA1 is required for the maintenance of chromosomal stability and DNA damage resistance (for review see [80–82]). The nuclear phosphoprotein BRCA1 contains tandem C-terminal BRCT motifs, a conserved protein sequence found in a large number of DNA damage-response proteins [83]. The integrity of the BRCT motifs is required for the role of BRCA1 in DSB repair [82] and HR [84–87]. Tumor-predisposing missense and deletion mutations in the BRCA1 BRCT domain, all of which render BRCA1 defective in its DSB repair function, also disrupt the ability of BRCA1 to bind FANCJ. A role of FANCJ helicase in DSB repair was suggested by the observation that overexpression of a FANCJ allele (K52R) carrying a mutation in its ATP-binding pocket that inactivates its ATPase/helicase function [62] resulted in a marked decrease in the ability of cells to repair DSBs, and that this dominant negative phenotype depended on a specific interaction between FANCJ and BRCA1 [8]. These results suggested that the role of FANCJ in DSB repair operates in a manner dependent on its association with BRCA1. Structural evidence demonstrated that two intact BRCA1 BRCT repeats as well as phosphorylation of FANCJ are critical for BRCA1 binding to FANCJ [88–90]. The FANCJ-BRCA1 interaction is cell cycle regulated and required for the G2/M checkpoint control in response to DNA damage [90, 91]. The importance of FANCJ helicase activity in tumor suppression was supported by the identification of two naturally occurring FANCJ missense mutations genetically linked to cancer that impair the DNA unwinding activity of the enzyme [8, 62]. Thus, the tumor suppressor role of FANCJ likely involves both its helicase activity and interaction with BRCA1. The preferential binding of BRCA1 to forked DNA structures [92] may help FANCJ load on to a critical DNA repair intermediate such as D-loop structure that the enzyme can catalytically unwind.
BRCA1-FANCJ Protein Complexes Exist in Cells
FANCJ was initially identified through Far Western blotting using phosphorylated BRCT repeats of BRCA1 [8]. It was subsequently reported that phosphoserine 990 of FANCJ is critical for FANCJ binding to BRCT motif of BRCA1 [88–90]. Furthermore, phosphorylated serine at 990 of FANCJ also associates with the BRCT motifs of Topoisomerase IIβ binding protein 1 (TopBP1) and BRCA1-associated RING domain protein (BARD1) [90, 91]. So far, only a limited number of proteins are reported to associate with FANCJ, as listed in Table (3).
Table 3.
Proteins that interact with FANCJ
| FANCJ Interacting Proteins |
Physical Association | Functional/Genetic Interaction |
DNA Damage Response | References |
|---|---|---|---|---|
| BRCA1 | FANCJ binds to the BRCT domain of BRCA1 |
Physical interaction required for DSB repair |
DSB and HR repair | [8, 48, 88–91] |
| MLH1 - PMS2 | MLH1 binds to helicase domain of FANCJ (128–158 aa) |
Physical interaction required for ICL repair |
ICL repair | [47] |
| RPA | FANCJ binds to RPA70 subunit |
Stimulation of FANCJ helicase activity |
Co-localization after MMC, IR or HU |
[50] |
| TOPBP1 | FANCJ associates with BRCA1 |
ATR activation | DNA damage signal transduction |
[49, 90] |
| BARD1 | FANCJ associates with BRCA1 |
Required for ubiquitin- ligase activity of BRCA1 |
DSB repair | [48, 90] |
An important area of study has been the formation of DNA damage-induced protein complexes that mediate the DNA damage response. Since BRCA1 plays a key role in the constellation of protein complexes, a number of studies have focused on the assembly of DNA repair centers with BRCA1 as their focal point. Greenberg and colleagues reported that BRCA1 is required for the transport of FANCJ, BARD1, BRCA2, and Rad51 to sites of DNA damage where other proteins such as the MRE11-RAD50-NBS1 complex associate [49]. The assembly of a BRCA1/FANCJ/BARD1 complex enables the interaction of BRCA1/FANCJ with TopBP1, a factor that plays an important role in the execution of the S phase checkpoint [49]. Modulation of DNA damage checkpoint function by this complex may contribute to tumor suppression.
FANCJ protein complexes were identified with mismatch repair factors (discussed further below) and with BRCA1/BRCA2/BARD1, the latter of which shows an increased association with chromatin during S phase [48]. The FANCJ complex is distinct from that of BRCA1-BRCA2-containing complex (BRCC), as it does not contain either BRCC36 (a 36 kDa protein in BRCC complex) or BRCC45/BRE (a 45 kDa protein in BRCC complex)[48]. Understanding the assembly, activities, and functions of FANCJ protein complexes in DNA damage response pathways remains a formidable challenge and worthy endeavor.
