

Abstract
By limiting cell cycle progression following detection of DNA damage, checkpoints are critical for cell survival and genome stability. Methylated DNA damage, when combined with inhibition of PARP activity, results in an ATR-dependent S phase delay of the cell cycle. Here, we demonstrate that another checkpoint kinase, ATM, also is involved in the DNA damage response following treatment with a sub-lethal concentration of MMS combined with the PARP inhibitor 4-AN. Both ATM and PARP activity are important for moderating cellular sensitivity to MMS. Loss of ATM activity, or that of its downstream effector Chk2, limited the duration of the S phase delay. The combination of MMS and 4-AN resulted in ATM and Chk2 phosphorylation and the time course of phosphorylation for both kinases correlated with the S phase delay. Chk2 phosphorylation was reduced in the absence of ATM activity. The Chk2 phosphorylation that remained in the absence of ATM appeared to be dependent on ATR and DNA-PK. The results demonstrate that, following initiation of base excision repair and inhibition of PARP activity, ATM activation is critical for preventing the cell from progressing through S phase, and for protection against MMS-induced cytotoxicity.
Keywords: PARP-1, PARP inhibitor, Methyl methanesulfonate, ATM, Chk2, Cell cycle
1. Introduction
DNA damage arising from spontaneous base loss or genotoxic agents that modify bases (reviewed in [1]) is repaired by base excision repair (BER). Both single-nucleotide and long-patch BER pathways have been identified. In the preferred single-nucleotide repair pathway, only one base is replaced, whereas two or more bases are replaced in long-patch repair. Single-nucleotide repair can be initiated when the damaged base is removed by a damage-specific monofunctional DNA glycosylase to create an abasic site. Cleavage of the abasic site by apurinic/apyrimidinic (AP) endonuclease creates a gap with a 5′-deoxyribosephosphate (dRP) terminus. DNA polymerase β (pol β) performs gap-filling synthesis, the dRP group is removed by the dRP lyase activity of pol β, and the nick is sealed by a DNA ligase.
In the absence of pol β activity, cells exhibit an increased sensitivity to the base methylating agent methyl methanesulfonate (MMS) that has been attributed to accumulation of intermediates of repair (e.g. the dRP group) [2-4]. The MMS hypersensitivity phenotype observed in pol β null mouse embryonic fibroblasts can be reversed by complementation with a pol β mutant lacking polymerase activity but still retaining the dRP lyase function [3]. The DNA damage surveillance protein poly(ADP-ribose) polymerase-1 (PARP-1) is known to bind gaps and nicks in DNA, including the dRP-containing intermediate of BER [5], and becomes activated. PARP-1 activation is important for recruitment of BER proteins to sites of BER and poly(ADP-ribosyl)ation appears to also have a role in modifying chromatin structure (reviewed in [6]). Cells treated with the PARP inhibitor 4-amino-1,8-naphthalimide (4-AN) are extremely sensitized to MMS [5,7], indicating that activation of PARP has a protective function against cytotoxic BER intermediates.
Investigations focused on the impact of PARP inhibition are proving to be an important tool in understanding DNA damage responses and cell cycle checkpoint pathways. PARP inhibition by 4-AN will prevent PARP autoribosylation. Under these conditions PARP remains bound to DNA [8], hindering access of repair proteins and preventing completion of BER [9,10] (Aya Masaoka, personal communication). We have shown that PARP inhibition in cells treated with a sub-lethal dose of MMS results in an ATR and Chk1-dependent accumulation of S phase cells [11,12]. Generally, ATR is activated in response to replication fork stalling and single-strand breaks (SSBs), and signals through Chk1 kinase to slow S phase (reviewed in [13]). One explanation for our observations is that persistence of PARP-bound DNA results in replication fork stalling, and thus S phase delay may be due to the persistence of SSBs and/or the inability of PARP-1 to dissociate from the DNA lesion (reviewed in [6]). Eventually, cells treated with MMS and 4-AN progress through S phase and accumulate in G2/M [11]. That PARP-1 -/- cells treated with MMS and 4-AN bypass the S phase delay and arrest directly in G2/M [12] suggests that inactivated PARP is a critical component of this model.
