

Abstract
The herpes simplex virus type 1 (HSV-1) thymidine kinase (TK) promoter contains elements involved in both constitutive and induced expression. We determined that phorbol 12-myristate 13-acetate (PMA) induces the HSV-1 TK promoter in HEK293 cells. However, PMA did not induce expression from the promoter in HeLa cells and did not result in a globally increased gene expression in HEK293 cells. Induction of HSV-1 TK promoter required activation of both of JNK and ERK pathways. However, activation of the two pathways alone was not sufficient for induction of HSV-1 TK promoter. By transiently transfecting into HeLa cells the adenoviral E1A gene, which exists as an integrant in HEK293 genome, we demonstrated that E1A proteins are necessary for induction of HSV-1 TK promoter by PMA. We propose mechanisms by which signaling pathways activated by the tumor-promoter PMA cooperate with the oncogene E1A to stimulate a eukaryotic promoter, namely the HSV-1 TK promoter.
Keywords: PMA, JNK, ERK, E1A, Thymidine kinase
Introduction
The 5′ non-transcribed region of the HSV-1 TK gene contains three cis-acting elements that are involved in both constitutive and induced expression of the TK gene, which is a member of the delayed early or β genes of HSV-1: a proximal element containing a TATA box and located at −12 to −29 (to which the general transcription factor TFIID binds), the first distal element located at −47 to −61 and the second distal element situated at −80 to −105 [1-3]. The first distal element has a GC-box, a binding site for the transcription factor Specificity Protein-1 (sp1), and the second distal element contains binding sites for sp1 and CCAAT-transcription factor (CTF, also known as nuclear factor-I or NF-I) [2, 4]. Constitutive expression of TK takes place when a complete copy of the TK gene is microinjected into Xenopus laevis oocytes or is transfected into mammalian cells [1, 5-7]. Induced expression occurs during productive viral infection and this effect is mediated in trans by some of the products of the immediate early (α genes) of HSV-1 which are expressed within 2-4 hours after infection [2]. The α genes that stimulate the HSV-1 TK promoter include Infected Cell Polypeptide-4 (ICP-4) and ICP-0, and the mechanism of promoter induction appears to be modulation of the activities of cellular transcription factors [2, 8, 9]. The expression of TK reaches peak rates at 5-7 hours after infection. In addition to the products of the immediate early genes of HSV-1, certain cellular proteins and chemical treatments have been reported to alter the constitutive activity of the TK promoter. The TK promoter can be induced by the Human T-lymphotrophic Virus type 1 (HTLV-1) Tax protein [10], the androgen analog R1881 [11], and the transcription factors GATA4 and GATA6 [12]. The TK promoter can be suppressed by wild type p53 [13], dexamethasone [14], and nuclear receptors such as Chicken Ovalbumin Upstream Promoter Transcription Factor-1 (COUP-TFI) and Peroxisome Proliferator Activator Receptor alpha (PPARα) [15].
By virtue of its constitutive expression in mammalian cells the HSV-1 TK promoter has been adopted for use to drive the expression of reporter genes used as internal controls in reporter gene assays. One such system is the Renilla luciferase reporter gene developed by Promega Corporation [16]. Various Renilla luciferase-expressing plasmids are available, one of the most widely used being the pRL-TK plasmid. The Renilla luciferase coding region in the pRL-TK plasmid is placed downstream of a promoter region that consists of 753 base pairs from the HSV-1 genome; the first 724 base pairs are from the −724 to −1 region of the HSV-1 TK gene and the last 29 base pairs are from the +1 to +29 region of the TK gene [16]. While investigating the effects of vitamin D on the activities of various cell signaling pathways in HEK293 cells using luciferase-based reporter gene assays, we noticed that the activity of the Renilla luciferase expressed from the pRL-TK plasmid used as an internal control was markedly induced when the cells were treated with PMA. An extensive search of the Medline did not reveal any report of a similar observation. Therefore, we decided to further investigate the effect of PMA on the expression of the Renilla luciferase from the pRL-TK plasmid.
PMA, also known as 12-O-tetradecanoylphorbol 13-acetate (TPA), is an ester form of phorbol. Phorbol is an organic compound that is isolated from croton oil, which in turn is derived from the seeds of the plant Croton tiglium, a shrub that is found in Southeast Asia [17-19]. Various phorbol esters have been used in research as potent tumor promoters [17]. PMA mimics diacylglycerol and appears to function by directly activating several of the protein kinase C (PKC) isozymes intracellularly [17, 20, 21]. Activated PKC subsequently activates the Nuclear Factor-kappaB (NF-κB), Jun N-terminal Kinase (JNK) and Extracellular Signal-Regulated Kinase (ERK) signal transduction pathways which regulate a diverse set of cellular processes [22-25].
In this article, we report that PMA treatment induces expression from the HSV-1 TK promoter in the HEK293 cell line and this effect depends on the activation of PKC and two of its downstream effectors, namely JNK and ERK. Moreover, we demonstrate that the activating effect of PMA on the HSV-1 TK promoter requires the presence of the adenoviral E1A protein, thus providing an example of a tumor promoter and an oncogene cooperating to activate a eukaryotic promoter.
Materials and Methods
Cells
HEK293 (Human Embryonic Kidney-293) and HeLa (human cervical carcinoma) cell lines were maintained in Dulbecco's Modified Eagle's Medium (DMEM) (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Invitrogen), 100 U/ml penicillin (Mediatech) and 100 μg/ml streptomycin (Mediatech). Cells were grown in an incubator at 37°C and 5% CO2. PMA was used at a final concentration of 50 ng/ml to treat cells, when indicated, except in dose-response studies. When Tumor Necrosis Factor α (TNFα) was used to treat cells, it was used at 10 ng/ml.
Plasmids
The pcDNA3.1/myc-His(−)A (hereinafter referred to as pcDNA3) plasmid and the pUC19 plasmid were from Invitrogen. The pGreenLantern-1 plasmid expressing green fluorescent protein (GFP) under the CMV enhancer/promoter (pGFP) was from GIBCO. The plasmid pCH110, which expresses β-galactosidase under the control of the Simian Virus-40 (SV40) early enhancer/promoter (pSV40-β-galactosidase), was from Amersham Biosciences. The plasmid pRL-TK that expresses Renilla luciferase under the HSV-1 TK promoter was from Promega. The sources of the pAP-1-Luc (Activator Protein-1, AP-1, luciferase) and pIgκ-Luc (NF-κB luciferase) were reported previously [26]. The pAP-1-Luc construct, containing the luciferase gene driven by six copies of the AP-1 consensus binding site, was kindly provided by Dr. Roya Khosravi-Far (Harvard Medical School). The pIgκ-Luc was kindly provided by Dr. Bruce Horwitz (Whitehead Institute at the Massachusetts Institute of Technology). The plasmid expressing the S32/36A mutant IκBα has been reported before [27].
The pGL2-TK plasmid which expresses the firefly luciferase gene under the HSV-1 TK promoter was constructed by excising the HSV-1 TK promoter from the pRL-TK plasmid and cloning it in front of the firefly luciferase gene in the pGL2 Basic plasmid (Promega) as described here (Fig. 1A). First, both plasmids were digested with BglII (Promega) and HindIII (Promega). The pGL2 Basic digest was then treated with shrimp alkaline phosphatase (Roche). The digests were purified by agarose gel electrophoresis and the purified fragments were ligated using the Rapid T4 DNA Ligase Kit (Roche). The plasmids were then propagated in DH5α competent E. coli (Invitrogen) and purified using the Qiagen HiSpeed Kit. The presence of the insert was confirmed by restriction digestion of the pGL2-TK plasmid with XhoI (Invitrogen) and SacII (New England BioLabs).
Fig. 1.

Construction of the pGL2-TK and pE1A-pcDNA3 plasmid vectors. A: The pGL2-TK plasmid was made by cloning the HSV-1 TK promoter, isolated after BglII and HindIII double digestion of the pRL-TK plasmid, in front of the firefly luciferase ORF in the pGL2 Basic plasmid which lacks any promoter/enhancer element. B: The pE1AcDNA3 plasmid was generated by first amplifying by PCR the E1A region (consisting of part of the 5′ non-transcribed region and the whole coding region) from the genomic DNA of HEK293 cells using primers to which linkers containing restriction sites for NheI and BamHI were added, and then cloning the E1A amplicon into the pcDNA3.1.myc-His(−)A plasmid downstream of the CMV promoter.
