

Abstract
Immobilization of a target molecule to a solid support is an indispensable step in phage display library sorting. Here we describe an immobilization method that addresses shortcomings of existing strategies. Our method is based on the use of a polyhistidine-tagged (His-tagged) target molecule and BTtris-NTA, a high-affinity capture reagent for His-tags that also contains a biotin moiety. BTtris-NTA provides a stable and reversible linkage between a His-tag and a streptavidin-coated solid support. Because His-tags are the de facto standard for recombinant protein purification, this method dramatically simplifies target preparation for phage display library sorting. Here, we demonstrate the utility of this method by selecting high-affinity binding proteins based on the fibronectin type III (FN3) scaffold to two His-tagged protein targets, yeast small ubiquitin-like modifier and maltose-binding protein. Notably, a significant number of FN3 clones binding either targets selected using the new immobilization method exhibited only very weak binding when the same target was immobilized by coating on a polystyrene surface. This suggests that the His-tag-mediated immobilization exposes epitopes that are masked by commonly used passive adsorption methods. Together, these results establish a method with the potential to streamline and enhance many binding-protein engineering experiments.
Keywords: directed evolution, high-throughput selection, monobodies, synthetic binding proteins
Phage display is a widely used method for the selection of protein variants from a large library based, most commonly, on their ability to bind to a target molecule of interest (Smith, 1985; Sidhu and Koide, 2007). In order to separate phages that bind to the target from those that do not, the target molecule needs to be immobilized on a solid support (Sidhu et al., 2000). When one wishes to select ‘binders’ to the native form of a target protein, it is important to immobilize the target in such a way that it is not denatured and most of its surfaces remain accessible. To maximize the recovery of phages that bind to the immobilized target, but not to other materials present in the apparatus, it is advantageous for the linkage between the target and the solid support to be conditionally reversible, so that phages specifically bound to the target can be selectively released from the solid support (Fellouse et al., 2007).
A common immobilization method is passive, non-specific adsorption to a modified polystyrene-based surface, usually in the format of the wells of a microtiter plate. Despite its widespread use and proven success in generating binding proteins, it is not clear what form the immobilized protein assumes on the polystyrene surface, and it is possible that some proteins are denatured upon immobilization and/or immobilized in a certain preferred orientation, which render a subset of epitopes inaccessible (Butler et al., 1992). Moreover, phages bound to the immobilized target on polystyrene surfaces are usually recovered by disrupting the molecular interactions with harsh solutions such as acid or denaturant (Sidhu et al., 2000). This type of elution is non-specific, and recovered phages include those that bind to the solid support. Thus, non-specific elution may lead to the enrichment of ‘background’ binders that do not specifically bind to the target, thereby reducing the overall efficiency of identifying true target-binding clones.
One way to address the limitations imposed by passive adsorption is to attach a biotin moiety to the target protein in a manner that does not affect the structural integrity of the target. Such modified targets can then be immobilized on a surface coated with a biotin-binding protein such as streptavidin or avidin. This method exploits the exceptionally high affinity and rapid kinetics of the biotin–(strept)avidin interaction (Stayton et al., 1999). Once formed, the interaction is essentially irreversible. This tight interaction allows for phage display selection at a very low target concentration, which is useful for the selection of high-affinity binders.
Biotin is most commonly attached to a protein either through chemical modification, commonly at amine or sulfhydryl groups, or through the construction of a fusion protein with an enzymatic biotinylation tag. Chemical biotinylation reagents include those that can later be cleaved with a reducing reagent, enabling specific release of an immobilized target (Griffiths et al., 1994; Fellouse et al., 2007). Utilizing such a cleavable biotinylation reagent, we have previously established a high-throughput pipeline for the selection of phage-displayed antibodies (Fellouse et al., 2007; Koide et al., 2007). However, in chemical biotinylation, one has to optimize reaction conditions, so that the target protein is biotinylated at a small number of sites, or ideally at a single site. Biotinylation of a protein at many sites may drastically change the target's surface properties, alter its conformation or result in the selection of binding proteins that recognize only an over-biotinylated form of the target. Further, biotinylation often reduces the solubility of the target protein due to the hydrophobic nature of the biotin moiety. Also, the cleavable biotinylation reagents that are currently commercially available utilize reductive cleavage of a disulfide bond. This necessitates storage and handling of a protein biotinylated in this manner under oxidizing conditions. Accordingly, these reagents are incompatible with a target protein that contains free cysteine residues that need to be kept reduced, and such reactive cysteines that are non-essential must be eliminated via chemical modification or mutagenesis prior to biotinylation. Therefore, although we found our procedures utilizing a cleavable biotinylation reagent to be highly effective in producing high-affinity binders, they require multiple steps and careful optimization of target modification as summarized in Fig. 1A.