One important role for PARP activation is in preventing the progression of SSB damage to double-strand breaks (DSBs). Increased amounts of γ-H2A.X, an early marker for DSBs, are observed following oxidative damage in cells with reduced levels of PARP-1 protein [14], or in cells treated with the combination of MMS and 4-AN [11]. Although ATR was required for the S phase delay and the phosphorylation of Chk1 in response to treatment with MMS and 4-AN, the increase in γ-H2A.X was only partially diminished when ATR was inhibited (unpublished observation). This suggested that additional checkpoint kinases are activated in response to MMS and 4-AN. One of these kinases, ATM, has been shown to be involved in cell cycle arrest (reviewed in [15]) and phosphorylation of H2A.X [16] in response to ionizing radiation (IR)-induced DSBs. It is known that replication forks stalled at SSBs are capable of collapsing and forming DSBs (reviewed in [13]). In the absence of the repair protein XRCC1, treatment of cells with MMS alone results in an S phase delay that requires ATR and ATM [17]. XRCC1 is thought to serve as a SSB sensor and/or scaffold protein for the assembly of BER factors at sites of damage (reviewed in [18]). Presumably, it is the accumulation of BER intermediates as a result of inefficient repair that eventually leads to fork stalling. Similarly, inhibition of PARP activity also leads to an accumulation of BER intermediates and triggers an S phase delay. However, a full understanding of the mechanism behind this PARP inhibition-induced cell cycle delay in the context of base damage remains unclear. Considering the emerging use of combination chemotherapy with PARP inhibitors and DNA methylating agents [19] (reviewed in [6,20]), understanding the mechanisms underlying this treatment strategy is important. We now determine whether ATM and its downstream effector kinase Chk2 are activated in response to MMS-induced DNA damage combined with inhibition of PARP.
2. Materials and methods
2.1. Cell lines
ATRkd cells are SV40-transformed human fibroblasts expressing an ATR kinase dead (kd) mutant under the control of a Tetracycline (Tet) inducible promoter [21], and were obtained from Fred Hutchinson Cancer Research Center. The AT cells are SV40-transformed human fibroblasts derived from an Ataxia-telangiectasia (AT) patient. The AT-complemented (AT-comp) cell line has an episomally maintained plasmid encoding a wild-type copy of the ATM gene [22]. As a control, the AT cell line contains the plasmid vector used in the complementation. Both AT cell variants were obtained from Coriell Cell Repository.
2.2. Cytotoxicity studies
ATRkd, AT or AT-comp cells were seeded at a density of 20,000 cells per well in six-well plates. The next day, cells were treated for 1 h with a range of MMS (Sigma-Aldrich) concentrations, plus 4-AN (10 μM) (Acros) or ATM kinase inhibitor (2-morpholin-4-yl-6-thianthren-1-yl-pyran-4-one) (ATMi) (25 μM) (EMD Biosciences) [23] as indicated. Cells were then washed with Hanks' balanced salt solution (HBSS) (HyClone), and incubated with control medium, or medium containing 4-AN and/or ATMi at the concentrations indicated above. Incubations were continued for a total of 24 h for ATMi and 48 h for 4-AN. The medium was then removed, cells were washed with HBSS, and further incubated in control medium for an additional 7-10 days until untreated control cells were 80% confluent. Cells (triplicate wells for each drug concentration) were counted by a cell lysis procedure [24], and results were expressed as the number of cells in drug-treated wells relative to control wells (% control growth).
2.3. Flow cytometric cell cycle analysis
Cells were seeded in 100-mm dishes at 3.3 ×106 cells per plate. The following day cells were treated for 1 h with MMS (0.5 mM for ATRkd cells, and 0.1 mM for AT and AT-comp cells), 4-AN (10 μM), ATMi (25 μM), caffeine (1 mM) (Sigma-Aldrich), Chk2 inhibitor II (2-(4-(4-chlorophenoxy)phenyl)-1H-benzimidazole-5-carboxamide) (1 μM) (Sigma-Aldrich) [25] and/or Chk2 inhibitor (5-(2-Amino-5-oxo-1,5-dihydroimidazol-4-ylidine)-3,4,5,10-2H-azepino[3,4-b]indol-1-one) (0.2 μM) (EMD Biosciences) as indicated. After exposure to MMS, cells were washed with HBSS and incubated with fresh medium alone or medium containing 4-AN, ATMi, caffeine and/or Chk2 inhibitor at the concentrations indicated above for up to 24 h. Before the times specified (2-24 h) after the beginning of exposure to MMS, 10 μM BrdUrd (Sigma-Aldrich) was added to the dishes for 2 h (ATRkd cells) or 1 h (AT and AT-comp cells) to pulse-label the cells. Cells were washed with phosphate-buffered saline (PBS), harvested by trypsinization, and then washed a second time with PBS. The cell pellet obtained after centrifugation was resuspended in 100 μl of cold PBS, and the cells were dropped slowly into 70% ethanol and allowed to fix at 4°C overnight. The samples were washed, suspended in 2 N HCl containing 0.5% Triton X-100 and incubated for 30 min at room temperature to denature DNA. The cell samples were pelleted, resuspended in 0.1 M sodium borate, pH 8.5, to neutralize the acid, and then washed with PBS. Cells were then incubated at 4°C overnight with 20 μl of anti-BrdUrd fluorescein isothiocyanate (FITC)-conjugated antibody (BD Biosciences) in PBS containing 0.5% Tween 20 and 1% bovine serum albumin (BSA) and 5 μl of 10 mg/ml RNase (Sigma) stock solution. The following day the cells were pelleted, washed with PBS, and resuspended in 1 ml of PBS containing 5 μg/ml of PI (Sigma-Aldrich). The samples were analyzed by flow cytometry using ModFit LT software (Verity Software House, Inc.). Data represent the mean of at least three independent experiments.
