

Abstract
A transition as a function of increasing temperature from harmonic to anharmonic dynamics has been observed in globular proteins by using spectroscopic, scattering, and computer simulation techniques. We present here results of a dynamic neutron scattering analysis of the solvent dependence of the picosecond-time scale dynamic transition behavior of solutions of a simple single-subunit enzyme, xylanase. The protein is examined in powder form, in D2O, and in four two-component perdeuterated single-phase cryosolvents in which it is active and stable. The scattering profiles of the mixed solvent systems in the absence of protein are also determined. The general features of the dynamic transition behavior of the protein solutions follow those of the solvents. The dynamic transition in all of the mixed cryosolvent–protein systems is much more gradual than in pure D2O, consistent with a distribution of energy barriers. The differences between the dynamic behaviors of the various cryosolvent protein solutions themselves are remarkably small. The results are consistent with a picture in which the picosecond-time scale atomic dynamics respond strongly to melting of pure water solvent but are relatively invariant in cryosolvents of differing compositions and melting points.
X-ray diffraction, dynamic neutron scattering, and various spectroscopies have demonstrated a quantitative change in the nature of internal motions of proteins, at ≈200–220 K (1–8). Below this transition, the internal motions are essentially harmonic whereas above it anharmonic dynamics contribute and, at physiological temperatures, dominate the internal fluctuations. The anharmonic motions may involve confined continuous diffusion (6, 9) and/or jump diffusion between potential energy wells associated with “conformational substates” of slightly different structure in which proteins are trapped below the transition (2, 10–12).
Correlations have been made between some protein functions (such as ligand binding, electron transfer, and proton pumping) and the presence of equilibrium anharmonic motion (8, 13–17). Recent x-ray diffraction work on carbon monoxy-myoglobin showed that, below 180 K, photodissociated ligands migrate to specific sites within an internal cavity of an essentially immobilized, frozen protein, from which they subsequently rebind by thermally activated barrier crossing (18). On photodissociation above 180 K, ligands escape from the distal pocket, aided by protein fluctuations that transiently open exit channels.
The increased flexibility conferred by anharmonic dynamics may indeed be required for some proteins to rearrange their structures to achieve functional configurations. However, the forms and time scales of the anharmonic motions required for function are in general unknown. In particular, it was recently shown that, in cases of enzyme activity in a cryosolvent, the rate-limiting step is independent of the ≈220 K dynamic transition (19, 20). Furthermore, it was demonstrated that dynamic transition temperatures in a cryosolvent solution of glutamate dehydrogenase are strongly dependent on the time scale of the motions involved (21).
To further understand the nature of dynamic transitions in proteins, it is particularly important to characterize solvent effects. Solvent can in principle affect protein dynamics by modifying the effective potential surface of the protein and/or by frictional damping (22, 23). Solvent viscosity is known to influence fast, diffusion-limited reaction rates in, for example, ligand binding to myoglobin (24). Changes in the structure and internal dynamics of proteins as a function of solvent conditions at physiological temperatures have been found by using several experimental techniques. In the absence of minimal hydration, lysozyme does not function (25), and related NMR measurements of exchangeability of the main chain amide hydrogens suggest a hydration-related increase in conformational flexibility of the protein (26), which may be coupled to (probably small) Raman-detected conformational changes that appear to be necessary before activity (26, 27). Rayleigh scattering of Mössbauer radiation demonstrated an increase of dynamic amplitudes on hydration of myoglobin (28). This increase was also found in subsequent neutron work on lysozyme powders (29), and qualitatively similar results have been found in more recent neutron work on parvalbumin, lysozyme, myoglobin, and the bovine pancreatic trypsin inhibitor (23, 30–33).
It is clear from the above and other work that solvent affects protein dynamics at physiological temperatures. Therefore, a solvent dependence of the dynamic transition might be expected. Indeed, measurements on CO binding to myoglobin indicate that dynamic behavior of the protein is correlated with a glass transition in the surrounding solvent (34, 35), and a recent molecular dynamics analysis of hydrated myoglobin also indicates a major solvent role in protein dynamic transition behavior (36). Several recent neutron studies have been made of environmental effects on the dynamic transition in proteins. These studies indicate that gross changes in the environment, such as dehydration or trehalose embedding, can abolish the transition (14, 37, 38).