Fig. 1.

(A) A flowchart outlining steps required for target preparation using a chemical biotinylation reagent (Fellouse et al., 2007; Koide et al., 2007). Note that the method presented in this work (B) completely eliminates these steps. (C) The chemical structure of BTtris-NTA (Reichel et al., 2007). The X's denote Ni ligands such as His and imidazole.
Enzymatic biotinylation provides an alternative to these chemical biotinylation methods. Biotin ligase recognizes a specific sequence (‘biotin acceptor domain’) and attaches biotin to a Lys residue in the sequence (Schatz, 1993). This process can be exploited either in vivo or in vitro, but producing a new fusion protein adds an extra step in target protein production. Also the biotin moiety attached by biotin ligase is not cleavable, making it difficult to specifically release the immobilized target.
It is clear from these considerations that although biotinylation is a highly effective immobilization method that can retain the structural integrity of a target protein, it has significant limitations. Therefore, a new, reversible immobilization method that does not involve covalent modification but retains a high-affinity linkage would significantly improve the efficiency of phage-display library selection and potentially expand the range of target proteins that can be used in such projects.
Polyhistidine tags (His-tags) have become the standard fusion tag for protein purification by immobilized metal ion affinity chromatography. For example, virtually all proteins in large-scale structural genomics centers are produced, at least initially, as a His-tag fusion (Graslund et al., 2008). The tag's small size and efficacy in a wide range of solution conditions are ideally suited for many applications. Thus, it would be highly beneficial if a method were available for directed high-affinity immobilization of His-tag proteins. The standard His-tag ligands, such as the nitrilotriacetic acid (NTA) moiety loaded with Ni(II) ions, bind with low affinity (∼10 µM) (Dorn et al., 1998). Stable immobilization of His-tagged proteins on NTA surfaces requires a high density of the NTA molecule (Lata et al., 2005) to allow simultaneous interactions of the His-tag with several chelated metal ions. These high densities of immobilized transition metal ions, however, lead to increased non-specific interactions with the excess immobilized Ni(II) ions (Lata et al., 2005). Although there are anti-His-tag antibodies, which circumvent this problem, their high costs and relatively low affinity limit their effectiveness.
Recently, Lata et al. (2005) developed a series of His-tag ligands that have multiple NTA moieties. These ligands can form stoichiometrically well-defined multivalent interactions with a His-tag, and thus offer high affinity binding in solution with a Kd in the low nanomolar range. Among these, tris-NTA contains three NTA moieties that can simultaneously bind to a maximum of six histidine side chains with a Kd of ∼2 nM for a 6-histidine tag and 0.14 nM for a 10-histidine tag (Lata et al., 2005). Further, Reichel et al. (2007) developed a biotinylated form of tris-NTA called BTtris-NTA with the initial motivation of labeling a His-tag protein with biotin for downstream detection. The high-affinity linkage between a His-tagged protein and BTtris-NTA in solution allows for the capture of the complex with a solid support presenting a biotin-binding protein, such as streptavidin-coated magnetic beads. By this strategy, a His-tagged target can be specifically captured, while avoiding presentation of excess of immobilized Ni(II) ions susceptible to non-specific interaction as in other His-tag capture methods. Moreover, the association of the biotin–streptavidin complex is much faster than that of the His-tag with Ni-NTA, making capture of the BTtris-NTA labeled target more rapid and efficient than immobilization directly through the His-tag.
The ability of BTtris-NTA to rapidly capture a His-tag protein on solid support with high affinity and selectivity is an attractive feature for its potential use in phage display selection (Fig. 1B). Furthermore, the linkage between tris-NTA and a His-tag can be disrupted under physiological conditions with imidazole or EDTA, providing a convenient means for mild and selective release of the captured protein. Because low target concentration is necessary for the selection of high-affinity binders and selection processes involve vigorous washing, it was not clear whether the non-covalent BTtris-NTA label could be used in such selection experiments. Moreover, selection with BTtris-NTA might enrich sequences that are rich in histidine that bind to the tris-NTA moiety but not to the actual target. Therefore, in this work, we tested whether BTtris-NTA could be used for the selection of binding proteins from a phage display library.