2.4. Western blot analysis of ATM and phosphorylated Chk2
For Chk2 analysis, 3.3 × 106 cells were plated on 100-mm dishes one day prior to treatment with MMS and 4-AN. For ATM immunoprecipitation experiments 7.5 × 106 cells were plated on 150-mm dishes. In some experiments, the ATR kinase dead mutant protein was induced in ATRkd cells by treatment with doxycycline (Dox) (1 μg/ml) for 48 h prior to any other treatments. One day after plating, or following the induction of the ATRkd mutant, cells were treated for 1 h with MMS (0.5 mM for ATRkd cells, 0.1 mM for AT and AT-comp cells), and with 4-AN (10 μM), Dox (1 μg/ml), ATMi (10 μM), DNA-PK inhibitor NU7026 (10 μM) (Sigma-Aldrich) or caffeine (2 mM) as indicated. Cells were then washed once with HBSS, and incubated with medium alone or in medium containing inhibitors at the concentration indicated above for up to a total of 20 h. For IR exposure, cells were plated as described above one day prior to exposure. Before irradiation, cold medium was added to the cells and ice-cold cells were irradiated in a 137Cs irradiator (8 Gy for ATRkd, or 15 Gy for AT and AT-comp cells). Afterwards, cells were returned to warm medium and incubated for 2 h.
For preparation of whole cell extracts for analysis of Chk1 and Chk2, cell monolayers were rinsed with PBS, scraped, collected into PBS and centrifuged. Cell pellets were resuspended in Buffer I (10 mM Tris-HCl, pH 7.8, 0.2 M KCl, 25 mM NaF and complete protease inhibitor cocktail (Roche)). An equal volume of Buffer II (10 mM Tris-HCl, pH 7.8, 0.2 M KCl, 25 mM NaF, 2 mM EDTA, 40% glycerol, 0.2% NP-40 and 2 mM dithiothreitol (DTT)) was then added as described previously [26]. The suspension was rotated for 1 h at 4°C and extracts were clarified by centrifugation in a microcentrifuge at full speed for 10 min at 4°C. Total protein concentration of extracts was determined by the Bradford assay using BSA as protein standard.
Whole cell extracts for analysis of ATM were prepared by washing cells twice in PBS, suspending in lysis buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 25 mM NaF, 0.1 mM sodium orthovanadate, 0.2% Triton X-100, 0.3% NP-40 and complete protease inhibitors (Roche) and incubation for 30 min on ice. Extracts were then clarified by centrifugation in a microcentrifuge at full speed for 30 min at 4°C. [27].
Extract protein samples for Chk1 and Chk2 analysis (20 μg) were loaded onto 4-12% Bis-Tris NuPAGE gels (Invitrogen) and electrophoresed in MOPS running buffer (50 mM MOPS, 50 mM Tris base, 0.1% SDS and 1 mM EDTA). Proteins were transferred to nitrocellulose filters at 25 V overnight. Following transfer, filters were blocked in 5% nonfat dry milk in PBS with 0.1% Tween 20 (PBST) at room temperature for 1 h. Filters were then incubated overnight at 4°C with primary antibody, either rabbit polyclonal antibody directed against Chk2 phosphorylated at threonine 68 (T68) (1:500 dilution in PBST, Santa Cruz Biotechnology or 1:1,000 dilution in 1% nonfat dry milk in PBST, Cell Signaling), Chk1 phosphorylated at serine 345 (S345) (1:1,000 dilution in 5% BSA (PBST), Cell Signaling), rabbit antibody directed against bulk Chk2 (1:500 dilution, Santa Cruz Biotechnology), mouse antibody directed against bulk Chk1 (1:1,000 dilution, Santa Cruz Biotechnology) or mouse monoclonal antibody directed against tubulin (1:20,000 dilution, Sigma-Aldrich). The following day filters were blotted with anti-rabbit or anti-mouse IgG-horseradish peroxidase (HRP) conjugated secondary antibody (1:10,000-50,000 dilution, Bio-Rad) and visualized using Super Signal chemiluminescent detection (Thermo Scientific) according to the manufacturer's instructions. In experiments where blots were probed with multiple primary antibodies, they were stripped for 10 min at 37° C and 5 min at room temperature in Restore Western Blot Stripping Buffer (Thermo Scientific), then washed twice in PBST.