Here, we examine how modification of the composition of the solvent affects dynamic transition behavior by using neutron scattering, which provides a means of directly probing atomic fluctuations in protein solutions (32, 39). The dynamic information is obtained from the elastic scattering, which can be used to derive the cross section-weighted average mean-square displacement 〈u2〉 of the atoms in the solution. The 〈u2〉 contains contributions from all motions resolvable by the instrument used, which in the present experiments corresponds to those faster than ≈100 ps. It is found that, in pure water, the dynamics reflect melting but are invariant in cryosolvents of strongly different composition and phase transition behavior.
Methods
The experiments were performed on a thermophilic xylanase enzyme in a dry powder, in D2O, and in single-phase water-solvent mixtures consisting of 40% CD3OD/60% D2O, 70% CD3OD/30% D2O, 40% DMSO/60% D2O, and 80% DMSO/20% D2O (all solvent percentages are by volume). The corresponding mixed solvent solutions in the absence of protein were also examined. These cryosolvents were chosen because they are all well-characterized and maintain enzyme activity. The xylanase was chosen because it is a relatively simple, single subunit enzyme that is stable under all of the experimental conditions used in this work and active down to at least 190 K. The enzyme was purified and assayed as described elsewhere (19, 20). For the 40% CD3OD/60% D2O and 70% CD3OD/30% D2O solvents, xylanase activity data exist (40, 41). As found in previous work (20), all of the xylanase activity was recovered at the end of the present experiments, confirming that the enzyme is stable under all of the experimental conditions used here.
The dynamic neutron scattering measurements were performed on the IN6 time-of-flight spectrometer at the Institut Laue-Langevin, Grenoble. The incident neutron wavelength was 5.12 Å, and energy resolutionwas 50 μeV. All data were collected with the sample holder oriented at 135o relative to the incident beam. The samples were contained in aluminum flat-plate cells of 0.7 mm thickness for the protein–cryosolvent and 2 mm thickness for the cryosolvent solutions. The following protein samples were run (with their measured transmissions in parentheses): 68 mg/ml xylanase in 70% CD3OD/30% D2O (0.89); 68 mg/ml xylanase in 40% CD3OD/60% D2O (0.88); 51 mg/ml xylanase in 80% DMSO/20% D2O (0.87); 68 mg/ml xylanase in 40% DMSO/60% D2O (0.90); 100 mg/ml dry powder xylanase (0.98); and 100 mg/ml xylanase in D2O (0.90).
The dry powder was prepared by freeze drying and then holding at 100 milliTorr for 2 days at 200°C. The following perdeuterated solvents were run: 70% CD3OD/30% D2O (0.80); 40% CD3OD/60% D2O (0.79); 80% DMSO/20% D2O (0.80); 40% DMSO/60% D2O (0.86); and pure D2O (0.82).
Samples were first cooled to 110 K and then heated back up to 300 K in steps of 10 K. Raw data were corrected in identical fashion. The elastic intensity was determined by integrating the detector counts over the energy range of the instrumental resolution. The detectors were calibrated by normalizing with respect to a standard vanadium sample. The cell scattering was subtracted, taking into account attenuation of the singly scattered beam. Finally, the scattering was normalized with respect to that at the lowest measured temperature, 110 K.
The quantity measured in neutron scattering is the dynamic structure factor, S(q,ω), which is a function of the scattering wave vector q and the energy transfer ω (32, 29). S(q,ω) has two components: incoherent (which arises from self-correlations in the atomic positions) and coherent (which arises from self- and cross-correlations in the atomic positions). The incoherent scattering cross section (scattering power) for hydrogen is an order of magnitude greater than any other cross section in biological systems. Therefore, the hydrogen incoherent scattering dominates the signal from the protein. Because hydrogens are evenly distributed in a protein, the protein scattering gives a global view of the protein motions. To maximize the contribution from the protein motions under controlled conditions, the present experiments were performed by using fully deuterated solvents and hydrogen/deuterium exchanged proteins (twice dissolved in D2O and freeze-dried). This deuteration procedure serves to ensure that the solvent is fully deuterated (and not partially hydrogenated by exchange of hydrogen from the enzyme) and that no change in the deuteration of the enzyme or solvent occurs during the experiment. The consequent partial deuteration of the enzyme is limited to the exchangeable hydrogens, which means that the hydrogens left are essentially all in the protein. Knowledge of the isotopic scattering cross sections and isotopic compositions of the samples then indicates that, for the protein solutions, about 50% of the scattering is incoherent and 50% coherent. Seventy-five percent of the incoherent scattering originates from the protein, because of the strong hydrogen contribution. Therefore, 25% originates from the solvent atoms and will influence the observed profile in a manner depending on the solvent dynamics and the region of q,ω space examined.