RPA Interacts with FANCJ in a DNA Damage-Inducible Manner and Stimulates its Helicase Activity
FANCJ was observed to efficiently unwind a short duplex of 20 bp, but severely limited in unwinding a 47 bp substrate, suggesting that the enzyme is not very processive [50]. However, in the presence of the single-stranded DNA binding protein RPA, FANCJ unwinds the longer duplex substrate much more efficiently. Stimulation of FANCJ helicase activity by RPA is specific since heterologous single-stranded binding proteins do not increase FANCJ helicase activity to any significant extent. The mechanism for RPA stimulation of FANCJ helicase activity may be more complex than simply a role of RPA to coat the unwound ssDNA tracts left behind the advancing helicase. Consistent with this idea, FANCJ directly binds with high affinity to the RPA70 subunit of the heterotrimer. FANCJ and RPA in human nuclear extracts were shown to coimmunoprecipitate with each other and colocalized after DNA damage induced by ionizing radiation (IR) or MMC or replicational stress induced by hydroxyurea [50]. Since FANCJ is implicated in a downstream event of FANCD2 monoubiquitination, the possibility that a mutation in an upstream member (core complex protein FANCA) of the FA pathway might affect the interaction of FANCJ and RPA was investigated. FANCJ and RPA were co-immunoprecipitated with each other from extracts of FA-A cells, suggesting that the interaction is not dependent on upstream events of the FA pathway. Likewise, FANCJ and RPA were coimmunoprecipitated with each other from extracts of FA-D2 mutant cells, suggesting that the FANCJ-RPA interaction can occur independently of the classic FA pathway altogether.
The Interaction of MutLα̣ with FANCJ Is Essential for Cross-Link Repair
Although the interaction of FANCJ and BRCA1 was found to be important for DSB repair [8], Cantor and colleagues determined that FANCJ binding to BRCA1 was not required to correct the ICL-induced cell cycle progression defect in FA-J cells [47]. Thus, FANCJ operates independent of BRCA1 to correct FA-J cells, a result that supports the finding that FANCJ’s role in the FA pathway is independent of BRCA1 in chicken cells [93]. In an effort to identify other FANCJ protein interactions that might be important for the ICL response, Peng et al. used a two-step immunoaffinity strategy with human cells expressing a double-tagged FANCJ construct to identify novel protein partners [47]. FANCJ was found to exist in a protein complex with the mismatch repair proteins MLH1-PMS2 (MutLα). Like FANCJ, MutLα was found to function downstream of FANCD2 monoubiquitination. FANCJ was shown to directly interact with MLH1 independent of BRCA1, and this DNA-independent interaction was mapped to a defined region (residues 128–158) within the FANCJ helicase domain. Disruption of the native MLH1/FANCJ interaction by expression of an eGFP-tagged FANCJ128–158 fusion protein resulted in poorer cell survival after MMC treatment compared to expression of eGFP. Further mapping revealed that FANCJ residues Lys 141 and Lys142 were critically important for MLH1 binding, but not FANCJ helicase activity, and were essential to correct the sensitivity of FA-J cells to agents that induce ICL and consequently interfere with cell cycle progression, resulting in 4N DNA accumulation [47]. In sum, genetic studies demonstrated that FANCJ helicase activity and MLH1 binding, but not BRCA1 binding, is required to correct the cross-link response in FA-J cells. Although mismatch repair proteins have been implicated in the sensing and/or processing of ICLs, this study provided the first evidence for a direct link between FA and mismatch repair, leading to the prediction that FANCJ has a broader role in DNA damage signaling independent of BRCA1 [47].
It was speculated that the MutLα-associated mismatch repair complex serves to mobilize or regulate FANCJ DNA unwinding at the site of DNA damage, which facilitates the repair of the lesion. Alternatively, FANCJ may catalyze protein displacement and remove the mismatch repair complex from DNA, enabling the repair machinery access to the damaged DNA and also alleviate prolonged G2/M arrest.
How Might FANCJ Helicase Operate in ICL Through HR Repair?
The seminal demonstration that FANCJ interacts with BRCA1 and that cells harboring a FANCJ mutant defective in its interaction with BRCA1 fail to repair DSBs suggests that the helicase functions in DSB repair by promoting HR [8]. FA-J cells, like other FA mutant cells, are hypersensitive to agents that induce DNA ICLs. Of the thirteen gene products implicated in the FA pathway, only FANCJ, FANCD1 (BRCA2) and the binding partner of FAND1 (FANCN) operate downstream of FANCD2 monoubiquitination. It was proposed that FANCJ may operate with BRCA2 during an early step of HR repair to set up proper Rad51 protein filament formation on ssDNA as an early step in HR repair. Consistent with this possibility, FANCJ interacts with RPA, a protein that assembles on processed ssDNA ends at sites of DSBs prior to Rad51 filament formation. A recent review that focuses on the mechanisms of HR in DSB and ICL repair and DNA damage tolerance suggests a potential role for a 5’ to 3’ helicase in gap repair to resolve lagging strand blockage [94]. In this model, Li and Heyer proposed that recombinational repair of the gap on the lagging strand involves dislodging of the blocked strand by a 5’ to 3’ helicase, assembly of Rad51 on the ssDNA, and formation of the D-loop. However, contradictory to the model that FANCJ operates upstream of Rad51 is the observation that Rad51 focus formation after hydroxyurea (HU) exposure is not dependent on FANCJ [22]. It is conceivable that a predicted early step of FANCJ in HR repair is restricted to a specific pathway(s) such as that elicited by a replication fork encounter with an ICL. Other possible roles for FANCJ in the metabolism of ICLs exist such as the facilitation of translesion synthesis by promoting loading of a translesion polymerase through unwinding of DNA structures associated with stalled replication forks or dissociation of protein-DNA complexes [2].