For western blot analysis of ATM from extracts, samples containing 100 μg of total extract protein each were run on 6% Tris-glycine SDS-PAGE. Proteins were transferred to nitrocellulose at 25 V overnight. Following transfer, the filter was blocked in 5 % nonfat dry milk in Tris-buffered saline with 0.1% Tween 20 (TBST). The filter was then incubated overnight at 4°C with mouse monoclonal antibody directed against ATM (1:500 dilution, Genetex). The following day after blotting with anti-mouse IgG-HRP conjugated secondary antibody (1:10,000 dilution, Bio-Rad), protein was visualized by chemiluminescent detection.
For Flag immunoprecipitation of ATM, monolayers were rinsed with PBS, scraped, collected into PBS and centrifuged. Cells were suspended in 100 μl extraction buffer (20 mM HEPES-KOH, pH 7.9, 25% glycerol, 450 mM NaCl, 50 mM NaF, 0.2 mM EDTA, 1μg/ml leupeptin, 1μg/ml aprotinin, 1μg/ml pepstatin, 0.25 mg/ml AEBSF, and 0.5 mM DTT), snap frozen in liquid nitrogen and thawed on ice. Freeze/thaw cycles were repeated two more times. Extracts were clarified by centrifugation in a microcentrifuge at full speed for 10 min at 4°C. Extracts were then supplemented with NP-40 to 0.1% and mixed with 10 μl Flag M2-agarose (Sigma-Aldrich) overnight at 4°C. The resin was then washed with 1 ml high salt wash buffer (20 mM HEPES-KOH, pH 7.9, 10% glycerol, 450 mM NaCl, 50 mM NaF, 0.2 mM EDTA, 0.1% NP-40, 1μg/ml leupeptin, 1μg/ml aprotinin, 1μg/ml pepstatin, 0.25 mg/ml AEBSF, and 0.5 mM DTT). The resin was collected by centrifugation and the wash buffer was discarded. This high salt wash was repeated two more times. The resin was then washed in low salt buffer (20 mM HEPES-KOH, pH 7.9, 10% glycerol, 100 mM NaCl, 50 mM NaF, 0.2 mM EDTA, 0.1% NP-40, 1μg/ml leupeptin, 1μg/ml aprotinin, 1μg/ml pepstatin, 0.25 mg/ml AEBSF, and 0.5 mM DTT) twice in the same manner.
Samples were run on SDS-PAGE and western blotted as described above. Filters were incubated overnight at 4°C with mouse monoclonal antibody directed against ATM phosphorylated at serine 1981 (S1981; 1:1,000 dilution, Santa Cruz Biotechnology). After blotting with anti-mouse IgG-HRP secondary antibody (1:10,000 dilution), chemiluminescent detection was performed. Filters were then stripped using Restore Western Blot Stripping Buffer as described above, and after washing twice in PBST, filters were incubated overnight at 4°C with rabbit polyclonal antibody directed against bulk ATM (1:500 dilution, Santa Cruz Biotechnology). Following blotting with anti-rabbit IgG-HRP secondary antibody (1:10,000 dilution), protein was detected by chemiluminescence.
3. Results
Previous observations indicate that, following inhibition of PARP activity combined with MMS-mediated base damage, cells accumulate in S phase in an ATR-dependent manner [11]. Increased levels of γ-H2A.X were detected suggesting that treatment resulted in formation of DSBs. Since ATM is known to phosphorylate H2A.X and to mediate cell cycle responses to DSB DNA damage, we propose an as yet unknown role for ATM and its downstream effectors in this cell cycle checkpoint response to treatment with MMS and 4-AN.
3.1 Cellular sensitivity to MMS and PARP inhibition is enhanced in the absence of ATM
It is well known that cells have increased sensitivity to MMS when it is combined with the PARP inhibitor 4-AN, thus highlighting the importance of PARP activity in the response to MMS-mediated DNA damage. If inhibition of PARP activity following base damage leads to the formation of DSBs, then the disruption of ATM-mediated signaling would impact the cellular sensitivity to the MMS and PARP inhibitor combination. To address this possibility, MMS cell survival assays were conducted using cells treated with an inhibitor of ATM or cells that are defective in ATM expression.