The pE1A-pcDNA3 plasmid was constructed by amplifying the adenovirus (Ad) type 5 E1A gene from the genomic DNA of HEK293 cells by PCR and then cloning it into pcDNA3.1/myc-His(−)A as described here (Fig. 1B). Genomic DNA was isolated from HEK293 cells grown to confluency on a 100-mm dish using the GenElute Mammalian Genomic DNA Miniprep Kit (Sigma). PCR was carried out on the genomic DNA using the PfuUltra High-Fidelity DNA Polymerase System (Stratagene) in a thermal cycler (Bio-Rad MyCycler). The following forward and reverse primers, respectively, were used: 5′-ctaactgctagcgggcgggtgacgtagtag-3′ and 5′-agtgctaggatccttgcctgggatgagcc-3′. The reaction conditions were as follows: an initial denaturation at 95°C for 5 mins; 30 cycles of denaturation at 95°C for 60 s, annealing at 60°C for 60 s and extension at 72°C for 8 mins; and a final extension at 72°C for 10 mins. The PCR amplicon and the pCDNA3 vector plasmid were digested with NheI (Promega) and BamHI (New England BioLabs) after which the vector digest was treated with shrimp alkaline phosphatase. The digests were then purified by agarose gel electrophoresis and the digested amplicon was ligated into the pCDNA3 vector backbone. The plasmid construct was then propagated and purified as mentioned above. The success of the cloning was confirmed by performing restriction digestion with NheI and BamHI.
Reagents
Chemicals were obtained as follows: GF 109203X and U0126 (both resuspended in DSMO) from Biomol; JNK Inhibitor II (dissolved in DMSO) from Calbiochem; ionomycin (dissolved in ethanol) from Calbiochem; PMA (dissolved in DMSO) from Sigma; and TNFα (resuspended in phosphate buffered-saline, PBS) from R&D Systems. Non-conjugated antibodies were obtained as follows: Ad type 5 E1A murine monoclonal antibody (mAb) from Lab Vision; c-Fos, sp1, ERK1 and ERK2 rabbit polyclonal antibodies (pAbs) from eBioscience; phospho-p44/42 MAP Kinase (T202/Y204) rabbit pAb from Cell Signaling Technology; c-Jun, phospho-c-Jun (S63), and β-tubulin rabbit pAbs from Santa Cruz Biotechnology; β-actin mouse mAb from Abcam. Horseradish peroxidase (HRP)-conjugated anti-mouse IgG secondary Ab was from GE Healthcare and HRP-conjugated anti-rabbit IgG Ab was from Invitrogen. Lipofectamine was from Invitrogen. Complete Protease Inhibitors Cocktail was from Roche.
Immunoblotting
Cells were seeded on a six-well plate at a density of 500,000 cells per well one day prior to treatment. On the second day, the cells were treated as appropriate and harvested by centrifugation. The cells were lysed by resuspending the cell pellet in 150 μl of Cell Extraction Buffer (Invitrogen) to which Complete Protease Inhibitors Cocktail tablets were added and then incubated on ice for 30 minutes. The lysates were then cleared by centrifugation. Total protein was measured using the bicinchoninic acid (BCA) Protein Assay Kit (Pierce) and the protein concentration was normalized across the cell lysates. SDS-PAGE was carried out by loading 7.5 μg of total protein diluted in an equal volume of 2× SDS Sample Buffer (Invitrogen) per lane on a 12% Tris-HCl pre-cast gel (Bio-Rad). After electrophoresis, the separated proteins were transferred to a 45-micron nitrocellulose membrane (GE Healthcare), which was then incubated with primary Ab followed by incubation with secondary Ab. The membrane was developed using the Western Blotting Luminol Reagent (Santa Cruz Biotechnology) and exposed to an autoradiography film. For immunoblotting experiments the data shown are representative of 2-3 independent experiments with similar results. Protein bands were, when necessary, digitally scanned and quantified with the ImageJ Gel Analyzer (NIH).
Transient transfection
For all transfections, except for the RT-PCR experiments, HEK293 or HeLa cells were seeded on 6-well plates (300, 000 cells per 962 mm2 well) one day prior to transfection. Cells were transfected using Lipofectamine (Invitrogen) according to the instructions of the manufacturer. For each culture well, 6 μl of Lipofectamine and a total of 1.5 μg total DNA, both diluted in 100 μl of Opti-MEM (Invitrogen), were used. In most transfections, 100 ng of pGFP plasmid was included to monitor transfection efficiency. Total DNA was normalized to 1.5 μg per well by adding an empty vector plasmid, which was pcDNA3 unless indicated otherwise. The transfection mixture was replaced with complete medium after 5 hours. For RT-PCR experiments, cells were seeded on 100-mm plates (2 million cells per plate) one day prior to transfection and they were transfected with a total DNA of 9 μg and 30 μL of Lipofectamine, both diluted in 600 μL of Opti-MEM.
Reporter gene assays
For luciferase assays, cells were transfected with the appropriate reporter gene plasmids. The amount of the pRL-TK plasmid used in the luciferase reporter gene assays was 20 ng per well of a six-well plate in all cases. Cells were harvested after the appropriate interval of culture and treatment and lysed in 200 μl of Passive Lysis Buffer (Promega). After two freeze-thaw cycles, the lysate was pre-cleared by centrifugation and 100 μl of the lysate was used for luciferase assay. Luciferase assays were carried out on the LUMIstar Galaxy microplate reader (BMG Labtech) using the requisite substrate: Stop & Glo Substrate for Renilla luciferase and Luciferase Assay Buffer II for firefly luciferase, both from Promega. For Renilla luciferase assays, normalization of the luciferase values was carried out against the total protein concentration measured on 10 μl of the cell lysates. In the other cases, normalization was performed using an internal control reporter gene. Fold induction was calculated by dividing the normalized value of the luciferase activity of the sample in question with the normalized value of the luciferase activity of the appropriate control. For the β-galactosidase assay, cells were transfected with the pSV40-β-galactosidase plasmid and were harvested after the appropriate duration of culture and treatment. The assay was conducted using the β-Gal Assay Kit (Invitrogen) according to the instructions provided by the manufacturer. The cells were resuspended in 100 μl of 1X β-galactosidase assay lysis buffer and after two freeze-thaw cycles and pre-clearing, the assay was performed on 20 μl of lysate. The results were normalized against the total protein concentration.
Semiquantitative RT-PCR
Following the appropriate treatment, cells were harvested and half of the cell lysates were used for each RNA extraction. Total RNA was extracted using the Promega SV total RNA Isolation System. DNAse digestion was performed on-column. The RNA was eluted in 100 μl RNAse-free water. The integrity of the RNA extract was confirmed by agarose gel electrophoresis. Synthesis and amplification of cDNA was carried out using the SuperScript III One-Step RT-PCR System with Platinum Taq DNA Polymerase (Invitrogen). The following forward and reverse primers, respectively, were used to amplify a portion of the Renilla luciferase mRNA: 5′-agagaaaggtgaagttcgtcgtcc-3′ and 5′-gagaactcgctcaacgaacgatt-3′. The following forward and reverse primers, respectively, were used to amplify a portion of the β-actin mRNA: 5′-cctcgcctttgccgatcc-3′ and 5′-ggatcttcatgaggtagtcagtc-3′. Amplification of Renilla luciferase and β-actin was conducted in duplex. The reactions were carried out in the following sequence: 1 cycle of cDNA synthesis for 30 min at 55°C; 1 cycle of denaturation for 2 mins at 94°C; 25 cycles of PCR amplification (94°C for 15 s; 50°C for 30 s and 68°C for 60 s); 1 cycle of final extension for 5 mins at 68°C. For each reaction, 100 ng of total RNA was used. The PCR products were electrophoresed on an agarose gel and the bands were quantified with the ImageJ Gel Analyzer.