Materials and methods
BTTris-NTA synthesis
BTTris-NTA was synthesized as described previously (Lata et al., 2005; Reichel et al., 2007).
Protein preparation
MBP and yeast small ubiquitin-like modifier (ySUMO) were expressed as His10- and His6-tagged proteins, respectively, using pHFT1 and pHFT2 vectors (Huang et al., 2006; Koide et al., 2007). Selected monobodies (FN3-based binding proteins) were also expressed as His10-tagged proteins. These proteins were purified using Ni-affinity chromatography (Ni-chelating Sepharose, Amersham). For surface plasmon resonance (SPR) measurements, the His-tag of the target proteins was cleaved using TEV protease and removed using Ni-affinity chromatography.
Phage display library sorting
General procedures for phage preparation and analysis of individual clones with phage ELISA have been described previously (Sidhu et al., 2000; Koide and Koide, 2007). A phage display library of monobodies was similar to that described previously (Gilbreth et al., 2008), but two surface loops (the BC and FG loops) were diversified using the following amino acid composition: Tyr, 30%; Ser, 15%; Gly, 10%; Phe, 5%; Trp, 5% and all other except for Cys, 2.5% each.
Library sorting was performed at room temperature essentially following published procedures (Fellouse et al., 2007; Koide et al., 2007; Gilbreth et al., 2008) except that His-tagged targets complexed with BTtris-NTA were used. His-tagged proteins were mixed with BTtris-NTA in TBS (50 mM Tris–HCl buffer, pH 7.5, 150 mM NaCl) and incubate at room temperature for 30 min before use.
In the first round, a target-BTtris-NTA complex was mixed with a sufficient amount of streptavidin-conjugated magnetic beads (Streptavidin MagneSphere Pramagnetic Particles; Promega) in TBS. To this target solution, 1012–13 phagemids suspended in 1 ml TBS plus 0.5% BSA were added, and the solution was mixed and incubated for 15 min at room temperature. After washing the beads twice with TBS containing 0.05% Tween20 (TBST), the beads suspension containing bound phagemids was added to fresh Escherichia coli XL1-Blue culture. Phagemids were amplified in the presence of 0.1 mM IPTG. In the second and later rounds, library sorting was performed using a Kingfisher instrument (Thermo Scientific) (Fellouse et al., 2007; Koide et al., 2007). Phagemids amplified from the previous round were incubated with a target-BTtris-NTA complex in TBS containing 0.5% BSA, and then captured by streptavidin-conjugated magnetic beads. After a total of five washing steps with 100 µl TBST with BSA (20 s per step), phagemids bound to the target protein were eluted by incubating the beads with 400 mM imidazole in TBS or 10 mM EDTA in TBS for 10 min. Recovered phages were then amplified.
SPR measurements
A monobody containing a His10 tag was immobilized on a NTA-chip (BIAcore) in a BIAcore 2000 instrument, and binding of targets (10–100 nM) was monitored. Sensorgrams were analyzed with the BIAEvaluation program (BIAcore).
Results and discussion
BTTris-NTA effectively immobilizes His-tagged proteins
We first performed a set of mock selection experiments to examine if the interaction of BTtris-NTA and a histidine-tagged protein is strong enough to survive the vigorous washing steps used in our procedures. We used His10-tagged maltose-binding protein (MBP) as a target and phages displaying an MBP-binding protein, monobody MBP74 (Koide et al., 2007). Monobodies are synthetic binding proteins based on the fibronectin type III (FN3) scaffold (Koide et al., 1998) (Fig. 2A). Four types of mock selection were performed, with and without BTtris-NTA and with and without His-tag-MBP. The MBP-binding phages and control phages were, respectively, incubated with these targets, captured with streptavidin-coated magnetic beads, washed vigorously and released with imidazole. Phages displaying the MBP-binding monobody were recovered efficiently only when both BTtris-NTA and His-tag MBP were included (Fig. 3). These results demonstrate that the interaction between BTtris-NTA and the His-tag-MBP was maintained throughout the selection process and thus the combination of BTtris-NTA and a His-tagged protein may be used as an alternative to a covalently biotinylated protein.