Inhibition of ATM activity in ATRkd cells without Dox induction that have wild-type levels of ATR, as well as ATM (Fig. 1B), resulted in sensitization to MMS (Fig. 1A, compare diamonds (+ATMi) to circles). This increased MMS sensitivity in the presence of the ATM inhibitor alone suggests a role for ATM in response to methylated DNA damage. The combination of PARP and ATM inhibitors further enhanced MMS sensitivity (Fig. 1A, triangles). These results indicate that both PARP and ATM activities are critical for protection against the cytotoxicity of methylated DNA damage, in addition to the importance of ATM in mediating the DSB response.
Fig. 1.

Increased cellular sensitivity to MMS in the absence of ATM and PARP activity. (A) Growth inhibition of ATRkd cells (without Dox induction) treated with MMS alone for 1 h (control, circles), MMS and ATMi (10 μM for 24 h) (+ATMi, diamonds), MMS and 4-AN (10 μM for 48 h) (+4-AN, squares) or MMS with ATMi and 4-AN (+4-AN+ATMi, triangles). (B) Western blot analysis of extracts from AT cells transfected with complementation vector without cDNA insert (AT), AT cells complemented with a wild-type ATM cDNA (AT-comp) and ATRkd cells. (C) Growth inhibition of the AT-comp cells (left panel, closed symbols) or AT cells (right panel, open symbols) when treated with MMS alone (control, circles), MMS and ATMi (+ATMi, diamonds), MMS and 4-AN (+4-AN, squares) or MMS with ATMi and 4-AN (+4-AN+ATMi, triangles) as in (A). Experiments were conducted as described in ‘Materials and Methods.’ Results represent mean ± S.E.M of three independent experiments.
To more directly measure the importance of ATM in the cellular response to PARP inhibition during DNA damage, we performed similar cell survival experiments using a human fibroblast cell line (AT) that carries an Ataxia-telangiectasia mutation, and a complemented cell line expressing the wild-type ATM gene (AT-comp) [22]. As shown in Fig. 1B, ATM is undetectable in the AT cells (lane 1), while complementation with wild-type ATM gene yields levels of ATM protein comparable to levels detected in the positive control ATRkd cell line (compare lanes 2 and 3).
Consistent with observations made above with ATRkd cells, inhibition of either ATM or PARP activities increased the sensitivity of the AT-comp cells to MMS (Fig. 1C, left panel, compare circles with diamonds (+ATMi) or squares (+4-AN)). Cells lacking ATM exhibited sensitivity to MMS comparable to the sensitivity observed in the AT-comp cells treated with ATMi (Fig. 1C, compare open circles, right panel with closed diamonds, left panel). This suggests that ATM plays a role in the response to base damage created by MMS. A similar hypersensitivity to MMS in AT cells has been described previously [28]. Both panels in Fig. 1C represent results obtained from the same set of experiments. Treatment with ATMi had no effect on sensitivity to MMS in AT cells lacking ATM (right panel, compare circles with diamonds (+ATMi)). The increase in MMS sensitivity in AT cells treated with 4-AN was nearly identical to the sensitivity observed in the AT-comp cells treated with 4-AN and ATMi (compare open squares, right panel with closed triangles, left panel) confirming the specificity of the ATMi. This result highlights two important concepts. It suggests that inefficient BER as a result of inhibition of PARP activity promotes accumulation of DSBs and additionally demonstrates the importance of ATM in minimizing the cytotoxic effects of impaired BER and DSB production.
3.2 Role of ATM in checkpoint response to PARP inhibition during BER
In order to investigate the role of the ATM checkpoint response in PARP inhibited cells with MMS-mediated base damage, we compared the accumulation of ATM-deficient and -proficient cells in S and G2/M phases of the cell cycle. In untreated cell populations, the proportion in S phase ranged from 40-50% (Fig. 2, left panel, diamonds), while in cell populations treated with MMS and 4-AN as many as 70% were in S phase (squares and circles). AT-comp cells treated with MMS and 4-AN accumulate in S phase between 4 and 20 h after treatment, after which point the accumulation declines (closed squares). In the presence of ATMi, accumulation of S phase cells occurs at around 5 h in the same manner as in cells with active ATM. However, the proportion of S phase cells begins to decline at a much earlier time, between 8-16 h (closed circles). Similarly, AT-deficient cells treated with MMS and 4-AN exhibit a peak in S phase accumulation around 8 h after treatment that begins to decline at times between 8-16 h (open squares). In the presence of caffeine, which is known to inhibit the phosphorylation of substrates and checkpoint pathways that depend on ATM or ATR activity [11,29-33], both AT and AT-comp cells failed to exhibit any substantial accumulation in S phase over the same time period (data not shown). Our results suggest that ATR-dependent pathways are important for initiation of the S phase delay [11] while ATM is required to maintain the duration of this delay. Interestingly, it has been observed that both ATM and ATR are required for maintaining an S phase delay in XRCC1-deficient cells 24 h after treatment with MMS [17].