Flow cytometry
For E1A flow cytometry, 300,000 cells were seeded per well on a 6-well plate one day prior to staining. On the second day, the cells were treated as indicated and harvested after the appropriate interval of time. The cells were washed in Stain Buffer (2% FBS in PBS) and then fixed by incubation for 20 minutes in Intracellular Fixation Buffer (eBioscience). After fixation, the cells were incubated for 1 hour in Ad type 5 E1A murine mAb diluted in Permeabilization Buffer (eBioscience) to a concentration of 2 μg/ml. Finally, the cells were incubated in FITC-conjugated goat anti-mouse IgG (Southern Biotech) diluted in Permeabilization Buffer to 2 μg/ml. The cells were then resuspended in 500 μl of Stain Buffer and flow cytometry was carried out. For GFP flow cytometry, cells were transfected with the pGFP plasmid, harvested after the appropriate treatment and then resuspended in 500 μl of PBS. Flow cytometry data was collected on a FACS Calibur Flow Cytometer (BD Biosciences) using CellQuest software (BD Biosciences). The data were analyzed using the WinMDI software (The Scripps Institute).
Statistical analysis
Statistical analysis was conducted using the two-tailed Student's t test to compare the arithmetic means of two non-paired samples with unequal variance. A p value of less than 0.05 was taken as indicating a statistically significant difference. A minimum sample size of 3 was employed to make statistical comparisons between two samples.
Results
PMA induces expression of Renilla luciferase from the pRL-TK plasmid in HEK293 cells
While investigating the effects of vitamin D on the activities of various cellular signal transduction pathways in HEK293 cells using luciferase reporter gene assays, we observed that the activity of the Renilla luciferase expressed from the pRL-TK plasmid used as an internal control was induced nearly five-fold when the cells transfected with the pRL-TK plasmid were treated with a combination of PMA and ionomycin (Fig. 2A). Further testing determined that this induction was caused solely by PMA and that ionomycin did not induce expression from the pRL-TK plasmid (Fig. 2B). For comparison, the proinflammatory cytokine TNFα was tested and it did not induce Renilla luciferase (Fig. 2A); when TNFα was used in combination treatment with PMA, there was a slight reduction of the induction, but the reduction was not statistically significant (Fig. 2A). To confirm that the effect of PMA is characteristic of the HEK293 cell line, we repeated the experiment in HEK293 cells obtained from another laboratory. As shown in Fig, 2C, the effect of PMA was reproducible in HEK293 cells obtained from the other source (HEK293*, second pair of columns). In addition, there was no induction of Renilla luciferase by PMA in HeLa cells, suggesting that the effect of PMA could be unique to HEK293 cells. A similar lack of induction of Renilla luciferase by PMA has been reported for HeLa cells previously by Daubeuf et al [28].
Fig. 2.

PMA induces expression of Renilla luciferase from the pRL-TK plasmid in HEK293 cells. Cells were transfected with the pRL-TK plasmid and treatment as indicated was added at 24 hours post-transfection. The cells were then harvested at the appropriate times and Renilla luciferase assays were carried out. A and B: HEK293 cells transfected with the pRL-TK plasmid were left non-treated or treated as shown for 8 hours; ionomycin was used at 1 μM.; ethanol and DMSO were added to a final concentration of 0.1% (v/v). Fold change was calculated by setting the normalized Renilla luciferase relative luminescence units (RLU) for the non-treated sample as 1. The results shown are the mean and SD of three independent experiments. C: HEK293 and HeLa cells (HEK293* indicates HEK293 cells obtained from another laboratory) transfected with the pRL-TK plasmid were left non-treated or treated with PMA for 8 hours. Fold change was calculated by setting the normalized Renilla luciferase RLU for the non-treated sample of the corresponding cell line as 1. The results shown are the mean and SD of three independent experiments. D: HEK293 cells transfected with the pRL-TK plasmid together with a GFP-expressing plasmid and an empty vector plasmid (either pcDNA3 or pUC19) as shown were left non-treated or treated with PMA for 8 hours. Fold change was calculated by setting the normalized Renilla luciferase RLU for the non-treated sample transfected with pGFP/pcDNA3 as 1. The results shown are the mean and SD of three independent experiments. E: HEK293 cells transfected with the pRL-TK plasmid were treated with PMA for various durations of time as shown. The results shown are the mean and SD of four independent experiments. F: HEK293 cells transfected with the pRL-TK plasmid were treated with various concentrations of PMA as shown for 8 hours. Fold change was calculated by setting the normalized Renilla luciferase RLU for the non-treated sample (0 ng/ml) as 1. The results shown are the mean and SD of three independent experiments.
Because co-transfected plasmids could affect the expression of another plasmid as previously reported for the β-galactosidase reporter gene [29, 30], we tested the effect of PMA when various combinations of plasmids were co-transfected. In our transfection experiments we typically co-transfected the pRL-TK plasmid with a pGFP plasmid (to monitor for transfection efficiency) and pCDNA3 (empty vector plasmid to normalize for the amount of DNA). When pRL-TK was transfected with a different empty vector plasmid (pUC19) together with a GFP plasmid or only with pCDNA3 there was no significant difference on the induction of Renilla luciferase by PMA ruling out any possible effect of the co-transfected plasmids pGFP or pcDNA3 (Fig. 2D). In time-course and dose-response studies, the induction of Renilla luciferase by PMA increased with increasing duration of treatment from 4 to 12 hours with a fixed dose of PMA (Fig. 2E) and also with increasing doses of PMA from 1 ng/ml to 50 ng/ml (Fig. 2F). Therefore, based on the findings shown above, we conclude that PMA induces the expression of Renilla luciferase from the pRL-TK plasmid in HEK293 cells and this property appears to be unique to HEK293 cells as it does not occur in HeLa cells.
The induction of Renilla luciferase expression by PMA is not due to a global increase in gene expression in HEK293 cells
To rule out the possibility that the induction of Renilla luciferase from the pRL-TK plasmid could be part of an enhanced general gene expression that could have been caused by PMA, we tested the effect of PMA on expression of exogenous genes from two viral promoters, namely the CMV promoter and the SV40 promoter. For the CMV promoter, we used GFP as the reporter gene placed downstream of the CMV promoter. HEK293 cells were transfected with the GFP plasmid and the cells were treated with PMA for 8 hours beginning at 24 hours after transfection. Flow cytometry showed that there was only a slight and statistically non-significant increase in the mean fluorescence intensity (MFI) of GFP induced by PMA (Fig. 3A upper right and lower left histograms and Fig. 3B) and also the percentage of GFP-positive cells was similar (Fig. 3A lower right histogram). For the SV40 promoter, we used the β-galactosidase gene placed downstream of the SV40 promoter as the reporter gene. As shown in Fig. 3C, PMA treatment did not produce a significant increase in the expression of the β-galactosidase gene from the SV40 promoter in HEK293 cells, as determined by the β-galactosidase assay. In addition, PMA treatment did not cause an increase in the expression of the endogenous housekeeping genes β-actin and β-tubulin in HEK293 cells (Fig. 3D). Other groups have reported the induction of some of these promoters in certain cell types; the discrepancy with our observations could be due to cell type-specific differences and/or due to major disparities in the conditions of cell treatment [31-33]. Our results indicate that the effect of PMA on the expression of Renilla luciferase from the HSV-1 TK promoter in HEK293 cells is likely to be a specific effect rather than being part of a globally enhanced gene expression.
Fig. 3.

The induction of Renilla luciferase expression by PMA is not due to a global increase in gene expression in HEK293 cells. A and B: HEK293 cells transfected with 200 ng of a plasmid expressing GFP under the CMV promoter were left untreated or treated with PMA for 8 hours beginning at 24 hours post-transfection. The cells were then harvested and flow cytometry was carried out with excitation and emission at 488 nm and 530 nm, respectively. Representative histograms of live cells gated based on light scatter profiles are shown in panel A. The upper left histogram is from cells transfected with an empty vector plasmid. The mean and SD of the MFI of the GFP signal in the GFP-positive cells from three independent experiments are shown in panel B. C: HEK293 cells transfected with 100 ng of a plasmid expressing β-galactosidase under the SV40 promoter were left non-treated or were treated with PMA for 8 hours. The cells were then harvested β-galactosidase assay was performed. The results show the mean and SD of three independent experiments. D: Overnight cultures of HEK293 cells were left non-treated or were treated with PMA for 8 hours. At the end of the treatment, the cells were harvested and immunoblotting was performed for β-actin and β-tubulin.