A compelling model was proposed to explain the biological significance of the BRCA1-FANCJ interaction and to reconcile the observations that Rad51 foci are formed in FANCJ-deficient cells [22] whereas BRCA1 is required for subnuclear assembly of Rad51 foci after DNA damage [95, 96]. Cantor and Andreassen suggested that FANCJ functions to disrupt Rad51 protein-DNA filaments to complete HR, and BRCA1 regulates FANCJ by preventing the motor ATPase from prematurely removing Rad51 polymer from ssDNA involved in HR, which in turn would reduce chromosomal instability [97]. According to the authors, this model predicts that inhibition of FANCJ in BRCA1-deficient cells may lead to increased cell survival after DNA damage. This possibility should be readily testable, and if true, would suggest that targeting FANCJ as a mechanism to enhance DNA damaging anti-cancer therapy in BRCA1-deficient cells is potentially problematic.
A ROLE OF FANCJ IN DNA REPLICATION
There has been some discussion that in addition to a role in ICL repair, some FA proteins may function to stabilize the replication fork during conditions of replicational stress [98, 99]. A recent study using Xenopus laevis oocyte extracts shows that FANCL is required to stabilize the replication fork [100]. As a DNA helicase, FANCJ is a likely candidate to operate in this capacity, potentially extending its range of function beyond cross-link repair. We will explore this idea further, presenting recent discoveries in support of a function for FANCJ in DNA replication.
FANCJ Is Required for Timely Progression Through S Phase
A recent paper by Kumaraswamy and Shiekhattar experimentally addressed the hypothesis that FANCJ plays a role during S phase [48]. They observed that acute depletion of FANCJ by RNA interference resulted in an increased number of cells in the G1 phase of the cell cycle 6 hr after release from the DNA polymerase inhibitor aphidicolin. Furthermore, bromodeoxyuridine (BrdU) incorporation studies demonstrated reduced DNA synthesis in the FANCJ-depleted cells after release from aphidicolin, suggesting that FANCJ is necessary for timely progression through S phase. Consistent with this, FANCJ was found to show enhanced association with chromatin and display elevated DNA-dependent ATPase activity, activated by FANCJ dephosphorylation, as cells progressed through S phase [48].
To assess the role of the FANCJ ATPase or its BRCA1-interacting domains, cells expressing a catalytically inactive FANCJ ATPase mutant (K52R) or BRCA1-interactive defective mutant (S990A) were examined with the hypothesis that the FANCJ domain mutants might exert a dominant negative effect [48]. Cell lines expressing FANCJ mutants defective in either ATP hydrolysis or interaction with BRCA1 displayed a defect in S phase progression, increased IR sensitivity, and activation of the DNA damage checkpoint 53BP1 and the presence of DSBs as demonstrated by elevated γH2AX phosphorylation [48]. Altogether, these observations led the authors to propose that FANCJ may have a role in resolving difficult structural motifs encountered by the replication forks during DNA replication. Defects in FANCJ helicase activity would lead to stalled replication forks, which in turn impede S phase progression. The interrupted replication forks would ultimately give rise to DSBs and chromosomal instability.
FANCJ Resolves G-Quadruplex DNA Structures that Impede Replication and Pose a Source of Genomic Instability
The requirement for activation of FANCJ helicase activity to achieve timely progression through S phase of the cell cycle suggested a role of FANCJ in some aspect of replication [48]; however, its precise functions remain to be understood. Failure to replicate DNA in a timely and faithful manner can result in mutations, genomic instability, and cellular dysfunction manifested by cell transformation, senescence, or death. To deal with replication blocking lesions, proteins associated with the DNA replication fork or involved in distinct DNA damage response pathways facilitate fork progression or the accurate repair and restart of damaged or broken replication forks.
It was proposed that the peculiar type of genomic instability in mutants of C. elegans dog-1, a FANCJ homologue that also functions in ICL repair [101], was due to the accumulation of secondary DNA structures in G-rich tracts [56]. A recent study showed that sequences which match the G4 DNA signature are deleted in dog-1 mutants [102]. Another study showed that there is not only PolyG/C tract deletions in dog-1 mutants, but also large chromosomal rearrangements, including duplication, translocation and deficiencies [103]. However, demonstration that DOG-1 is a bonfide ATP-dependent helicase on G-quadruplexes or even duplex DNA substrates remains to be shown. DOG-1 may interact with accessory factors that modulates its activity. FANCJ G4 DNA unwinding was found to be differentially modulated by DNA repair factors that directly interact with FANCJ [76]. The single-stranded DNA binding protein RPA stimulated FANCJ G4 unwinding in a specific manner, whereas the mismatch repair complex MSH2/6 (MutSα), which interacts with G4 DNA with high affinity [72], inhibited FANCJ G4 helicase activity. The strong co-localization of FANCJ and RPA in response to nucleotide depletion by hydroxyurea [50] suggests that the two proteins collaborate in situations of replicational stress, a condition that arises when the replication fork is impeded by a G4 structure.