Fig. 2.

(A) A schematic drawing of the monobody scaffold. β-Strands A–G and the three loops that are diversified in the library are indicated. (B) The amino acid sequences and affinity of monobodies obtained from library selection. The sequences for the three loops are shown, with the numbering of Main et al. (1992). Kd values determined using SPR are shown. (C and D) SPR sensorgrams for the interactions of MBP and monobody M4 (C) and ySUMO and monobody Y12. The His-tagged monodies were immobilized on an NTA-chip and the association and dissociation of MBP and ySUMO were monitored.
Fig. 3.
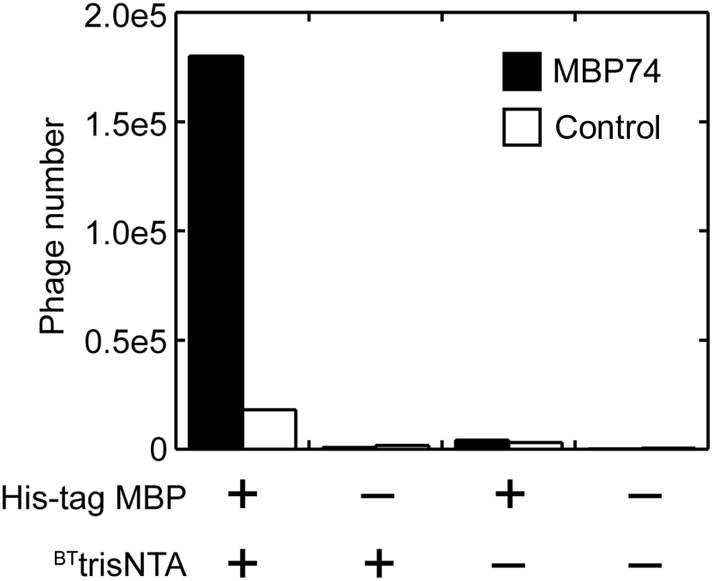
Recovery of phages displaying an MBP-binding monobody (filled bar) from selection with His-tagged MBP immobilized to magnetic beads through BTtris-NTA (the leftmost column). As a negative control, phages displaying a non-functional, ‘shaved’ FNfn10 in which residues in the BC and FG loops have been replaced with Ser (Koide et al., 2007) were used (open bar). The numbers of recovered phages are shown. The three right columns represent control experiments in which BTtris-NTA and/or the target were omitted.
BTTris-NTA can be used in phage-display library sorting
We then proceeded to selection of monobodies from a naïve library. His10-tagged MBP and His6-tagged ySUMO were used as targets. We have previously selected monobodies to these targets using chemically biotinylated proteins (Koide et al., 2007). We have adapted the selection method established previously (Koide et al., 2007; Gilbreth et al., 2008) with small modifications. In place of chemically biotinylated proteins, the His-tagged proteins were mixed with the stoichiometric amount of BTtris-NTA, and instead of recovering the target-phage complex by cleaving the biotinylation reagent with dithiothreitol (DTT), we eluted the target-phage complex with 400 mM imidazole. The target concentration was 200 nM for the first round and 100 nM for the second through fourth round. In the fifth round, we performed selection using 100, 20 nM and no target, and determined the number of recovered phages. The ratio of the phage number recovered in the presence of target over that in the absence of target, termed ‘enrichment ratio’, is a good indicator of whether the selection succeeded or not. If the enrichment is 10 or higher, usually a majority of the clones from the enriched pool bind to the target (Fellouse et al., 2007; Koide et al., 2007). For the selection with MBP, we obtained an enrichment ratio of 12 and 11 with 100 and 20 nM MBP, respectively. For the selection with ySUMO, the enrichment was 7.7 and 1.4 with 100 and 20 nM ySUMO, respectively.
Next, we randomly selected 12 clones each from the enriched pool sorted with 20 nM MBP and that with 100 nM ySUMO. Ten of 12 clones from the MBP-binding pool and 9 of 12 clones of the ySUMO-binding pool bound to their respective target as tested using phage ELISA. This was consistent with the good enrichment ratios observed for these sorting experiments. Sequence determination revealed one dominant clone and a second, additional clone for MBP and five clones for ySUMO (Fig. 2B). Importantly, the recovered phage clones bound to their cognate targets but not to BTtris-NTA and no clones with more than two His residues in the diversified loops were found, although the library design can encode oligo His sequences.