PMA induces Renilla luciferase expression by stimulating the HSV-1 TK promoter
To determine if the effect of PMA took place at the level of transcription, we first performed a semi-quantitative RT-PCR assay for Renilla luciferase. HEK293 cells transfected with the pRL-TK plasmid were treated either with PMA or, for comparison purposes, with TNFα for 4 hours beginning at 24 hours post-transfection. Then, total cellular RNA was isolated and subjected to duplex RT-PCR using primers specific for the Renilla luciferase mRNA and the human β-actin mRNA. As presented in Fig. 4A, there was an increased amount of Renilla luciferase mRNA in the cells treated with PMA as compared to the non-treated controls or those treated with TNFα thus localizing the effect of PMA at the level of transcription. PMA caused a nearly three-fold increase in the amounts of Renilla luciferase mRNA when the band intensities of Renilla luciferase were normalized to the corresponding β-actin band (Fig. 4B). To establish that the effect of PMA is related to the HSV-1 TK promoter and not to another DNA sequence within the pRL-TK plasmid, we cloned the HSV-1 TK promoter from the pRL-TK plasmid in front of the firefly luciferase gene in the pGL2 Basic plasmid which lacks any promoter/enhancer sequences and used the construct in luciferase assays. There was only minimal expression of firefly luciferase, even after PMA treatment, when HEK293 cells were transfected with the pGL2 Basic plasmid (Fig. 4C, first and second columns). When HEK293 cells were transfected with the pGL2 plasmid into which the HSV-1 TK promoter was cloned (pGL2-TK), there was an increased basal activity of firefly luciferase which could be attributed to the presence of a promoter element (Fig. 4C, third column). When this construct was transfected into HEK293 cells and the cells were then treated with PMA, there was a nearly three-fold induction of the firefly luciferase gene (Fig. 4C, fourth column). These results confirmed that the effect of PMA on Renilla luciferase takes place by enhancing transcription from the HSV-1 TK promoter.
Fig. 4.

PMA induces Renilla luciferase expression by stimulating the HSV-1 TK promoter. A and B: HEK293 cells grown on 100-mm plates were transfected with 120 ng of pRL-TK plasmid and they were then left non-treated or treated with PMA or TNFα for 5 hours beginning at 24 hours after transfection. Total RNA was isolated and duplex RT-PCR was carried out on 100 ng of total RNA using primers specific for Renilla luciferase and β-actin. The PCR products were resolved by agarose gel electrophoresis. A representative image of the bands is shown in panel A. The mean and SD of the band intensities of Renilla luciferase bands normalized to those of the corresponding β-actin bands from three independent experiments are shown in panel B; fold change was calculated by setting the value for the non-treated samples as 1. C: HEK293 cells transfected with 100 ng of either the pGL2 Basic or the pGL2-TK plasmid were left non-treated or treated with PMA for 8 hours beginning at 24 hours post-transfection. The cells were then harvested and firefly luciferase assays were performed. The results show the mean and SD of the normalized RLU values from three independent experiments.
The activation of JNK and ERK is necessary for the induction of the HSV-1 TK promoter by PMA
PMA affects various cellular processes largely by activating PKC which in turn activates the JNK, ERK and NF-κB signaling pathways [17, 20-25]. Therefore, we wanted to know if those pathways are required for the induction of the HSV-1 TK promoter by PMA in HEK293 cells. To determine the requirement of PKC, JNK and ERK, HEK293 cells transfected with the pRL-TK plasmid were pre-treated for 60 minutes with a specific inhibitor beginning at 23 hours post-transfection followed by PMA treatment for 8 hours, after which the cells were lysed and luciferase assays were performed. The inhibitors used were GF 109203X, JNK Inhibitor II and U0126 to inhibit, respectively, PKC, JNK and ERK. GF 109203X is a potent and selective PKC inhibitor [34]. JNK Inhibitor II (also known as SP600125) blocks the catalytic activities of the JNK enzymes thus inhibiting the activation of the JNK pathway without significantly affecting the NF-κB and ERK pathways [35]. U0126 blocks the phosphorylation of ERK1/2 thus inhibiting the activation of the ERK pathway and does not affect the stimulation of the NF-κB and JNK pathways [36-38]. When HEK293 cells were pre-treated with any one of those inhibitors there was a marked inhibition of the induction of Renilla luciferase by PMA, by 2.5 to 3.5 fold (Fig. 5A). Maximal inhibition was obtained with the PKC inhibitor GF 109203X.
Fig. 5.

The activation of JNK and ERK is necessary for the induction of the HSV-1 TK promoter by PMA. A: HEK293 cells were transfected with the pRL-TK plasmid and at 23 hours post-transfection, some of the samples were pre-treated in duplicates for 1 hour a shown with one of the following inhibitors: GF 109203X 2 μM, JNK Inhibitor II 5 μg/ml, or U0126 10 μM. At the end of the pre-treatment, one set of the duplicates was treated with PMA for 8 hours, while the other set was left without any additional treatment; then the cells were harvested and Renilla luciferase assays were performed. Fold change was calculated by setting the normalized Renilla luciferase RLU for the nonpre-treated, non-treated sample as 1. The results shown are the mean and SD of three independent experiments. B: HEK293 cells were transfected with the pRL-TK plasmid and some of the samples were cotransfected with 100 ng per well of the S32/36A IκBα plasmid. The cells were treated with PMA for 8 hours beginning at 24 hours post-transfection, after which the cells were harvested and Renilla luciferase assays were performed. Fold change was calculated by setting the normalized Renilla luciferase RLU for the non-IκBα-transfected, non-treated sample as 1. The results shown are the mean and SD of three independent experiments. C and D: Overnight cultures of HEK293 cells were taken and some of the cultures were pre-treated with JNK Inhibitor II (C) or U0126 (D) for 1 hour. At the end of the pre-treatment, the cells were left without additional treatment or were treated with PMA for 15 minutes. Then, the cells were harvested and immunoblotting for p-c-Jun (C) or p-ERK1/2 (D) was performed. E: HEK293 cells were transfected with 100 ng of the NF-κB-luciferase plasmid and, as an internal control, with 100 ng of the SV40-β-galactosidase plasmid. Some of the samples were also cotransfected with 100 ng per well of the S32/36A IκBα plasmid. At 24 hours post-transfection, the cells were either left without treatment or were treated with PMA for 8 hours after which the cells were harvested and firefly luciferase and β-galactosidase assays were performed. The firefly luciferase RLU values were normalized to the values of β-galactosidase assays. Fold change was calculated by setting the normalized firefly luciferase RLU for the non-treated samples as 1. The results show the mean and SD of three independent experiments.
To determine the requirement of the NF-κB pathway, HEK293 cells were cotransfected with the pRL-TK plasmid and a plasmid expressing the S32/36A mutant IκBα, which is a potent and specific inhibitor of the activation of NF-κB [27]. The cells were then treated with PMA for 8 hours beginning at 24 hours post-transfection, after which the cells were lysed and luciferase assays were carried out. Transfection of the S32/36A IκBα plasmid did not cause a significant inhibition of the induction of Renilla luciferase by PMA (Fig. 5B) indicating that the NF-κB pathway is not necessary for this effect of PMA. The efficacy of the inhibitors in blocking the activation of the PKC-mediated pathways by PMA is shown in Fig. 5C (for JNK), Fig 5D (for ERK) and Fig. 5E (for NF-κB),
Additional evidence for the requirement that each of the JNK and ERK pathways needs to be activated for PMA to induce the HSV-1 TK promoter was provided by the fact that treatment of HEK293 cells by the proinflammatory cytokine TNFα did not induce the HSV-1 TK promoter (Figs. 2A, 4A and 4B). In our experiments, TNFα induced the phosphorylation of ERK1/2 following the treatment of HEK293 cells for 15 minutes, albeit to a lesser degree than that attained after PMA treatment (data not shown). However, the induction of the AP-1 luciferase gene, a reporter of JNK activity, after treatment of the cells with TNFα for 8 hours was very low (data not shown). These observations indicate that the activation of ERK alone was not sufficient to induce the HSV-1 TK promoter. The results presented above demonstrate that the activation of PKC and the subsequent activation of each of the JNK and ERK pathways are required for the induction of the HSV-1 TK promoter by PMA in HEK293 cells.