Fig. 2.

Role of ATM in the cell cycle responses to PARP inhibition and MMS-mediated DNA damage. S phase (left panel) and G2/M (right panel) accumulation of AT cells and AT-comp cells either mock-treated or treated with MMS (0.1 mM for 1 h) and 4-AN (10 μM continuously), or AT-comp cells treated additionally with ATMi (25 μM continuously). Results represent mean ± S.E.M of three independent experiments. Experiments were conducted as described in ‘Materials and Methods’.
As the proportion of cells in S phase decreased at later times, there is a parallel accumulation in G2/M (Fig. 2, right panel). The proportion of untreated cells in G2/M is maintained in the range of 20-30% throughout the experiment (diamonds). The fraction of MMS and 4-AN treated cells in G2/M reaches 60-70% at 24 h (squares and circles). For AT-comp cells, this accumulation in G2/M occurs between 16-20 h and is maintained until at least 24 h after the start of treatment (closed squares). When ATM activity is inhibited, approximately 35% of cells have accumulated in G2/M by 16 h (closed circles), while the majority remain in S phase at 16 h where ATM is active (Fig. 2, left panel, closed squares). AT cells exhibit a similar earlier onset of G2/M accumulation (Fig. 2, right panel, open squares) corresponding with their earlier decline in S phase accumulation (compare Fig. 2, right and left panel open squares). Accumulation of AT cells in S phase showed no change when treated with ATMi (data not shown), indicating that the effects of ATMi on the cell cycle in the AT-comp cells are occurring through a specific inhibition of ATM. Interestingly, G2/M accumulation in cells treated with MMS and 4-AN reached the same level at 24 h regardless of whether ATM activity is present. This indicates that while ATM is important for sustaining the S phase delay in response to MMS and 4-AN treatment, it is not necessary for accumulation of G2/M cells. It is possible that other checkpoint kinases redundant with ATM allow ATM-deficient cells to reach the same level of G2/M accumulation as ATM-proficient cells. Future studies will determine whether the G2/M accumulation involves activation of other damage response pathways.
The dependence of the duration of the S phase accumulation on ATM activity led us to question whether Chk2, a downstream effector of ATM-mediated signaling, also plays any role. To determine the contribution of Chk2, we compared the accumulation of S phase cells treated with MMS and 4-AN in the absence and presence of an inhibitor of Chk2 kinase activity [25]. As shown in Fig. 3A, left panel, treatment of AT-comp cells with Chk2 inhibitor II (white bars) results in a more rapid decline in the number of cells in S phase between 16 and 24 h in comparison to cells without inhibitor (gray bars). This disappearance of S phase cells was accompanied by a corresponding earlier accumulation in G2/M (Fig. 3A, compare left and right panels). Similar results were observed with an alternate Chk2 inhibitor (5-(2-amino-5-oxo-1,5-dihydroimidazol-4-ylidine)-3,4,5,10-2H-azepino[3,4-b]indol-1-one) (data not shown). These data indicate that Chk2, like ATM, is important for maintaining the S phase delay initiated by inhibiting PARP during BER. Interestingly, in AT cells, the Chk2 inhibitor resulted in a still earlier disappearance of S phase cells (between 8 and 16 h) (Fig. 3B, left panel). This further reduction in the duration of the S phase delay in AT cells under conditions of Chk2 inhibition suggests that checkpoint kinases in addition to ATM may contribute to the activation of Chk2 and sustaining the S phase delay.
Fig. 3.