FANCJ-depleted cells were found to be sensitive to telomestatin (TMS) which selectively interacts with G-quadruplex DNA [104]. FANCJ-depleted cells treated with TMS displayed impaired proliferation, elevated apoptosis and DNA damage compared to siRNA control cells [76], suggesting that G4 DNA represents a physiological substrate of FANCJ. Although the FA pathway has been classically described in terms of ICL repair, the cellular defects associated with FANCJ mutation may extend beyond the reduced ability to repair ICLs and involve other types of DNA structural roadblocks to replication (Fig. (3)).
SUMMARY AND PERSPECTIVE
An in-depth understanding of the molecular pathogeneis of FA and breast cancer is only beginning to emerge. As a tumor suppressor and genome caretaker, FANCJ stands at the crossroads of these diseases. Understanding the role of FANCJ in DNA repair and the maintenance of genomic stability is a formidable challenge, but one of great importance. Future work will likely lead to a more detailed comprehension of the role of FANCJ in the classic FA pathway as well as its involvement in the response to replicational stress. Furthermore, dissection of the molecular functions and interactions of FANCJ in cellular DNA metabolism should lead to new insights for FANCJ and FANCJ-like helicases in chromosomal integrity and disease-related pathways.
ACKNOWLEDGMENTS
This work was supported by the Intramural Research program of the NIH, National Institute on Aging and the Fanconi Anemia Research Fund (RMB).
Abbreviations
- ATM
ataxia-telangiectasia mutated
- ATR
ATM and rad3 related
- BARD1
BRCA1-associated RING domain protein
- BLM
Bloom’s syndrome helicase
- BRCC
BRCA1-BRCA2-containing complex
- BRCC36
a 36 kDa protein in BRCC complex
- BRCC45/BRE
a 45 kDa protein in BRCC complex correspond to the brain- and reproductive organs-specific gene, BRCT: Breast cancer C-terminal BRE
- BrdU
bromodeoxyuridine
- DinG
damage-inducible G helicase
- DSB
double strand break
- dog-1
deletions of guanine-rich DNA
- FA
Fanconi anemia
- H2AX
Histone isoform 2AX
- HJ
Holliday Junction
- HR
homologous recombination
- HU
hydroxyurea
- ICLs
interstrand cross-links
- IR
ionizing radiation
- MMC
mitomycin C
- NLS
nuclear localization sequence
- nt
nucleotide
- NER
nucleotide excision repair
- RPA
Replication Protein A
- Rtel
Regulator of telomere length
- TFIIH
Transcription factor IIH
- TMS
Telomestatin
- TopBP1
Topoisomerase IIβ binding protein 1
- XPD
Xeroderma pigmentosum group D
REFERENCES
- 1.Levitus M, Joenje H, de Winter JP. Cell Oncol. 2006;28:3–29. doi: 10.1155/2006/974975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang W. Nat. Rev. Genet. 2007;8:735–748. doi: 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
- 3.Taniguchi T, D'Andrea AD. Blood. 2006;107:4223–4233. doi: 10.1182/blood-2005-10-4240. [DOI] [PubMed] [Google Scholar]
- 4.Andreassen PR, D'Andrea AD, Taniguchi T. Genes Dev. 2004;18:1958–1963. doi: 10.1101/gad.1196104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, III, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 6.Pichierri P, Rosselli F. EMBO J. 2004;23:1178–1187. doi: 10.1038/sj.emboj.7600113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang X, Kennedy RD, Ray K, Stuckert P, Ellenberger T, D'Andrea AD. Mol. Cell Biol. 2007;27:3098–3108. doi: 10.1128/MCB.02357-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, Wahrer DC, Sgroi DC, Lane WS, Haber DA, Livingston DM. Cell. 2001;105:149–160. doi: 10.1016/s0092-8674(01)00304-x. [DOI] [PubMed] [Google Scholar]
- 9.Gari K, Decaillet C, Stasiak AZ, Stasiak A, Constantinou A. Mol. Cell. 2008;29:141–148. doi: 10.1016/j.molcel.2007.11.032. [DOI] [PubMed] [Google Scholar]