The activation of JNK and ERK alone is not sufficient for the induction of the HSV-1 TK promoter by PMA
Having established that the activation of each of the JNK and ERK pathways is required for the induction of the HSV-1 TK promoter by PMA, then we wanted to know if the activation of these two pathways is solely sufficient for the induction of the HSV-1 TK promoter. Because PMA induced the HSV-1 TK promoter in HEK293 cells but not in HeLa cells (Fig. 2C), we examined if there is a difference in the activation of the two signaling pathways in these two cell lines following treatment with PMA. The cells were transfected with a firefly luciferase gene placed downstream of a promoter that has binding sites for AP-1 (AP-1 luciferase) and at 24 hours after transfection the cells were exposed to PMA treatment for 8 hours, after which luciferase assays were carried out. The degree of induction of JNK activity as determined by the AP-1 luciferase assay was more or less similar between the two cell types (Fig. 6A). We examined the activation of the ERK pathway by performing immunoblotting for phosphorylated ERK1/2 in cell lysates of PMA-treated cells. The results demonstrated that PMA induced the phosphorylation of ERK1/2 in both cell types even though the degree of activation was slightly higher in HeLa cells (Fig. 6B). According to these observations, PMA activated the JNK and ERK pathways in both HEK293 cells and HeLa cells. However, because of the fact that PMA failed to induce the HSV-1 TK promoter in HeLa cells (Fig. 2C), despite the activation of JNK and ERK, we conclude that activation of the two pathways in itself is not sufficient for the stimulation of the HSV-1 TK promoter. This suggested that the possibility of the existence of a cellular factor or process in HEK293 cells, but missing in HeLa cells, which enabled PMA to induce the HSV-1 TK promoter.
Fig. 6.

The activation of JNK and ERK is not solely sufficient for the induction of the HSV-1 TK promoter by PMA. A: HEK293 cells were transfected with 100 ng of AP-1 luciferase plasmid and, as an internal control, with 100 ng of SV40-β-galactosidase plasmid and HeLa cells were transfected with 100 ng of AP-1 luciferase plasmid and, as an internal control, with 20 ng of pRL-TK plasmid. At 24 hours post-transfection the cells were left non-treated or were treated with PMA for 8 hours. At the end of the treatment, the cells were harvested and firefly luciferase, Renilla luciferase and β-galactosidase assays were performed as appropriate. The firefly luciferase RLU values for HEK293 cells were normalized to those the β-galactosidase assay. The firefly luciferase RLU values for HeLa cells were normalized using the values for Renilla luciferase as a standard control. Fold change was calculated by setting the normalized RLU of the luciferase gene for the non-treated sample of the corresponding cell line to 1. The data shown are the mean and SD of three to five independent experiments. B: Overnight cultures of HEK293 and HeLa cells were either left non-treated or were treated with PMA for 15 minutes. At the end of the treatment the cells were harvested and immunoblotting for p-ERK1/2, ERK1 and ERK2 was carried out. C and D: Overnight cultures of HEK293 and HeLa cells were left non-treated or were treated with PMA for 8 hours. At the end of the treatment the cells were harvested and immunoblotting was performed for c-Jun, c-Fos and sp1.
PMA has been reported to increase the expression of the transcription factors c-Jun and c-Fos [33, 39, 40], which are two of the major components of the AP-1 transcription factor complex activated by JNK [41]. Therefore, we wanted to examine if PMA induced a differential expression of c-Jun or c-Fos between HEK293 cells and HeLa cells. We treated HEK293 and HeLa cells with PMA for 8 hours and then performed immunoblotting for c-Jun c-Fos. The results presented in Fig. 6C showed that the basal levels of c-Jun were much less in HeLa cells compared to that in HEK293 cells; however, PMA treatment increased the amounts of c-Jun protein in both cell types, even though the amount of c-Jun protein in HeLa cells was still lower than that in HEK293 cells. Because PMA was able to induce c-Jun expression in both HEK293 and HeLa cells (Fig. 6C), whereas it did not induce the HSV-1 TK promoter in HeLa cells (Fig. 2C), it is unlikely that the induction of c-Jun expression alone was responsible for the induction of the HSV-1 TK promoter in HEK293 cells, although it could contribute to the activation of the JNK pathway. The basal expression of c-Fos was similar between the two cell types and PMA treatment did not increase the levels of the protein. We also examined if PMA treatment alters the expression of sp1 protein, a transcription factor which has binding sites on the HSV-1 TK promoter [2, 4]. The results presented in Fig. 6D showed that the basal levels of sp1 were similar between the two cell types and PMA treatment for 8 hours did not alter the levels of the protein. Based on the observations presented above, we concluded that the activation of JNK and ERK was not in itself sufficient for the induction of the HSV-1 TK promoter; also the induction of the promoter by PMA could not be explained by changes in expression levels of the transcription factors c-Jun, c-Fos or sp1.
The adenoviral E1A proteins are involved in the induction of the HSV-1 TK promoter by PMA
Because one of the adenoviral E1A proteins has been reported to induce the expression of Renilla luciferase in Saos-2 human osteosarcoma cells [42] and because a portion of the Ad type 5 genome encompassing the E1A gene is integrated into the genome of HEK293 cells [43, 44], we wanted to examine if the presence E1A proteins is responsible for the PMA induction of the HSV-1 TK promoter in HEK293 cells. First, we performed immunoblotting for E1A to assess if PMA increased the expression of E1A in HEK293 cells, which could then have induced the HSV-1 TK promoter, since PMA has been reported to cause a transient increase in the levels of E1A mRNA [45-47]. Our results, presented in Fig. 7A and Fig. 7B, showed that PMA treatment for 6 hours slightly increased the levels of E1A proteins in HEK293 cells, by about 1.8-fold and similar treatment with TNFα increased E1A levels by about 1.3-fold; those changes were not statistically significant. Next, we quantitated the amounts of E1A proteins in HEK293 cells by performing intracellular staining followed by flow cytometry. The results demonstrated that PMA treatment for 8 hours slightly increased the expression of E1A (Fig. 7C). Based on these observations, we concluded that PMA did slightly increase the expression of E1A in HEK293 cells. But it is questionable if this small increase in E1A was responsible for the induction of the HSV-1 TK promoter, especially so because TNFα which caused some increase in E1A did not induce the HSV-1 TK promoter at all.
Fig. 7.
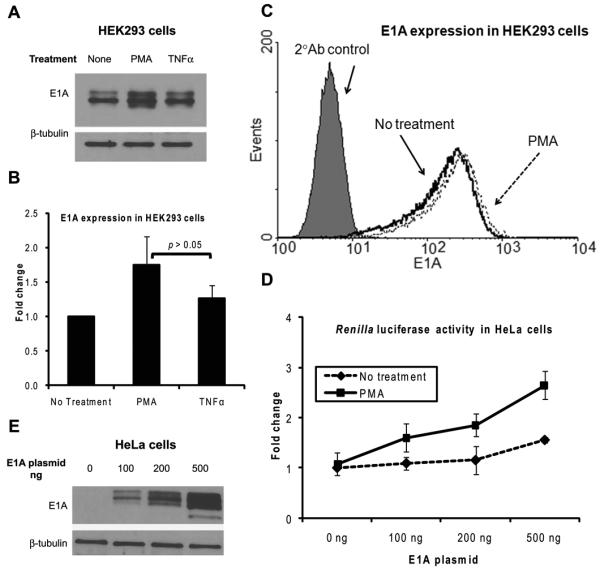
The adenoviral protein E1A is involved in the induction of the HSV-1 TK promoter by PMA. A, B and C: Overnight cultures of HEK293 cells were either left non-treated or were treated with PMA for 6 hours. At the end of the treatment the cells were harvested and subjected to either cell lysis followed by immunoblotting for E1A (A) and (B) or intracellular staining for E1A followed by flow cytometry (C). A representative immunoblot is shown in panel A; panel B shows mean and SD of the fold change of the immunoblot band intensities of E1A normalized to those of β-tubulin from three independent experiments with the normalized band intensity for the non-treated samples set as 1. Panel C shows an overlay of representative histograms. D and E: HeLa cells were co-transfected with the pRL-TK plasmid and various amounts of the pE1A-cDNA3 plasmid. At 24 hours post-transfection, the cells were either left non-treated or were treated with PMA for 8 hours, at the end of which the cells were harvested and Renilla luciferase assays (D) and immunoblotting for E1A (E) were carried out. Fold change for the Renilla luciferase assay was calculated by setting as 1 the normalized Renilla luciferase RLU value of the non-treated non-E1A-transfcted samples. The results in panel D show the mean and SD of three independent experiments. The immunoblot shown in panel E is from the non-treated samples.