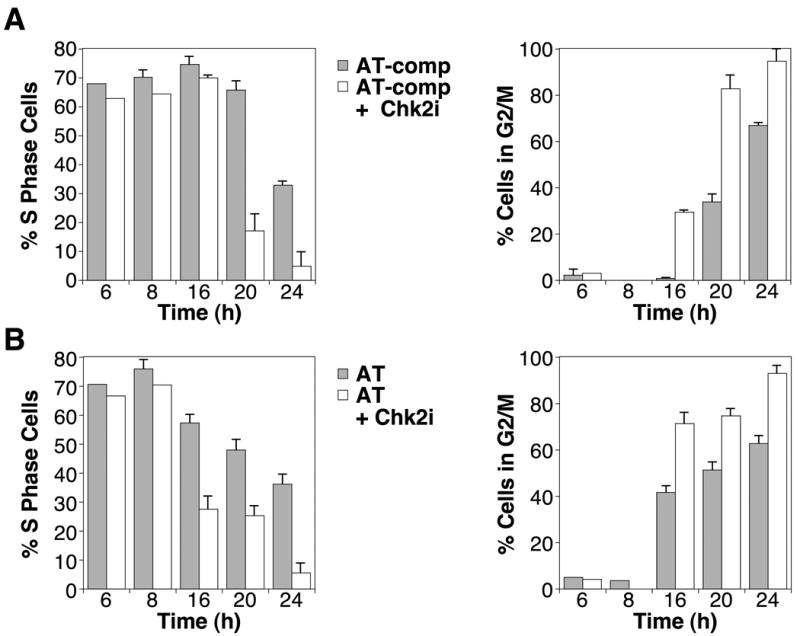
Role of Chk2 in the cell cycle responses to PARP inhibition and MMS-mediated DNA damage. (A) S phase accumulation (left panel) and G2/M accumulation (right panel) of AT-comp cells after treatment with MMS (0.1 mM for 1 h) + 4-AN (10 μM continuously) without (gray bars) or with Chk2 inhibitor II (1 μM continuously) (white bars). (B) S phase accumulation (left panel) and G2/M accumulation (right panel) of AT cells after treatment with MMS + 4-AN without (gray bars) or with Chk2 inhibitor II (white bars) as defined in panel A. Experiments were conducted as described in ‘Materials and Methods.’ Results represent mean ± S.E.M of three independent experiments.
As shown in Fig. 2 and Fig. 3, ATM-deficient and -proficient cells display similar fractions in S phase and G2/M 24 h after MMS and 4-AN. However, treatment of both cell lines with the Chk2 inhibitor resulted in a lower proportion of cells in S phase and a corresponding greater fraction in G2/M by 24 h. This difference in cell cycle profiles at 24 h with the Chk2 inhibitor may reflect cross talk between multiple kinases serving redundant roles in signaling checkpoint responses through Chk2. The inability to activate the shared downstream kinase, Chk2, compromises the capacity to maintain the S phase delay and as a result cells accumulate earlier in G2/M.
3.3. Phosphorylation of ATM and Chk2 in response to DNA base damage and PARP inhibition
The cell cycle experiments presented above (Fig. 3) are consistent with an ATM-independent contribution to activation of Chk2 in our system. However, it is well-documented that, when ATM is activated through autophosphorylation at S1981 (reviewed in [15]), it signals for cell cycle checkpoint by phosphorylating T68 of Chk2 (reviewed in [34]). As shown in Fig. 4A, ATM is phosphorylated at S1981 in AT-comp cells as early as 1 h after the start of treatment with MMS and 4-AN (compare lanes 1 and 2, upper panel), and reaches a constant level by 4 h (lane 4). Bulk levels of ATM remained constant through the course of the experiment (Fig. 4, lower panel). Upon closer examination, it appears that phosphorylation of ATM begins as early as 30 min after beginning treatment with MMS and 4-AN (Fig. 4B, compare lanes 1 and 2). As expected, neither phosphorylated nor bulk levels of ATM were detectable in AT cells (Fig. 4C, lanes 1 and 3). Thus it appears that ATM is rapidly activated in response to treatment with MMS and 4-AN.
Fig. 4.

Phosphorylation of ATM in MMS + 4-AN-treated cells. (A and B) Flag antibody immunoprecipitation of extracts from AT cells complemented with Flag-tagged ATM prepared at the indicated timepoints after treatment with MMS (0.1 mM for 1 h) + 4-AN (10 μM continuously). Samples were immunoblotted with antibody directed against phospho-S1981 ATM (upper panel) and bulk ATM (lower panel). (C) Flag immunoprecipitation of extracts from AT cells (-ATM) or AT cells complemented with Flag-tagged ATM (AT-comp, +ATM) 20 h after mock-treatment or treatment with MMS (0.1 mM for 1 h) + 4-AN (10 μM continuously). Samples were immunoblotted with antibodies as described in panel A.