These MBP- and ySUMO-binding monobodies were then prepared as free proteins and their binding affinity was measured using SPR. The dissociation constants for these monobodies are in the low and mid nanomolar ranges (Fig. 2), clearly demonstrating the ability to select high-affinity binding proteins using BTtris-NTA and a His-tag protein.
His10-tag is more effective than His6-tag
In the above experiments, selection using 20 nM ySUMO did not yield significant enrichment of binding clones. In contrast, MBP binders were selected with 20 nM target. The failure to enrich ySUMO binders was not due to a lack of high-affinity clones in the naïve library, because we selected ySUMO binders with a Kd as low as 28 nM from sorting with 100 nM ySUMO (Fig. 2B). Such strong binders should have been selected with 20 nM target. A major difference between the MBP and ySUMO targets is that MBP had a His10-tag whereas ySUMO had a His6-tag. We suspected that the linkage of BTtris-NTA and the His6-tag was not stably maintained at the concentration of 20 nM, leading to the release of BTtris-NTA from the target-binder complexes during sorting. Although the affinity of BTtris-NTA and His6 in Kd was determined to be 2 nM (Lata et al., 2005), the complex may dissociate during vigorous washing steps in selection.
In order to investigate if the His6- and His10-tags give different results in library sorting, we performed selection with His6-ySUMO and His10-ySUMO. The Kd of BTtris-NTA and His10 is 0.14 nM (Lata et al., 2005), which is ∼15 times lower than that of BTtris-NTA and His6. We also included covalently biotinylated ySUMO as a control whose biotin does not dissociate during binding and washing steps. Selection of ySUMO-binders was performed using target concentration of 200 nM for the first round and 100 nM for the second and third rounds. In the fourth round, we performed selection using 100, 20 nM and no target, and determined the enrichment ratios. In this set of selections, we used 10 mM EDTA in TBS as an elution reagent because this method reduces the recovery of non-specific binders compared with imidazole elution. For chemically biotinylated ySUMO, we used 100 mM DTT in 20 mM Tris–Cl (pH8) that cleaves a disulfide linkage in the biotinylation reagent.
The His10- and His6-tags showed a significant difference in the effectiveness in library sorting. The outcome of the three sets of sorting was evaluated by determining the enrichment ratio. The pool sorted with chemically biotinylated-ySUMO gave enrichment ratios of 340 and 230 with 100 and 20 nM target concentrations, respectively. The enrichment ratios were 130 and 100 with 100 and 20 nM His10-ySUMO. In contrast, His6-ySUMO gave enrichment ratios of only 6 and 1. These differences in enrichment ratio indicate that, although both His6- and His10-tagged proteins can be used as targets in conjunction with BTtris-NTA, His10 is more efficient in immobilizing a target and accordingly in enriching binders over the course of a multiple-round selection.
The above data made it clear that over the course of a multiple-round selection, the use of a BTtrisNTA–His6-ySUMO complex resulted in reduced enrichment. It remained quantitatively unclear, however, how drastically the use of the BTtrisNTA–His6-ySUMO complex affected the output of a round of selection compared with the other immobilization methods. In an experiment complementary to the above library sorting, we compared the enrichment ratios obtained for each immobilization method when the input phage population and number were identical across all three methods. For this ‘mock fourth round’, we used the amplified phage after three rounds of selection with the chemically biotinylated ySUMO. Just as in the original selection, chemically biotinylated ySUMO gave enrichment of 340 and 230 with 100 and 20 nM target concentrations. His10-ySUMO gave identical enrichment ratios. In contrast, His6-ySUMO gave enrichment ratios of only 35 and 25, respectively. These results further confirmed that the low stability of the BTtrisNTA–His6-ySUMO complex resulted in a significantly lower efficiency in the recovery of target-binding clones, which is likely to have contributed to the very low level of binder enrichment when His6-ySUMO was used throughout multiple rounds of library sorting.