The isotopic coherent scattering cross sections and protein solution sample composition indicate that most of the coherent scattering originates from the solvent. The self-coherent contribution to this, which is strongest in the low q regime considered here (below 1 Å−1), is dynamic and identical in form to the incoherent scattering from the same atom (the amplitude is not identical because it is weighted by the coherent cross section) (39). In addition, there is a contribution originating from cross-correlations (i.e., distance distributions). The intramolecular “Bragg” part of this contribution lies at q values higher than those considered here. However, a structural “small-angle” scattering contribution can also exist in the same q-range as the self-coherent and incoherent scattering. The small-angle scattering can be distinguished from the dynamic self-coherent scattering by its q-dependence. To remove the temperature-independent small-angle scattering, the scattering profiles were normalized with respect to the intensities measured at 110 K. For the protein solution samples examined here, there was no evidence for additional temperature-dependent, small-angle coherent scattering.
We define 〈uL2〉 as the mean-square displacement of the Lth atom, where the brackets indicate an ensemble-average. If q2〈uL2〉 < 1, the elastic incoherent scattering is given by (39)
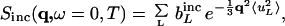 |
1 |
where bL is the scattering length of atom L. Assuming a uniform mean-square displacement 〈u2〉, this 〈u2〉 can be accessed by performing a plot of ln S(q,ω = 0,T) vs. q2 and taking the limiting slope as q→0 at each temperature. In the present work, the 〈u2〉 thus determined is equal to (〈u2〉T − 〈u2〉110), where 〈u2〉110 is the absolute mean-square displacement at 110 K. 〈u2〉 is analogous to that obtained by x-ray crystallographic temperature factor analysis except that it is molecule averaged, time resolved, and purely dynamic, i.e., without a static disorder contribution.
We introduce here the integrated elastic intensity SINT(T), obtained by summing S(q,ω = 0,T) over a range of small q values. SINT(T) provides a qualitative guide of dynamic transition behavior, with good counting statistical accuracy, and it is especially useful in cases where the ln S(q,ω = 0,T) vs. q2 plots are too noisy to extract meaningful 〈u2〉 data, such as was found for the pure solvent samples in the present work. The improved statistics are obtained by integrating over a range of q chosen to lie in the dynamic scattering region, i.e., the straight-line regions in the ln S(q,ω = 0,T) vs. q2 plots (0.35 < q < 1.03 Å−1 in Fig. 2). The approximate physical sense of SINT(T) can be appreciated by expanding the exponential in Eq. 1 to first order, integrating over q, and averaging over the atoms. One then obtains that SINT(T) ∝ −〈u2〉.
Figure 2.

Ln S(q,0) vs. q2 plot for xylanase in 70% CD3OD/D2O.
In certain pure solvent systems, significant temperature-dependent small-angle coherent scattering was found (see Results). The pure solvent SINT(T) should therefore be considered only as an approximate guide to the dynamic transition behavior of the pure solvent systems studied. Moreover, relatively poor counting statistics were obtained for the pure solvent samples, due in part to the absence of strongly scattering hydrogens, rendering impossible the extraction of statistically significant mean-square displacements for these samples.
Structural and Thermodynamic Measurements on the Solvents.
X-ray scattering experiments on the cryosolvents were carried out down to 110 K in Debye-Scherrer geometry on line 9.1 at the Daresbury Synchrotron Radiation Source (Daresbury, U.K.) using a curved image-plate detector system (41). The cryosolvents were in standard 0.7-mm, thin-walled glass capillary tubes. The cooling rate of 120 K/h was similar to that used in the neutron experiments, and the warming rate was 360 K/h. The scattering from the samples was observed on both cooling and warming at a number of intermediate temperatures. The presence of crystallization was identified by the appearance of powder diffraction peaks. Where no crystallization was observed, the changes to measured diffraction patterns were as would be expected from a liquid or glass on changing the temperature. Samples were also examined visually during image plate changes down to 110 K for any change in optical opacity that would have indicated possible changes, such as crystallization or phase separation. Differential scanning calorimetry (DSC) measurements were made by using a Shimadzu DSC-50 differential scanning calorimeter. Samples were sealed in 50 μl aluminum crucibles (sample weights between 25 and 40 mg) and cooled down by using a flowing nitrogen atmosphere. All data were collected in heating at a standard rate of 10 K/min (42).