- 10.Meetei AR, Medhurst AL, Ling C, Xue Y, Singh TR, Bier P, Steltenpool J, Stone S, Dokal I, Mathew CG, Hoatlin M, Joenje H, de Winter JP, Wang W. Nat. Genet. 2005;37:958–963. doi: 10.1038/ng1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meetei AR, de Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, van d V, Oostra AB, Yan Z, Ling C, Bishop CE, Hoatlin ME, Joenje H, Wang W. Nat. Genet. 2003;35:165–170. doi: 10.1038/ng1241. [DOI] [PubMed] [Google Scholar]
- 12.Meetei AR, Sechi S, Wallisch M, Yang D, Young MK, Joenje H, Hoatlin ME, Wang W. Mol. Cell Biol. 2003;23:3417–3426. doi: 10.1128/MCB.23.10.3417-3426.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Niedernhofer LJ, Lalai AS, Hoeijmakers JH. Cell. 2005;123:1191–1198. doi: 10.1016/j.cell.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 14.Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, Neveling K, Kelly P, Seal S, Freund M, Wurm M, Batish SD, Lach FP, Yetgin S, Neitzel H, Ariffin H, Tischkowitz M, Mathew CG, Auerbach AD, Rahman N. Nat. Genet. 2007;39:162–164. doi: 10.1038/ng1947. [DOI] [PubMed] [Google Scholar]
- 15.Ciccia A, Ling C, Coulthard R, Yan Z, Xue Y, Meetei AR, Laghmani eH, Joenje H, McDonald N, de Winter JP, Wang W, West SC. Mol. Cell. 2007;25:331–343. doi: 10.1016/j.molcel.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 16.Ling C, Ishiai M, Ali AM, Medhurst AL, Neveling K, Kalb R, Yan Z, Xue Y, Oostra AB, Auerbach AD, Hoatlin ME, Schindler D, Joenje H, de Winter JP, Takata M, Meetei AR, Wang W. EMBO J. 2007;26:2104–2114. doi: 10.1038/sj.emboj.7601666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, Ikeda H, Fox EA, D'Andrea AD. Science. 2002;297:606–609. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- 18.Xia B, Dorsman JC, Ameziane N, de VY, Rooimans MA, Sheng Q, Pals G, Errami A, Gluckman E, Llera J, Wang W, Livingston DM, Joenje H, de Winter JP. Nat. Genet. 2007;39:159–161. doi: 10.1038/ng1942. [DOI] [PubMed] [Google Scholar]
- 19.Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ, Livingston DM. Mol. Cell. 2006;22:719–729. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 20.Levitus M, Waisfisz Q, Godthelp BC, de Vries Y, Hussain S, Wiegant WW, Elghalbzouri-Maghrani E, Steltenpool J, Rooimans MA, Pals G, Arwert F, Mathew CG, Zdzienicka MZ, Hiom K, de Winter JP, Joenje H. Nat. Genet. 2005;37:934–935. doi: 10.1038/ng1625. [DOI] [PubMed] [Google Scholar]
- 21.Levran O, Attwooll C, Henry RT, Milton KL, Neveling K, Rio P, Batish SD, Kalb R, Velleuer E, Barral S, Ott J, Petrini J, Schindler D, Hanenberg H, Auerbach AD. Nat. Genet. 2005;37:931–933. doi: 10.1038/ng1624. [DOI] [PubMed] [Google Scholar]
- 22.Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, Andreassen PR, Cantor SB. Cancer Cell. 2005;8:255–265. doi: 10.1016/j.ccr.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 23.Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD. Mol. Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- 24.Dorsman JC, Levitus M, Rockx D, Rooimans MA, Oostra AB, Haitjema A, Bakker ST, Steltenpool J, Schuler D, Mohan S, Schindler D, Arwert F, Pals G, Mathew CG, Waisfisz Q, de Winter JP, Joenje H. Cell Oncol. 2007;29:211–218. doi: 10.1155/2007/151968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sims AE, Spiteri E, Sims RJ, III, Arita AG, Lach FP, Landers T, Wurm M, Freund M, Neveling K, Hanenberg H, Auerbach AD, Huang TT. Nat. Struct. Mol. Biol. 2007;14:564–567. doi: 10.1038/nsmb1252. [DOI] [PubMed] [Google Scholar]
- 26.Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, III, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D'Andrea AD, Elledge SJ. Cell. 2007;129:289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xue Y, Li Y, Guo R, Ling C, Wang W. Hum. Mol. Genet. 2008;17:1641–1652. doi: 10.1093/hmg/ddn054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanada K, Hickson ID. Cell Mol. Life Sci. 2007;64:2306–2322. doi: 10.1007/s00018-007-7121-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.D'Andrea AD, Grompe M. Nat. Rev. Cancer. 2003;3:23–34. doi: 10.1038/nrc970. [DOI] [PubMed] [Google Scholar]
- 30.Joenje H, Patel KJ. Nat. Rev. Genet. 2001;2:446–457. doi: 10.1038/35076590. [DOI] [PubMed] [Google Scholar]