Because many potential targets for phage display selection contain free cysteine residues, we also tested if BTtris-NTA can be used in library sorting in the presence of the common reducing agent DTT. Because DTT has metal chelating ability, it could potentially act as a competitor for a His-tag protein. One millimolar DTT did not significantly reduce the binding of MBP- and ySUMO-binding phages to their respective targets (Fig. 4), suggesting that our method can also be used to select binders to a His-tagged target containing a reactive Cys residue that needs to be kept reduced in the presence of a reducing reagent such as DTT.
Fig. 4.

Effect of DTT on the immobilization of His-tagged targets tested via binding of monobody-displaying phages. His-tagged MBP (for the M4 monobody) and ySUMO (for the Y12 monobody) were immobilized to neutravidin-coated wells of a microtiter plate and binding of MBP- and ySUMO-binding monobodies displayed on phages were tested in the presence and absence of 1 mM DTT. Bound phages were detected with an anti-phage antibody. The identities of the two monobody clones are shown in Fig. 2.
His-tag-mediated immobilization exposes epitopes masked in passive adsorption
To test if the His-tag-mediated immobilization and standard passive adsorption result in a qualitative difference in the state and accessibility of immobilized targets, we first tested binding of the two MBP-binding FN3 monobodies (Fig. 2B) to the antigen immobilized using different methods: (i) His-tagged MBP and BTtrisNTA, (ii) passive adsorption of His-tagged MBP and (iii) chemical biotinylation of His-tag cleaved MBP. Both monobodies showed strong binding to the antigen immobilized using His-tag and BTtrisNTA and to a lower degree to the chemically biotinylated antigen (Fig. 5A). In contrast, neither showed significant binding to the same antigen directly coated to the polystyrene surface. This lack of binding was not due to low levels of immobilization, because similar amounts of the antigen were detected by an anti-FLAG antibody (Fig. 5B). Also, the strong binding of the monobodies to the His-tag cleaved MBP eliminates the possibility that the monobody–MBP interaction is somehow mediated by the His-tag. Together, these results indicate that passive adsorption immobilizes MBP in a manner that renders the epitope of these monobodies inaccessible.
Fig. 5.

Comparison of binding of FN3 monobody clones to MBP and ySUMO immobilized via His-tag and BTtrisNTA and via direct coating. (A) Phage ELISA readout for binding of the MBP-1 and -2 monobodies (Fig. 2B) to His10-MBP directly coated to polystyrene surface (gray bars), to His10-MBP immobilized via BTtrisNTA (white bars) and to MBP immobilized via chemical biotinylation (striped bars). The black bars show binding data to the surface containing BTtrisNTA but not His10-MBP. (B) ELISA readout with anti-FLAG antibody showing the amounts of immobilized MBP. Symbols are the same as in (A). (C) Phage ELISA readout for 12 independent clones of ySUMO-binding FN3 monobodies binding to His10-ySUMO immobilized via direct coating (gray bars) and via BTtrisNTA (white bars). The black bars are the negative control as in (A).
Next, in order to see if the weak binding to passively immobilized antigen is a specific feature for MBP, we tested FN3 monobody clones that had been selected from the above ySUMO-sorting experiments (Fig. 5) using both His-tag-mediated and direct immobilization methods. Similar to the results with MBP, while all clones bound to His10-ySUMO immobilized through BTtrisNTA equally well, a subset exhibited very weak binding to His10-ySUMO directly coated on polystyrene surface (e.g. clone 3 in Fig. 5C). These results suggest that ySUMO is immobilized on polystyrene surface in a non-random fashion, which sequesters a subset of epitopes or that immobilization via adsorption somehow alters the ySUMO structure. Because BTtrisNTA immobilizes the His-tag portion but not ySUMO itself, the entire ySUMO surfaces should be accessible by binding proteins, which allows for the selection of broader types of binders.
In conclusion, our results have demonstrated that the combination of BTtris-NTA and a His-tagged target can significantly simplify the target preparation process for phage-display library selection. The method also presents an antigen in a manner that minimizes sequestration of epitopes. We have established a generic protocol for rapid and reversible biotinylation of target proteins, which does not require optimization for each target and enables mild and selective phage elution. Because the new method can be used in the presence of DTT, it also expands the range of targets to which phage display selection with reversible target immobilization can be applied. In our experience in performing high-throughput sorting of phage display libraries using chemical biotinylation, target preparation (Fig. 1A) is a major bottleneck. As most recombinant proteins are made with a His-tag, and are thus already properly formatted, we expect that the new sorting method using BTtris-NTA presented here will streamline library sorting. These results should be broadly applicable to all molecular display techniques, including phage display, mRNA/ribosome display (Lipovsek and Pluckthun, 2004) and yeast surface display (Boder and Wittrup, 2000).