So, we next examined if the presence of E1A confers cells the ability to respond to PMA stimulation by inducing the HSV-1 TK promoter. To answer this question we cotransfected an E1A-expressing plasmid and the pRL-TK plasmid into HeLa cells, which normally lack E1A, and then performed Renilla luciferase assays after PMA treatment for 8 hours. In HeLa cells transfected the E1A plasmid, the basal activity of Renilla luciferase was basically unchanged with 100 ng or 200 ng of E1A plasmid and was increased by 1.6-fold with 500 ng of E1A plasmid (Fig. 7D). When such cells were treated with PMA, the activity of Renilla luciferase was induced by 1.6-fold with 100 ng of E1A plasmid, and this increased to 2.6-fold with 500 ng of E1A plasmid. The degree of induction of the HSV-1 TK promoter obtained after PMA treatment in the transfected HeLa cells was lower than obtained in HEK293 cells. This could be due to differences in the degree of availability of transcription regulatory factors, such as c-Jun; for example, the degree of expression of c-Jun in HeLa cells was much less than that in HEK293 cells (Fig. 6C) and transfection of E1A plasmid did not increase the cellular levels of c-Jun in HeLa cells (data not shown). The degree of expression of E1A in transfected HeLa cells as determined by immunoblotting is shown in Fig. 7E. These observations established that the E1A proteins are involved in a cooperative manner with PMA in the induction of the HSV-1 TK promoter.
Discussion
Summary of findings
In this article, we provide empirical evidence that PMA induces the HSV-1 TK promoter in the presence of the adenoviral E1A proteins, whereas in the absence of E1A PMA caused little induction of the HSV-1 TK promoter. The induction occurred in a dose- and time-dependent manner and was not due to a global increase in gene expression caused by PMA. This induction required the activation of both the JNK and ERK pathways. However, the activation of the two pathways was not in itself sufficient to induce the promoter, as PMA activated the two pathways in HeLa cells that normally lack the E1A proteins but yet failed to induce the promoter in those cells. PMA did slightly increase the cellular levels of E1A proteins in HEK293 cells but that increase does not appear to be the sole reason for the induction of the HSV-1 TK promoter. Experimental evidence in support of this argument is provided by the fact that PMA treatment resulted in more than doubling of Renilla luciferase activity after only four hours of treatment. Furthermore, in HeLa cells transfected with the E1A plasmid, the increase in the amount of expressed E1A proteins was not paralleled by an increase in the activity of Renilla luciferase.
We believe that a synergistic effect between E1A and the pathways activated by PMA is responsible for the induction of the promoter. A similar synergism with transfected E1A has been reported before for the effect of PMA on the TNF promoter in Jurkat cells [48, 49], for the effect of PMA or LPS on the CMV major immediate early promoter in THP-1 cells and for the effect of PMA on the CMV promoter in Jurkat cells [50]. However, to the best of our knowledge, this is the first report of PMA inducing the HSV-1 TK promoter in the presence of the E1A proteins. An important contribution of these findings is that they provide an experimental system for investigating the biochemical and genetic interactions that take place when the signaling pathways activated by the tumor promoter PMA work in a synergistic manner with the oncogenic E1A proteins to induce a eukaryotic promoter. Proposed sites for these interactions are presented below following a brief discussion of PMA, E1A and the HSV-1 TK promoter.
PMA as an inducer of gene expression
PMA influences a number of cellular processes by acting chiefly through the activation of the PKC isozymes (a family of serine-threonine protein kinases that play a key role in carcinogenesis) which subsequently activate the NF-κB, JNK and ERK pathways [17, 20-25]. PMA treatment is known to induce the expression of certain cellular and viral genes and this induction appears to take place in a cell-specific manner, probably due to differences in the presence of particular PKC isozymes and/or other cellular proteins. These genes include the AP-1 proteins c-Fos and c-Jun [33, 39, 40], the prolactin gene [51], the metallothionein IIa gene [52], the SV40 promoter/enhancer in HepG2 cells [32] and the CMV immediate-early gene promoter and upstream elements and the murine sarcoma virus long terminal repeat (MSV LTR) in various cell lines [31].
E1A as a transactivator
The fact that PMA induced the HSV-1 TK promoter in HEK293 cells, but not in HeLa cells, led us to examine the possible roles of the E1A proteins, which are constitutively expressed in HEK293 cells. The HEK293 cell line contains a portion of the Ad5 DNA consisting of nucleotides 1 to 4344 integrated on chromosome 19 [43, 44]. This region of the Ad5 DNA codes for two early genes E1A (that codes for proteins that activate transcription and induce the cell to enter the S phase of the cell cycle) and E1B (which codes for anti-apoptotic proteins) and one delayed early gene pIX (a minor capsid protein) [43]. The E1A gene gives rise to two proteins due to alternative splicing; these two products, referred to as 12S (243 amino acid residues) and 13S (289 amino acid residues), share the same amino acid sequence except that the 13S protein has an additional segment composed of 46 amino acid residues in the middle of its sequence [53]. The 13S protein has three conserved regions (CR), namely CR1, CR2 and CR3, whereas the 12S protein lacks the CR3 domain. The E1A proteins do not directly bind to DNA but they increase the rate of transcription by interacting with a number of cellular general and specific transcription factors and other regulatory proteins such as the Rb-family proteins, the CRE-binding protein (CREB)-binding protein (CBP), the p300 histone acetylases, c-Jun and sp1 [53, 54].
Inducers of the HSV-1 TK promoter
TK activity, TK mRNA and TK protein exist at very low levels in quiescent cells; in dividing cells, it rapidly increases during late G1 and early S phases, reaches a maximum in mid S phase and declines during G2 and M phases [55]. The promoters of TK gene differ among various organisms but most have sp1 and CTF sites and some also have genuine or putative E2F sites. The HSV-1 TK gene (also known as UL23) codes for a protein with 376 amino acid residues that phosphorylates both purine and pyrimidine nucleosides and their analogs such as acyclovir [2]. TK is not essential for viral replication in replicating cell cultures since the dividing host cells provide TK [2, 56]. The enzyme is probably needed for viral replication in quiescent cells, such as neurons, to generate nucleoside triphosphate precursors to be used in the synthesis of DNA [2]. Methylation of cytosine residues around the CTF and sp1 sites inhibits basal transcription without blocking the binding of CTF and only slightly blocking the binding of sp1 [57]. The HSV-1 early gene product ICP4 activates TK expression by interacting with components of the general transcription machinery such as TFIIA [58] or by decreasing the affinity of binding of cellular negative transcription factors to TK promoter as proposed by Papavassiliou and Silverstein [59]. Other factors that activate the HSV-1 TK promoter include the varicella zoster virus protein p140 [60], HTLV-1 Tax protein [10], E1A protein [42], the androgen analog R1881 [11], and the transcription factors GATA4 and GATA6 [12].
Proposed mechanisms for the interactions between PMA, E1A and the HSV-1 TK promoter
We propose the following mechanisms to play various roles in the synergistic induction of the HSV-1 TK promoter by PMA and E1A (Fig. 8). The E1A proteins contain phosphorylation sites that are targets for a component of the ERK pathway and phosphorylation at those sites can enhance the transactivator functions of E1A, presumably by altering its interactions with other transcription factors [61]. JNK phosphorylates ATF-2 and c-Jun which then can activate TFIID which binds to the TATA box on the HSV-1 TK promoter [62].
Fig. 8.