- 31.Kennedy RD, D'Andrea AD. Genes Dev. 2005;19:2925–2940. doi: 10.1101/gad.1370505. [DOI] [PubMed] [Google Scholar]
- 32.Venkitaraman AR. Nat. Rev. Cancer. 2004;4:266–276. doi: 10.1038/nrc1321. [DOI] [PubMed] [Google Scholar]
- 33.Oyake T, Itoh K, Motohashi H, Hayashi N, Hoshino H, Nishizawa M, Yamamoto M, Igarashi K. Mol. Cell Biol. 1996;16:6083–6095. doi: 10.1128/mcb.16.11.6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, Chagtai T, Jayatilake H, Ahmed M, Spanova K, North B, McGuffog L, Evans DG, Eccles D, Easton DF, Stratton MR, Rahman N. Nat. Genet. 2006;38:1239–1241. doi: 10.1038/ng1902. [DOI] [PubMed] [Google Scholar]
- 35.Eelen G, Vanden BI, Verlinden L, Drijkoningen M, Smeets A, Neven P, Christiaens MR, Marchal K, Bouillon R, Verstuyf A. Oncogene. 2008;27:4233–4241. doi: 10.1038/onc.2008.51. [DOI] [PubMed] [Google Scholar]
- 36.Fan L, Fuss JO, Cheng QJ, Arvai AS, Hammel M, Roberts VA, Cooper PK, Tainer JA. Cell. 2008;133:789–800. doi: 10.1016/j.cell.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu H, Rudolf J, Johnson KA, McMahon SA, Oke M, Carter L, McRobbie AM, Brown SE, Naismith JH, White MF. Cell. 2008;133:801–812. doi: 10.1016/j.cell.2008.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wolski SC, Kuper J, Hanzelmann P, Truglio JJ, Croteau DL, Van HB, Kisker C. PLoS. Biol. 2008;6:e149. doi: 10.1371/journal.pbio.0060149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rudolf J, Makrantoni V, Ingledew WJ, Stark MJ, White MF. Mol. Cell. 2006;23:801–808. doi: 10.1016/j.molcel.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 40.Gupta R, Sharma S, Doherty KM, Sommers JA, Cantor SB, Brosh RM., Jr Nucleic Acids Res. 2006;34:6673–6683. doi: 10.1093/nar/gkl964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuo CF, McRee DE, Fisher CL, O'Handley SF, Cunningham RP, Tainer JA. Science. 1992;258:434–440. doi: 10.1126/science.1411536. [DOI] [PubMed] [Google Scholar]
- 42.Cunningham RP, Asahara H, Bank JF, Scholes CP, Salerno JC, Surerus K, Munck E, McCracken J, Peisach J, Emptage MH. Biochemistry. 1989;28:4450–4455. doi: 10.1021/bi00436a049. [DOI] [PubMed] [Google Scholar]
- 43.Guan Y, Manuel RC, Arvai AS, Parikh SS, Mol CD, Miller JH, Lloyd S, Tainer JA. Nat. Struct. Biol. 1998;5:1058–1064. doi: 10.1038/4168. [DOI] [PubMed] [Google Scholar]
- 44.Lukianova OA, David SS. Curr. Opin. Chem. Biol. 2005;9:145–151. doi: 10.1016/j.cbpa.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 45.Boon EM, Livingston AL, Chmiel NH, David SS, Barton JK. Proc. Natl. Acad. Sci. U. S. A. 2003;100:12543–12547. doi: 10.1073/pnas.2035257100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weiner BE, Huang H, Dattilo BM, Nilges MJ, Fanning E, Chazin WJ. J. Biol. Chem. 2007;282:33444–33451. doi: 10.1074/jbc.M705826200. [DOI] [PubMed] [Google Scholar]
- 47.Peng M, Litman R, Xie J, Sharma S, Brosh RM, Jr, Cantor SB. EMBO J. 2007;26:3238–3249. doi: 10.1038/sj.emboj.7601754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kumaraswamy E, Shiekhattar R. Mol. Cell Biol. 2007;27:6733–6741. doi: 10.1128/MCB.00961-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Greenberg RA, Sobhian B, Pathania S, Cantor SB, Nakatani Y, Livingston DM. Genes Dev. 2006;20:34–46. doi: 10.1101/gad.1381306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gupta R, Sharma S, Sommers JA, Kenny MK, Cantor SB, Brosh RM., Jr Blood. 2007;110:2390–2398. doi: 10.1182/blood-2006-11-057273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Voloshin ON, Camerini-Otero RD. J. Biol. Chem. 2007;282:18437–18447. doi: 10.1074/jbc.M700376200. [DOI] [PubMed] [Google Scholar]
- 52.Parish JL, Rosa J, Wang X, Lahti JM, Doxsey SJ, Androphy EJ. J. Cell Sci. 2006;119:4857–4865. doi: 10.1242/jcs.03262. [DOI] [PubMed] [Google Scholar]
- 53.Petronczki M, Chwalla B, Siomos MF, Yokobayashi S, Helmhart W, Deutschbauer AM, Davis RW, Watanabe Y, Nasmyth K. J. Cell Sci. 2004;117:3547–3559. doi: 10.1242/jcs.01231. [DOI] [PubMed] [Google Scholar]