Results
Dynamic Neutron Scattering.
In Fig. 1 are presented SINT(T) for the samples. For the protein samples, it was possible to fit the data to straight lines in semilog plots of ln S(q,ω = 0,T) vs. q2 (an example of such a fit is given in Fig. 2) so as to obtain corresponding mean-square displacement data, which are shown in Fig. 3. The dry powder SINT(T) profile in Fig. 1A shows only a relatively small decrease, with temperature over the range examined (from 110 K to 300 K), and there is a correspondingly small increase in 〈u2〉 in the data in Fig. 3A. The major component of this 〈u2〉 is expected to be due to small-amplitude, solid-like, vibrational dynamics, the physical nature of which has been explored (43) although one cannot rule out the existence of a small anharmonic contribution. The profile indicates that, in dehydrated powder form, xylanase does not possess a marked transition in the neutron scattering data, consistent with previous work on other proteins (14, 15, 28).
Figure 1.

Normalized integrated elastic intensity. The q integration range is 0.35 Å−1 < q < 1.03 Å−1 and qmean = 0.66 Å−1. (A) Xylanase powder, pure D2O, and xylanase in D2O. (B) 40% CD3OD/60% D2O solvent, 70% CD3OD/30% D2O solvent, xylanase in 40% CD3OD/60% D2O, and xylanase in 70% CD3OD/30% D2O. (C) 40% DMSO/60% D2O, 80% DMSO/20% D2O, xylanase in 40% DMSO/60% D2O, and xylanase in 80% DMSO/20% D2O.
Figure 3.

Mean-square displacement vs. temperature for (A) xylanase that is dry, in D2O, in 40% CD3OD/D2O, and in 70% CD3OD/D2O. (B) Xylanase in 40% DMSO/60% D2O and in 80% DMSO/20% D2O.
For the pure D2O sample in Fig. 1A, there is a significant decrease with temperature from 110 K to 280 K, without any clear transition but with a gradual increase in curvature. The profile is again consistent with predominantly vibrational fluctuations, but of larger amplitude than in the dry protein powder. Between 280 K and 290 K, there is a sharp transition occurring slightly above the thermodynamic melting point, the latter being at 277 K. This strongly nonlinear increase of 〈u2〉 with T is consistent with the activation of anharmonic picosecond dynamics.
The general form of SINT(T) for xylanase in D2O resembles that of the pure D2O, again with a sharp transition associated with melting of the solution. Fig. 3A indicates that the melting transition in the xylanase/D2O system is associated with an 8-fold increase in the mean-square displacement over the temperature range 270–290 K. In Fig. 1A, the drop starts earlier and is less steep than that of D2O, indicating that the transition in the protein solution is slightly more gradual and begins at a slightly lower temperature (≈270 K). This suggests that the protein acts to depress the dynamic transition temperature. Experiments performed on glutamate dehydrogenase and myoglobin in D2O (not shown) produced results similar to the present xylanase work.
A phenomenon seen in all of the systems is that, below the transition, SINT(T) is higher for xylanase solution than for the corresponding pure solvent. This indicates that, in the protein solution, the atomic fluctuations are decreased relative to those in the pure solvent. This may reflect the relatively high packing of the protein interior, which might lead to restricted vibrational motion relative to in-bulk solvent. Furthermore, the amplitude of the vibrational modes of the solvent molecules could be reduced by interaction with the protein.
The decrease of SINT(T) with T in all four enzyme–cryosolvent systems and the corresponding pure solvents (Fig. 1 B and C) is much more gradual than in the D2O samples. To a first approximation, the dynamics of the protein solutions follow that of the pure solvents. SINT(T) decreases continually over the whole temperature range examined. For some of the samples, unevenness in SINT(T) is seen at low temperatures (below 220 K). This is likely to be related to the thermodynamic and structural phase transitions in these samples discussed in the next section; some contamination from small-angle scattering cannot be ruled out. Apart from this contamination, no clear inflection points in SINT(T) are visible for the pure CD3OD/D2O samples whereas for the DMSO samples a change of slope is seen at ≈200–220 K.