A theoretical model showing the cooperation of PMA-activated signaling pathways and the E1A proteins in the induction of the HSV-1 TK promoter. Solid lines connecting two components show known interactions between those two components; broken lines indicate suspected interactions. The numbers below the horizontal line representing a portion of the HSV-1 TK gene indicate nucleotide positions in relation to the transcription start site which is at +1. The four known transcription factor binding sites on the HSV-1 TK promoter are shown by shaded boxes. The transcription factor E2F also appears to bind to the proximal GC box, even though not depicted in the diagram.
E1A could regulate the activity of the HSV-1 TK promoter by directly or indirectly interacting with the following transcription factors that are known or suspected to bind to the promoter: TFIID, sp1, CBF and E2F. E1A can directly bind to and activate TFIID; moreover, E1A can enhance the activities of ATF-2 and c-Jun (by increasing its transcription and phosphorylation) which then heterodimerize and activate TFIID [63-67]. E1A can interact with sp1 leading to transcriptional activation [62, 63, 68]. E1A could associate with CCAAT-binding factor (CBF, also known as nuclear factor-Y, NFY) [69] leading to transcriptional activation. It is not known if CBF binds to the CCAAT site on HSV-1 TK promoter, but it is plausible to suspect it does since it binds to similar sites in other genes and in the human TK gene [70, 71]. Shin et al demonstrated that E2F, more specifically E2F-1, binds to the HSV-1 TK promoter in the absence of consensus binding motifs and strongly activates the promoter; they showed that it binds to the GC elements apparently used by sp1 [72]. E1A induces the activation of E2F by promoting the dissociation of E2F from inhibitory complexes with the Rb family proteins; the released E2F then binds to promoter sites and stimulates the cell to enter the S phase, which includes the activation of the cellular TK gene [73]. The murine TK promoter has a genuine E2F site in close proximity to the sp1 site and there is a cooperative binding of sp1 and E2F to their respective binding sites leading to transcriptional activation of the murine TK [55]. In addition, there is a putative E2F site on the human TK promoter [55]. So, E1A could enhance expression from the HSV-1 TK promoter by inducing the release of E2F from inhibitory complexes. Therefore, as proposed in the model, the PMA-signaling pathways and E1A could interact in a number of possible ways to cause the induction of expression from the HSV-1 TK promoter.
Acknowledgements
Research in the laboratory of J.A.H. is supported by a grant from the Alliance for Lupus Research. A.S. was a Research Fellow supported by an NIAMS Rheumatology Training Grant, T32 AR050947.
Abbreviations
- HSV-1
herpes simplex virus type 1
- TK
thymidine kinase
- PMA
phorbol 12-myristate 13-acetate
- sp1
Specificity Protein-1
- CTF
CCAAT-transcription factor
- CBF
CCAAT-binding factor
- PKC
protein kinase C
- NF-κB
Nuclear Factor-kappaB
- JNK
Jun N-terminal Kinase
- ERK
Extracellular Signal-Regulated Kinase
- AP-1
Activator Protein-1
- HEK293
Human Embryonic Kidney-293
- GFP
green fluorescent protein.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McKnight SL, Kingsbury R. Science. 1982;217:316–324. doi: 10.1126/science.6283634. [DOI] [PubMed] [Google Scholar]
- 2.Roizman B, Knipe DM, Whitley RJ. In: Fields Virology. Knipe DM, Howley PM, editors. Lippincott Williams & Wilkins; Philadelphia: 2007. pp. 2502–2601. [Google Scholar]
- 3.Coen DM, Weinheimer SP, McKnight SL. Science. 1986;234:53–59. doi: 10.1126/science.3018926. [DOI] [PubMed] [Google Scholar]
- 4.Jones KA, Yamamoto KR, Tjian R. Cell. 1985;42:559–572. doi: 10.1016/0092-8674(85)90113-8. [DOI] [PubMed] [Google Scholar]
- 5.Perucho M, Wigler M. Cold Spring Harb. Symp. Quant. Biol. 1981;45(Pt 2):829–838. doi: 10.1101/sqb.1981.045.01.101. [DOI] [PubMed] [Google Scholar]
- 6.Zipser D, Lipsich L, Kwoh J. Proc. Natl. Acad. Sci. U. S. A. 1981;78:6276–6280. doi: 10.1073/pnas.78.10.6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McKnight SL, Gavis ER, Kingsbury R, Axel R. Cell. 1981;25:385–398. doi: 10.1016/0092-8674(81)90057-x. [DOI] [PubMed] [Google Scholar]
- 8.Kwun HJ, Yim SW, Lee DH, Jang KL. Mol. Cells. 1999;9:277–280. [PubMed] [Google Scholar]
- 9.Gelman IH, Silverstein S. Proc. Natl. Acad. Sci. U. S. A. 1985;82:5265–5269. doi: 10.1073/pnas.82.16.5265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kubota S, Mukudai Y, Hattori T, Eguchi T, Kondo S, Takigawa M. DNA Cell Biol. 2001;20:563–568. doi: 10.1089/104454901317094972. [DOI] [PubMed] [Google Scholar]
- 11.Mulholland DJ, Cox M, Read J, Rennie P, Nelson C. Prostate. 2004;59:115–119. doi: 10.1002/pros.20059. [DOI] [PubMed] [Google Scholar]
- 12.Ho CK, Strauss JF., III BMC. Biotechnol. 2004;4:10. doi: 10.1186/1472-6750-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan JN, Liu BH, Lee H, Shaw YT, Chiou ST, Chang WC, Lai MD. Biochem. Biophys. Res. Commun. 1993;191:662–668. doi: 10.1006/bbrc.1993.1268. [DOI] [PubMed] [Google Scholar]
- 14.Ibrahim NM, Marinovic AC, Price SR, Young LG, Frohlich O. Biotechniques. 2000;29:782–784. doi: 10.2144/00294st04. [DOI] [PubMed] [Google Scholar]
- 15.Everett LM, Crabb DW, Steroid J. Biochem. Mol. Biol. 1999;70:197–201. doi: 10.1016/s0960-0760(99)00109-0. [DOI] [PubMed] [Google Scholar]
- 16.Sherf BA, Navarro SL, Hannah RR, Wood KV. Promega Notes. 1996;57:2–8. [Google Scholar]
- 17.Blumberg PM, Jaken S, Konig B, Sharkey NA, Leach KL, Jeng AY, Yeh E. Biochem. Pharmacol. 1984;33:933–940. doi: 10.1016/0006-2952(84)90448-9. [DOI] [PubMed] [Google Scholar]
- 18.Van Duuren BL, Orris L. Cancer Res. 1965;25:1871–1875. [PubMed] [Google Scholar]
- 19.Hecker E. Cancer Res. 1968;28:2338–2349. [PubMed] [Google Scholar]
- 20.Blumberg PM. Cancer Res. 1988;48:1–8. [PubMed] [Google Scholar]
- 21.Castagna M, Takai Y, Kaibuchi K, Sano K, Kikkawa U, Nishizuka Y. J. Biol. Chem. 1982;257:7847–7851. [PubMed] [Google Scholar]
- 22.Parker PJ, Murray-Rust J. J. Cell Sci. 2004;117:131–132. doi: 10.1242/jcs.00982. [DOI] [PubMed] [Google Scholar]
- 23.Jaken S. Curr. Opin. Cell Biol. 1996;8:168–173. doi: 10.1016/s0955-0674(96)80062-7. [DOI] [PubMed] [Google Scholar]
- 24.Tan SL, Parker PJ. Biochem. J. 2003;376:545–552. doi: 10.1042/BJ20031406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ron D, Kazanietz MG. FASEB J. 1999;13:1658–1676. [PubMed] [Google Scholar]
- 26.Shifera AS, Friedman JM, Horwitz MS. Mol. Cell Biochem. 2008;310:181–190. doi: 10.1007/s11010-007-9679-z. [DOI] [PubMed] [Google Scholar]