- 54.Skibbens RV. Genetics. 2004;166:33–42. doi: 10.1534/genetics.166.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Inoue A, Li T, Roby SK, Valentine MB, Inoue M, Boyd K, Kidd VJ, Lahti JM. Cell Cycle. 2007;6:1646–1654. doi: 10.4161/cc.6.13.4411. [DOI] [PubMed] [Google Scholar]
- 56.Cheung I, Schertzer M, Rose A, Lansdorp PM. Nat. Genet. 2002;31:405–409. doi: 10.1038/ng928. [DOI] [PubMed] [Google Scholar]
- 57.Youds JL, O'Neil NJ, Rose AM. Genetics. 2006;173:697–708. doi: 10.1534/genetics.106.056879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ding H, Schertzer M, Wu X, Gertsenstein M, Selig S, Kammori M, Pourvali R, Poon S, Vulto I, Chavez E, Tam PP, Nagy A, Lansdorp PM. Cell. 2004;117:873–886. doi: 10.1016/j.cell.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 59.Friedberg EC, Bardwell AJ, Bardwell L, Feaver WJ, Kornberg RD, Svejstrup JQ, Tomkinson AE, Wang Z. Philos. Trans. R. Soc. Lond B Biol. Sci. 1995;347:63–68. doi: 10.1098/rstb.1995.0010. [DOI] [PubMed] [Google Scholar]
- 60.Hoeijmakers JH, Egly JM, Vermeulen W. Curr. Opin. Genet. Dev. 1996;6:26–33. doi: 10.1016/s0959-437x(96)90006-4. [DOI] [PubMed] [Google Scholar]
- 61.Sung P, Bailly V, Weber C, Thompson LH, Prakash L, Prakash S. Nature. 1993;365:852–855. doi: 10.1038/365852a0. [DOI] [PubMed] [Google Scholar]
- 62.Cantor S, Drapkin R, Zhang F, Lin Y, Han J, Pamidi S, Livingston DM. Proc. Natl. Acad. Sci. U. S. A. 2004;101:2357–2362. doi: 10.1073/pnas.0308717101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gupta R, Sharma S, Sommers JA, Jin Z, Cantor SB, Brosh RM., Jr J. Biol. Chem. 2005;280:25450–25460. doi: 10.1074/jbc.M501995200. [DOI] [PubMed] [Google Scholar]
- 64.Kaplan DL. J. Mol. Biol. 2000;301:285–299. doi: 10.1006/jmbi.2000.3965. [DOI] [PubMed] [Google Scholar]
- 65.Sharma S, Doherty KM, Brosh RM., Jr Biochem. J. 2006;398:319–337. doi: 10.1042/BJ20060450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kawaoka J, Jankowsky E, Pyle AM. Nat. Struct. Mol. Biol. 2004;11:526–530. doi: 10.1038/nsmb771. [DOI] [PubMed] [Google Scholar]
- 67.Eoff RL, Spurling TL, Raney KD. Biochemistry. 2005;44:666–674. doi: 10.1021/bi0484926. [DOI] [PubMed] [Google Scholar]
- 68.Tackett AJ, Morris PD, Dennis R, Goodwin TE, Raney KD. Biochemistry. 2001;40:543–548. doi: 10.1021/bi002122+. [DOI] [PubMed] [Google Scholar]
- 69.Jeong YJ, Levin MK, Patel SS. Proc. Natl. Acad. Sci. U. S. A. 2004;101:7264–7269. doi: 10.1073/pnas.0400372101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maizels N. Nat. Struct. Mol. Biol. 2006;13:1055–1059. doi: 10.1038/nsmb1171. [DOI] [PubMed] [Google Scholar]
- 71.Eddy J, Maizels N. Nucleic Acids Res. 2006;34:3887–3896. doi: 10.1093/nar/gkl529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Larson ED, Duquette ML, Cummings WJ, Streiff RJ, Maizels N. Curr. Biol. 2005;15:470–474. doi: 10.1016/j.cub.2004.12.077. [DOI] [PubMed] [Google Scholar]
- 73.Siddiqui-Jain A, Grand CL, Bearss DJ, Hurley LH. Proc. Natl. Acad. Sci. U. S. A. 2002;99:11593–11598. doi: 10.1073/pnas.182256799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Oganesian L, Moon IK, Bryan TM, Jarstfer MB. EMBO J. 2006;25:1148–1159. doi: 10.1038/sj.emboj.7601006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zahler AM, Williamson JR, Cech TR, Prescott DM. Nature. 1991;350:718–720. doi: 10.1038/350718a0. [DOI] [PubMed] [Google Scholar]
- 76.Wu Y, Shin-ya K, Brosh RM., Jr Mol. Cell Biol. 2008;28:4116–4128. doi: 10.1128/MCB.02210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mohaghegh P, Karow JK, Brosh JR, Jr, Bohr VA, Hickson ID. Nucleic Acids Res. 2001;29:2843–2849. doi: 10.1093/nar/29.13.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sun H, Karow JK, Hickson ID, Maizels N. J. Biol. Chem. 1998;273:27587–27592. doi: 10.1074/jbc.273.42.27587. [DOI] [PubMed] [Google Scholar]