Funding
This work was supported by the National Institutes of Health (R01-GM72688, U54 GM74946 and R21-CA132700) and the University of Chicago Cancer Research Center.
Acknowledgements
We thank Dr Anthony Kossiakoff for access to a BIAcore 2000 instrument and the University of Chicago Cancer Research Center DNA Sequencing Facility for DNA sequencing service.
Footnotes
Edited by Arne Skerra
References
- Boder E.T., Wittrup K.D. Methods Enzymol. 2000;328:430–444. doi: 10.1016/s0076-6879(00)28410-3. [DOI] [PubMed] [Google Scholar]
- Butler J.E., Ni L., Nessler R., Joshi K.S., Suter M., Rosenberg B., Chang J., Brown W.R., Cantarero L.A. J. Immunol. Methods. 1992;150:77–90. doi: 10.1016/0022-1759(92)90066-3. [DOI] [PubMed] [Google Scholar]
- Dorn I.T., Neumaier K.R., Tampe R. J. Am. Chem. Soc. 1998;120:2753–2763. [Google Scholar]
- Fellouse F.A., et al. J. Mol. Biol. 2007;373:924–940. doi: 10.1016/j.jmb.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Gilbreth R.N., Esaki K., Koide A., Sidhu S.S., Koide S. J. Mol. Biol. 2008;381:407–418. doi: 10.1016/j.jmb.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graslund S., et al. Nat. Methods. 2008;5:135–146. doi: 10.1038/nmeth.f.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths A.D., et al. EMBO J. 1994;13:3245–3260. doi: 10.1002/j.1460-2075.1994.tb06626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Koide A., Nettle K.W., Greene G.L., Koide S. Protein Expr. Purif. 2006;47:348–354. doi: 10.1016/j.pep.2005.10.021. [DOI] [PubMed] [Google Scholar]
- Koide A., Koide S. Methods Mol. Biol. 2007;352:95–109. doi: 10.1385/1-59745-187-8:95. [DOI] [PubMed] [Google Scholar]
- Koide A., Bailey C.W., Huang X., Koide S. J. Mol. Biol. 1998;284:1141–1151. doi: 10.1006/jmbi.1998.2238. [DOI] [PubMed] [Google Scholar]
- Koide A., Gilbreth R.N., Esaki K., Tereshko V., Koide S. Proc. Natl Acad. Sci. USA. 2007;104:6632–6637. doi: 10.1073/pnas.0700149104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lata S., Reichel A., Brock R., Tampe R., Piehler J. J. Am. Chem. Soc. 2005;127:10205–10215. doi: 10.1021/ja050690c. [DOI] [PubMed] [Google Scholar]
- Lipovsek D., Pluckthun A. J. Immunol. Methods. 2004;290:51–67. doi: 10.1016/j.jim.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Main A.L., Harvey T.S., Baron M., Boyd J., Campbell I.D. Cell. 1992;71:671–678. doi: 10.1016/0092-8674(92)90600-h. [DOI] [PubMed] [Google Scholar]
- Reichel A., Schaible D., Al Furoukh N., Cohen M., Schreiber G., Piehler J. Anal. Chem. 2007;79:8590–8600. doi: 10.1021/ac0714922. [DOI] [PubMed] [Google Scholar]
- Schatz P.J. Biotechnology (NY) 1993;11:1138–1143. doi: 10.1038/nbt1093-1138. [DOI] [PubMed] [Google Scholar]
- Sidhu S.S., Koide S. Curr. Opin. Struct. Biol. 2007;17:481–487. doi: 10.1016/j.sbi.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Sidhu S.S., Lowman H.B., Cunningham B.C., Wells J.A. Methods Enzymol. 2000;328:333–363. doi: 10.1016/s0076-6879(00)28406-1. [DOI] [PubMed] [Google Scholar]
- Smith G.P. Science. 1985;228:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- Stayton P.S., et al. Biomol. Eng. 1999;16:39–44. doi: 10.1016/s1050-3862(99)00042-x. [DOI] [PubMed] [Google Scholar]