The dynamic transition behavior of the protein–cryosolvent solutions is clearly visible in the 〈u2〉 vs. T plots in Fig. 3. In the protein–cryosolvent solutions, large-amplitude anharmonic dynamics are activated at significantly lower temperatures than in the protein/D2O sample, and the increase of 〈u2〉 with T is much more gradual, resembling that seen in hydrated protein powders (7). The 〈u2〉 data in the four cryosolvent–protein systems are virtually identical.
Structural and Thermodynamic Properties of the Solvents.
Published melting temperatures in the hydrogenated mixed solvents (44, 45) and data on the enzyme stability in the mixed solvents are given in Table 1. Three of the four solvents melt in the range 232–235 K whereas the 70% CH3OH remains liquid to below 190 K.
Table 1.
Thermodynamic and stability data
| Solvent | Solvent melting temperature, K | Half-life of xylanase in solvent |
|---|---|---|
| 70% CH3OH/30% H2O | 188 | 20 min at 50°C |
| 40% CH3OH/60% H2O | 233 | 18 h at 60°C |
| 80% DMSO/20% H2O | 235 | <2 min at 50°C |
| 40% DMSO/60% H2O | 232 | 6 h at 80°C |
The stability data were taken at temperatures well above that at which the present experiments were performed. The half-lives at the temperatures used in the present neutron work are very much longer than those given here. The half-life of xylanase in water is >20 h at 95°C.
The DSC and synchrotron x-ray diffraction measurements were performed to provide additional structural and confirmatory thermodynamic information on the perdeuterated pure solvents. Both methods were used to indicate the presence of phase changes, whereas the diffraction data additionally probed structural effects as evidenced by the appearance of Bragg peaks, using heating/cooling rates closely similar to those used in the neutron experiments. The DSC indicated that the melting temperature is at 170–190 K for the 70% CD3OD/30% D2O sample and ≈235 K for the 40% CD3OD/60% D2O sample, consistent with the above published results.
All solvents showed noticeable Bragg scattering at low temperatures at q > 0.8 Å−1. For the 70% CD3OD/30% D2O sample, this was visible at temperatures below 190 K, whereas the 40% CD3OD/60% D2O solution exhibited Bragg diffraction at 220 K but not 245 K (intermediate temperatures were not examined). These results are consistent with the above published melting temperatures and our DSC measurements. The 40% CD3OD/60% D2O sample also exhibited small-angle x-ray scattering at very low q (0.34 Å−1) at 160–180 K, suggesting some long-range periodicity accompanying the formation of microcrystallites at these temperatures in this sample. The above Bragg scattering is consistent with the SINT(T) data in Fig. 1B, which shows nonsmooth behavior below 235 K for the 40% CD3OD/60% D2O sample but smooth behavior in the 70% sample. The nonsmooth behavior may be due to dynamic effects accompanying structural phase transitions or due to contamination of SINT(T) by low-q diffraction in the sample. The x-ray data for both the 80% and 40% DMSO solvents showed complex solid-state phase behavior between 170 K and 210 K, which, for the 80% sample, could be related to the uneven SINT(T) and the peak in 〈u2〉 at 190 K and 200 K seen in Fig. 3B. The final melting temperature was between ≈230–245 K for both these samples, again consistent with the results of refs. 44 and 45.
Discussion
A number of investigations strongly suggest that the intrinsic anharmonicity of the protein potential surface will be modulated by qualitative changes in solvation conditions (see, for example, refs. 14, 16, 22–33, and 37). The present results indicate a large effect of solvent on dynamic transition behavior by showing that the fast picosecond fluctuations of concentrated protein solutions largely follow those of the corresponding pure solvents.
The compositional inhomogeneity of the mixed solvents may play a role in the nonlinear increase of 〈u2〉 in the protein–cryosolvent systems being much more gradual than in D2O (Fig. 3). This 〈u2〉 behavior is consistent with the presence of a distribution of energy barriers, with successively higher barriers being crossed on the picosecond time scale with increasing temperature. This is in contrast to the abruptness of the transition in D2O, which indicates a more cooperative dynamic transition, associated with the melting point. The cooperativity may in principle involve both the ice–water structural transition and associated whole-molecule diffusion.