- 79.Fackenthal JD, Olopade OI. Nat. Rev. Cancer. 2007;7:937–948. doi: 10.1038/nrc2054. [DOI] [PubMed] [Google Scholar]
- 80.Ting NS, Lee WH. DNA Repair (Amst) 2004;3:935–944. doi: 10.1016/j.dnarep.2004.03.026. [DOI] [PubMed] [Google Scholar]
- 81.Narod SA, Foulkes WD. Nat. Rev. Cancer. 2004;4:665–676. doi: 10.1038/nrc1431. [DOI] [PubMed] [Google Scholar]
- 82.Liu Y, West SC. Breast Cancer Res. 2002;4:9–13. doi: 10.1186/bcr417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bork P, Hofmann K, Bucher P, Neuwald AF, Altschul SF, Koonin EV. FASEB J. 1997;11:68–76. [PubMed] [Google Scholar]
- 84.Snouwaert JN, Gowen LC, Latour AM, Mohn AR, Xiao A, DiBiase L, Koller BH. Oncogene. 1999;18:7900–7907. doi: 10.1038/sj.onc.1203334. [DOI] [PubMed] [Google Scholar]
- 85.Scully R, Ganesan S, Vlasakova K, Chen J, Socolovsky M, Livingston DM. Mol. Cell. 1999;4:1093–1099. doi: 10.1016/s1097-2765(00)80238-5. [DOI] [PubMed] [Google Scholar]
- 86.Moynahan ME, Chiu JW, Koller BH, Jasin M. Mol. Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- 87.Jasin M. Oncogene. 2002;21:8981–8993. doi: 10.1038/sj.onc.1206176. [DOI] [PubMed] [Google Scholar]
- 88.Botuyan MV, Nomine Y, Yu X, Juranic N, Macura S, Chen J, Mer G. Structure. 2004;12:1137–1146. doi: 10.1016/j.str.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shiozaki EN, Gu L, Yan N, Shi Y. Mol. Cell. 2004;14:405–412. doi: 10.1016/s1097-2765(04)00238-2. [DOI] [PubMed] [Google Scholar]
- 90.Yu X, Chini CC, He M, Mer G, Chen J. Science. 2003;302:639–642. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- 91.Rodriguez M, Yu X, Chen J, Songyang Z. J. Biol. Chem. 2003;278:52914–52918. doi: 10.1074/jbc.C300407200. [DOI] [PubMed] [Google Scholar]
- 92.Paull TT, Cortez D, Bowers B, Elledge SJ, Gellert M. Proc. Natl. Acad. Sci. U. S. A. 2001;98:6086–6091. doi: 10.1073/pnas.111125998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bridge WL, Vandenberg CJ, Franklin RJ, Hiom K. Nat. Genet. 2005;37:953–957. doi: 10.1038/ng1627. [DOI] [PubMed] [Google Scholar]
- 94.Li X, Heyer WD. Cell Res. 2008;18:99–113. doi: 10.1038/cr.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bhattacharyya A, Ear US, Koller BH, Weichselbaum RR, Bishop DK. J. Biol. Chem. 2000;275:23899–23903. doi: 10.1074/jbc.C000276200. [DOI] [PubMed] [Google Scholar]
- 96.Godthelp BC, Wiegant WW, Waisfisz Q, Medhurst AL, Arwert F, Joenje H, Zdzienicka MZ. Mutat. Res. 2006;594:39–48. doi: 10.1016/j.mrfmmm.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 97.Cantor SB, Andreassen PR. Cell Cycle. 2006;5:164–167. doi: 10.4161/cc.5.2.2338. [DOI] [PubMed] [Google Scholar]
- 98.Thompson LH, Hinz JM, Yamada NA, Jones NJ. Environ. Mol. Mutagen. 2005;45:128–142. doi: 10.1002/em.20109. [DOI] [PubMed] [Google Scholar]
- 99.Howlett NG, Taniguchi T, Durkin SG, D'Andrea AD, Glover TW. Hum. Mol. Genet. 2005;14:693–701. doi: 10.1093/hmg/ddi065. [DOI] [PubMed] [Google Scholar]
- 100.Wang LC, Stone S, Hoatlin ME, Gautier J. DNA Repair (Amst) 2008 doi: 10.1016/j.dnarep.2008.08.005. PMID: 18786657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Youds JL, Barber LJ, Ward JD, Collis SJ, O'Neil NJ, Boulton SJ, Rose AM. Mol. Cell Biol. 2008;28:1470–1479. doi: 10.1128/MCB.01641-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kruisselbrink E, Guryev V, Brouwer K, Pontier DB, Cuppen E, Tijsterman M. Curr. Biol. 2008;18:900–905. doi: 10.1016/j.cub.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 103.Zhao Y, Tarailo-Graovac M, O'Neil NJ, Rose AM. DNA Repair (Amst) 2008 doi: 10.1016/j.dnarep.2008.07.011. PMID: 18708164. [DOI] [PubMed] [Google Scholar]
- 104.Shin-ya K. Nippon Rinsho. 2004;62:1277–1282. [PubMed] [Google Scholar]
- 105.Holloway L. Nucleic Acids Res. 2000;28:3056–3064. doi: 10.1093/nar/28.16.3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gerring SL, Spencer F, Hieter P. EMBO J. 1990;9:4347–4358. doi: 10.1002/j.1460-2075.1990.tb07884.x. [DOI] [PMC free article] [PubMed] [Google Scholar]