- 27.Brockman JA, Scherer DC, McKinsey TA, Hall SM, Qi X, Lee WY, Ballard DW. Mol. Cell Biol. 1995;15:2809–2818. doi: 10.1128/mcb.15.5.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daubeuf S, Duvoix A, Wellman-Rousseau M, Diederich M, Visvikis A. Biochem. Biophys. Res. Commun. 2004;313:300–307. doi: 10.1016/j.bbrc.2003.11.121. [DOI] [PubMed] [Google Scholar]
- 29.Huszar T, Mucsi I, Terebessy T, Masszi A, Adamko S, Jeney C, Rosivall L. J. Biotechnol. 2001;88:251–258. doi: 10.1016/s0168-1656(01)00277-2. [DOI] [PubMed] [Google Scholar]
- 30.Farr A, Roman A. Nucleic Acids Res. 1992;20:920. doi: 10.1093/nar/20.4.920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lebkowski JS, McNally MA, Okarma TB, Lerch LB. Nucleic Acids Res. 1987;15:9043–9055. doi: 10.1093/nar/15.21.9043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Imbra RJ, Karin M. Nature. 1986;323:555–558. doi: 10.1038/323555a0. [DOI] [PubMed] [Google Scholar]
- 33.Greenberg ME, Ziff EB. Nature. 1984;311:433–438. doi: 10.1038/311433a0. [DOI] [PubMed] [Google Scholar]
- 34.Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F. J. Biol. Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- 35.Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. Proc. Natl. Acad. Sci. U. S. A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. J. Biol. Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 37.Davies SP, Reddy H, Caivano M, Cohen P. Biochem. J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsieh HL, Sun CC, Wu CB, Wu CY, Tung WH, Wang HH, Yang CM. J. Cell Biochem. 2008;103:1732–1746. doi: 10.1002/jcb.21563. [DOI] [PubMed] [Google Scholar]
- 39.Unlap T, Franklin CC, Wagner F, Kraft AS. Nucleic Acids Res. 1992;20:897–902. doi: 10.1093/nar/20.4.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adunyah SE, Unlap TM, Wagner F, Kraft AS. J. Biol. Chem. 1991;266:5670–5675. [PubMed] [Google Scholar]
- 41.Hess J, Angel P, Schorpp-Kistner M. J. Cell Sci. 2004;117:5965–5973. doi: 10.1242/jcs.01589. [DOI] [PubMed] [Google Scholar]
- 42.Thavathiru E, Das GM. Biotechniques. 2001;31:528–30, 532. doi: 10.2144/01313st06. [DOI] [PubMed] [Google Scholar]
- 43.Hehir KM, Armentano D, Cardoza LM, Choquette TL, Berthelette PB, White GA, Couture LA, Everton MB, Keegan J, Martin JM, Pratt DA, Smith MP, Smith AE, Wadsworth SC. J. Virol. 1996;70:8459–8467. doi: 10.1128/jvi.70.12.8459-8467.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Louis N, Evelegh C, Graham FL. Virology. 1997;233:423–429. doi: 10.1006/viro.1997.8597. [DOI] [PubMed] [Google Scholar]
- 45.Gai Q, James CB. In Vitro Cell Dev. Biol. Anim. 2001;37:465–470. doi: 10.1290/1071-2690(2001)037<0465:IOAROT>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 46.Carter TH, Milovanovic ZZ, Babiss LE, Fisher PB. Mol. Cell Biol. 1984;4:563–566. doi: 10.1128/mcb.4.3.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carter T, James C, Chan E, Greene B. Cancer Res. 1987;47:803–808. [PubMed] [Google Scholar]
- 48.Metcalf JP. Am. J. Physiol. 1996;270:L535–L540. doi: 10.1152/ajplung.1996.270.4.L535. [DOI] [PubMed] [Google Scholar]
- 49.Sanchez TA, Booth JL, Metcalf JP. Am. J. Physiol Lung Cell Mol. Physiol. 2002;283:L619–L627. doi: 10.1152/ajplung.00342.2001. [DOI] [PubMed] [Google Scholar]
- 50.Metcalf JP, Monick MM, Stinski MF, Hunninghake GW. Am. J. Respir. Cell Mol. Biol. 1994;10:448–452. doi: 10.1165/ajrcmb.10.4.8136160. [DOI] [PubMed] [Google Scholar]
- 51.Supowit SC, Potter E, Evans RM, Rosenfeld MG. Proc. Natl. Acad. Sci. U. S. A. 1984;81:2975–2979. doi: 10.1073/pnas.81.10.2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Imbra RJ, Karin M. Mol. Cell Biol. 1987;7:1358–1363. doi: 10.1128/mcb.7.4.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Berk AJ. In: Fields Virology. Knipe DM, Howley PM, editors. Lippincott Williams & Wilkins; Philadelphia: 2007. pp. 2355–2394. [Google Scholar]
- 54.Frisch SM, Mymryk JS. Nat. Rev. Mol. Cell Biol. 2002;3:441–452. doi: 10.1038/nrm827. [DOI] [PubMed] [Google Scholar]
- 55.Wintersberger E. Biochem. Soc. Trans. 1997;25:303–308. doi: 10.1042/bst0250303. [DOI] [PubMed] [Google Scholar]
- 56.Tenser RB. Intervirology. 1991;32:76–92. doi: 10.1159/000150188. [DOI] [PubMed] [Google Scholar]
- 57.Ben-Hattar J, Beard P, Jiricny J. Nucleic Acids Res. 1989;17:10179–10190. doi: 10.1093/nar/17.24.10179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zabierowski S, DeLuca NA. J. Virol. 2004;78:6162–6170. doi: 10.1128/JVI.78.12.6162-6170.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Papavassiliou AG, Silverstein SJ. J. Biol. Chem. 1990;265:9402–9412. [PubMed] [Google Scholar]
- 60.Cabirac GF, Mahalingam R, Wellish M, Gilden DH. Virus Res. 1990;15:57–68. doi: 10.1016/0168-1702(90)90013-2. [DOI] [PubMed] [Google Scholar]
- 61.Whalen SG, Marcellus RC, Whalen A, Ahn NG, Ricciardi RP, Branton PE. J. Virol. 1997;71:3545–3553. doi: 10.1128/jvi.71.5.3545-3553.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jones N. Curr. Top. Microbiol. Immunol. 1995;199(Pt 3):59–80. doi: 10.1007/978-3-642-79586-2_4. [DOI] [PubMed] [Google Scholar]
- 63.Liu F, Green MR. Nature. 1994;368:520–525. doi: 10.1038/368520a0. [DOI] [PubMed] [Google Scholar]
- 64.Brockmann D, Esche H. Curr. Top. Microbiol. Immunol. 2003;272:97–129. doi: 10.1007/978-3-662-05597-7_4. [DOI] [PubMed] [Google Scholar]
- 65.Zantema A, Van der Eb AJ. Curr. Top. Microbiol. Immunol. 1995;199(Pt 3):1–23. doi: 10.1007/978-3-642-79586-2_1. [DOI] [PubMed] [Google Scholar]
- 66.Hagmeyer BM, Angel P, van DH. Bioessays. 1995;17:621–629. doi: 10.1002/bies.950170708. [DOI] [PubMed] [Google Scholar]
- 67.Hagmeyer BM, Konig H, Herr I, Offringa R, Zantema A, van der EA, Herrlich P, Angel P. EMBO J. 1993;12:3559–3572. doi: 10.1002/j.1460-2075.1993.tb06030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Parks CL, Shenk T. J. Virol. 1997;71:9600–9607. doi: 10.1128/jvi.71.12.9600-9607.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lum LS, Hsu S, Vaewhongs M, Wu B. Mol. Cell Biol. 1992;12:2599–2605. doi: 10.1128/mcb.12.6.2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lum LS, Sultzman LA, Kaufman RJ, Linzer DI, Wu BJ. Mol. Cell Biol. 1990;10:6709–6717. doi: 10.1128/mcb.10.12.6709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chang ZF, Liu CJ. J. Biol. Chem. 1994;269:17893–17898. [PubMed] [Google Scholar]
- 72.Shin EK, Tevosian SG, Yee AS. J. Biol. Chem. 1996;271:12261–12268. doi: 10.1074/jbc.271.21.12261. [DOI] [PubMed] [Google Scholar]
- 73.Nevins JR. Curr. Top. Microbiol. Immunol. 1995;199Pt 3:25–32. doi: 10.1007/978-3-642-79586-2_2. [DOI] [PubMed] [Google Scholar]