The results also indicate that, in solutions of proteins in markedly different cryosolvents, similar picosecond dynamic transition behavior is seen. The 〈u2〉 data for xylanase in all four cryosolvents are closely similar, with onset of anharmonicity seen at ≈230 K (Fig. 3). This is around the highest melting points of three of the four cryosolvents which are at 232–235 K. Therefore, the dynamical transition behavior of these three samples occurs at about the same temperature as their phase transition. However, although the highest melting point for 70% CD3OD/30% D2O is below 190 K, no associated activation of the picosecond dynamics is seen close to this temperature in the corresponding protein solution. The 230–235 K transition does remain, although perhaps at a slightly lower temperature than for xylanase in 40% CD3OD/60% D2O. Remarkably, then, there is little difference in the temperature dependence of the picosecond dynamics of protein–cryosolvent samples here that contain very different proportions of the mixed solvents and water contents and have very different highest melting points. Melting of the 70% CD3OD/30% D2O solvent system has no measurable effect on the fast dynamics. The present results also raise the interesting question as to how the dynamics will vary as the proportion of CD3OD (or DMSO) is decreased, i.e., whether there is a (perhaps narrow) concentration range where a “switch” to the sharp D2O transition behavior takes place.
Structural effects that might be related to the presently observed dynamic insensitivity to cryosolvent composition would be interesting to investigate. One possible explanation might be that the structure of the solvent shell of the enzyme may be qualitatively similar in all of the cryosolvents but different from pure water. What limited evidence we do have from crystallographic studies in mixed solvents suggests that the non-water solvent molecules do interact with certain parts of the protein surface directly (46–49). However, although xylanase is active in all of the cryosolvent mixtures used here, the mixtures do destabilize the protein relative to water, and they do so to differing degrees (See Table 1). Therefore, if the effect of a solvent on stability is mediated in part by perturbing the shell of molecules closest to the protein, then these stability results would indicate that the composition of the solvent shell is not similar in each sample.
Another possibility, suggested by a referee and consistent with the present and other data, is that the onset of the anharmonic protein motion takes place at a temperature, determined either by the solvent or by the protein, being governed by the component having the higher transition temperature. According to this scenario, the solvent would drive the transition in the pure D2O solution whereas the protein would drive it in the 70% CD3OD/30% H2O system, for which in the absence of protein the solvent melts at about 190 K whereas the onset of anharmonicity is seen at ≈230–240 K. For the other mixed solvent systems, the protein and solvent transition temperatures would essentially coincide. This explanation is consistent with molecular dynamics simulations of isolated myoglobin, for which an intrinsic protein dynamic transition was seen at ≈220 K (50) and is also in agreement with the “protein cold/solvent hot” and “protein hot/solvent cold” results in the recent molecular dynamics simulations of hydrated myoglobin by Vitkup et al. (36).
In the future, it will be important to determine the time scale dependence of the solvent influence on dynamic transition behavior. Although it is shown here that the phase transition behavior of xylanase in 70% CD3OD/30% D2O is decoupled from the picosecond dynamic transition behavior, it is likely to be coupled to slower motions. Furthermore, because the time scale accessed in the present experiments is also well sampled with modern computer power in molecular dynamics simulation, this computational technique can in principle be used to obtain a detailed decomposition of the dynamic transition behavior revealed here.
Acknowledgments
We thank the Institut Laue Langevin (Grenoble) for access to neutron beam facilities, the United Kingdom Engineering and Physical Sciences Research Council for access to the Daresbury Synchrotron Radiation Source, the University of London Intercollegiate Thermal Analysis Research Service for facilitating the DSC measurements, and C. Monk and D. Thompson for assistance with enzyme preparation. For part of this work, R.M.D. was supported by a James Cook fellowship from the Royal Society of New Zealand, and V.R. was a European Union-funded Marie Curie Research Fellow.
Abbreviation
- DSC
differential scanning calorimetry
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Parak F, Formanek H. Acta Crystallogr A. 1971;27:573–578. [Google Scholar]
- 2.Frauenfelder H, Petsko G A, Tsernoglou D. Nature (London) 1979;280:558–563. doi: 10.1038/280558a0. [DOI] [PubMed] [Google Scholar]
- 3.Keller H, Debrunner P G. Phys Rev Lett. 1980;45:67–68. [Google Scholar]
- 4.Parak F, Frolov E N, Mossbauer R L, Goldanskii V I. J Mol Biol. 1981;145:825–833. doi: 10.1016/0022-2836(81)90317-x. [DOI] [PubMed] [Google Scholar]
- 5.Cohen S G, Bauminger E R, Nowik I, Ofer S, Yariv J. Phys Rev Lett. 1981;46:1244–1248. [Google Scholar]
- 6.Knapp E W, Fischer S F, Parak F. J Phys Chem. 1982;86:5042–5047. [Google Scholar]
- 7.Doster W, Cusack S, Petry W. Nature (London) 1989;337:754–756. doi: 10.1038/337754a0. [DOI] [PubMed] [Google Scholar]
- 8.Rassmussen B F, Stock A M, Ringe D, Petsko G A. Nature (London) 1992;357:423–424. doi: 10.1038/357423a0. [DOI] [PubMed] [Google Scholar]
- 9.Kneller G R, Smith J C. J Mol Biol. 1994;242:181–185. doi: 10.1006/jmbi.1994.1570. [DOI] [PubMed] [Google Scholar]
- 10.Elber R, Karplus M. Science. 1987;235:318–321. doi: 10.1126/science.3798113. [DOI] [PubMed] [Google Scholar]
- 11.Lamy A, Souaille M, Smith J C. Biopolymers. 1996;39:471–478. [Google Scholar]
- 12.Frauenfelder H, Sligar S, Wolynes P. Science. 1991;254:1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 13.Parak F, Frolov E N, Kononenko A A, Mossbauer R L, Goldanskii V I, Rubin A B. FEBS Lett. 1980;117:368–372. doi: 10.1016/0014-5793(80)80982-3. [DOI] [PubMed] [Google Scholar]
- 14.Ferrand M, Dianoux A J, Petry W, Zaccai G. Proc Natl Acad Sci USA. 1993;90:9668–9672. doi: 10.1073/pnas.90.20.9668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fitter J, Lechner R E, Dencher N. Biophys J. 1997;73:2126–2137. doi: 10.1016/S0006-3495(97)78243-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lehnert U, Réat V, Weil M, Zaccai G, Pfister C. Biophys. J. 1998. 1945–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding X, Rasmussen B F, Petsko G A, Ringe D. Biochemistry. 1994;33:9285–9293. [PubMed] [Google Scholar]
- 18.Ostermann A, Waschipky R, Parak F G, Nienhaus G U. Nature (London) 2000;404:205–208. doi: 10.1038/35004622. [DOI] [PubMed] [Google Scholar]
- 19.Daniel R M, Smith J C, Ferrand M, Hery S, Dunn R, Finney J L. Biophys J. 1998;75:2504–2507. doi: 10.1016/S0006-3495(98)77694-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dunn R V, Réat V, Finney J L, Ferrand M, Smith J C, Daniel R M. Biochem J. 2000;346:355–358. [PMC free article] [PubMed] [Google Scholar]
- 21.Daniel R M, Finney J L, Réat V, Dunn R, Ferrand M, Smith J C. Biophys J. 1999;77:2184–2190. doi: 10.1016/S0006-3495(99)77058-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brooks C L, III, Karplus M. J Mol Biol. 1989;208:159–181. doi: 10.1016/0022-2836(89)90093-4. [DOI] [PubMed] [Google Scholar]
- 23.Smith J C, Cusack S, Tidor B, Karplus M. J Chem Phys. 1990;93:2974. [Google Scholar]
- 24.Frauenfelder H. Nature (London) 1989;338:623–624. [Google Scholar]
- 25.Rupley J A, Gratton E, Careri G. Trends Biochem. 1983;8:18–22. [Google Scholar]
- 26.Poole P L, Finney J L. Int J Biol Macromol. 1983;5:308–310. [Google Scholar]
- 27.Poole P L, Finney J L. J Biosci. 1985;8,Suppl.:25–35. [Google Scholar]
- 28.Krupyanskii Y U F, Parak F, Goldanskii V I, Mossbauer R L, Gaubman E E, Engelmann H, Suzdalev I P. Z Naturforsch C. 1982;37:57–62. doi: 10.1515/znc-1982-1-211. [DOI] [PubMed] [Google Scholar]
- 29.Smith J C, Cusack S, Poole P L, Finney J L. J Biomol Struct Dyn. 1987;4:583–588. doi: 10.1080/07391102.1987.10507662. [DOI] [PubMed] [Google Scholar]
- 30.Zanotti J-M, Bellissent-Funel M-C, Parello J. Biophys J. 1999;76:2390–2411. doi: 10.1016/S0006-3495(99)77395-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perez J, Zanotti J-M, Durand D. Biophys J. 1999;77:454–469. doi: 10.1016/S0006-3495(99)76903-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith J C. Q Rev Biophys. 1991;24:227–291. doi: 10.1017/s0033583500003723. [DOI] [PubMed] [Google Scholar]
- 33.Diehl M, Doster W, Schober H. Biophys J. 1997;73:2726–2732. doi: 10.1016/S0006-3495(97)78301-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iben I E, Braunstein D, Doster W, Frauenfelder H, Hong M K, Johnson J B, Luck S, Ormos P, Schulte A, Steinbach P J, et al. Phys Rev Lett. 1989;62:1916–1919. doi: 10.1103/PhysRevLett.62.1916. [DOI] [PubMed] [Google Scholar]
- 35.Meyer E. Biophys J. 1994;70:862–873. doi: 10.1016/S0006-3495(94)80547-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vitkup D, Ringe D, Petsko G A, Karplus M. Nat Struct Biol. 2000;7:34–38. doi: 10.1038/71231. [DOI] [PubMed] [Google Scholar]
- 37.Fitter J. Biophys J. 1999;76:1034–1042. doi: 10.1016/S0006-3495(99)77268-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cordone L, Ferrand M, Vitrano E, Zaccai G. Biophys J. 1999;76:1043–1047. doi: 10.1016/S0006-3495(99)77269-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lovesey S W. Theory of Neutron Scattering from Condensed Matter. Oxford: Clarendon; 1984. [Google Scholar]
- 40.Dunn R V. M. Sc. thesis. Hamilton, New Zealand: Univ. of Waikato; 1998. [Google Scholar]
- 41.Roberts M A, Finney J L, Bushnell-Wye G. Mater Sci Forum. 1998;278:318–322. [Google Scholar]
- 42.Réat V, Finney J L, Steer A, Roberts M A, Smith J C, Dunn R, Petersen M, Daniel R M. J Biochem Biophys Methods. 2000;42:97–103. doi: 10.1016/s0165-022x(99)00053-6. [DOI] [PubMed] [Google Scholar]
- 43.Melchers B, Knapp E W, Parak F, Cordone L, Cupane A, Leone M. Biophys J. 1996;70:2092–2099. doi: 10.1016/S0006-3495(96)79775-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Douzou P, Hui Bon Hoa G, Maurel P, Travers F. In: Handbook of Biochemistry and Molecular Biology. 3rd Ed. Fasman G D, editor. Vol. 1. Boca Raton, FL: CRC; 1976. pp. 520–539. [Google Scholar]
- 45.Weast R C, editor. Handbook of Chemistry and Physics. 55th Ed. Boca Raton, FL: CRC; 1974. [Google Scholar]
- 46.Bouquiere J P, Finney J L, Lehmann M S. J Chem Soc Faraday Trans. 1993;89:2701–2705. [Google Scholar]
- 47.Lehmann M S, Mason S A, McIntyre G J. Biochemistry. 1985;24:5862–5869. doi: 10.1021/bi00342a026. [DOI] [PubMed] [Google Scholar]
- 48.Lehmann M S, Stansfield R F D. Biochemistry. 1989;28:7028–7033. doi: 10.1021/bi00443a037. [DOI] [PubMed] [Google Scholar]
- 49.Ringe D. Curr Opin Struct Biol. 1995;5:825–829. doi: 10.1016/0959-440x(95)80017-4. [DOI] [PubMed] [Google Scholar]
- 50.Smith J C, Kuczera K, Karplus M. Proc Natl Acad Sci USA. 1990;87:1601–1605. doi: 10.1073/pnas.87.4.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]