To ascertain whether the ATM-Chk2 checkpoint pathway is activated in response to MMS and 4-AN, we looked for phosphorylation of Chk2. Following treatment with MMS and 4-AN, phosphorylation of Chk2 in AT-comp cells was observed as early as 1 h following the start of treatment with MMS and 4-AN (Fig. 5A, upper panel, compare lanes 2 and 4). In AT cells, phosphorylation of Chk2 was not detectable until approximately 8 h (lane 11). Extract from AT-comp cells exposed to 15 Gy IR was included as a positive control for phosphorylation of Chk2 (lane 13). To monitor bulk levels of Chk2, blots were reprobed with antibody that recognizes Chk2. Note that phosphorylation of Chk2 is visible as slower migrating bands in these Chk2 blots (Fig. 5A, middle panel). As a loading control, the blots were probed with antibody against tubulin (Fig. 5A, lower panel). The temporal pattern of Chk2 phosphorylation correlates with the requirement for ATM in maintaining S phase over the same period of time (Fig. 2, left panel). Following treatment of ATRkd cells with MMS and PARP inhibitor, phosphorylation of Chk2 at T68 is detectable as early as 4 h, and progressively increases over time (Fig. 5B, upper panel). Thus, the time course of Chk2 phosphorylation observed in MMS and 4-AN-treated ATRkd cells correlates with the accumulation of these cells in S phase [11].
Fig. 5.

ATM-dependent phosphorylation of Chk2 in MMS + 4-AN-treated cells. (A) Extracts were prepared at the indicated timepoints from AT cells (-ATM) or AT-comp (+ATM) cells treated with MMS (0.1 mM for 1 h) + 4-AN (10 μM continuously), or 2 h after IR (15 Gy) as a positive control. Samples were immunoblotted with antibody directed against phospho-T68 Chk2 (upper panel), bulk Chk2 (middle panel) and tubulin as a loading control (lower panel). (B) Extracts were prepared at the indicated timepoints from ATRkd cells treated with MMS (0.5 mM for 1 h) + 4-AN (10 μM continuously). Samples were immunoblotted as described in panel A.
We next confirmed that Chk2 phosphorylation was dependent on the combined treatment with MMS and 4-AN. Cells were treated with MMS and 4-AN together or individually for 1 h and then incubated for an additional 19 h in the absence or presence of 4-AN. As shown in Fig. 6A, phosphorylation of Chk2 observed in AT-comp cells exceeded that in AT cells with MMS plus 4-AN (compare lanes 1 and 2 with lanes 7 and 8). When either cell type was treated with MMS or 4-AN individually, the level of Chk2 phosphorylation was negligible and comparable to levels in ‘Mock’-treated cells (compare lanes 1 and 2 with lanes 3, 4, 5 and 6). Chk2 phosphorylation was also dependent on the combined treatment of MMS and 4-AN in ATRkd cells (Fig. 6B). These results indicate that the combination of PARP inhibition and low dose MMS-induced DNA damage is necessary for phosphorylation of Chk2 at T68.
Fig. 6.

Phosphorylation of Chk2 depends on combined MMS and 4-AN treatment. (A) Extracts were prepared from AT cells (-ATM) or AT-comp (+ATM) cells 20 h after mock-treatment, or treatment with MMS (0.1 mM for 1 h), continuous 4-AN (10 μM) alone, or MMS + 4-AN. Samples were immunoblotted with antibody directed against phospho-T68 Chk2 (upper panel), bulk Chk2 (middle panel) and tubulin as a loading control (lower panel). (B) Extracts were prepared from ATRkd cells 20 h after treatment with MMS (0.5 mM for 1 h), continuous 4-AN (10 μM) alone, MMS + 4-AN, or 2 h after IR (8 Gy) as a positive control. Samples were immunoblotted as described in panel A.
While levels of Chk2 phosphorylation were greater in AT-comp cells, a significant level of Chk2 phosphorylation was still detectable in AT cells 20 h after treatment with MMS and 4-AN (Fig. 6A, compare lanes 1 and 7). This indicates that other kinases are involved in this Chk2 response. ATRkd cells were used to determine the role of ATR. As shown in Fig. 7A, caffeine reduced the level of Chk2 phosphorylation (lane 5). A specific ATM inhibitor also resulted in a significant decrease in the levels of Chk2 phosphorylation following MMS and 4-AN treatment (compare lanes 2 and 4). Treatment of ATRkd cells with Dox induces expression of a kinase dead ATR and creates a dominant-negative phenotype [21]. Phosphorylation of Chk2 was only moderately impaired by treatment with Dox (compare lanes 2 and 3, upper and middle panels), suggesting a minor role for ATR in phosphorylation of Chk2 in response to MMS and 4-AN. As control for ATR activity, the same samples were probed for phosphorylation of Chk1. As shown previously [11] and here, Chk1 phosphorylation in response to MMS and 4-AN (Fig. 7B, compare lanes 1 and 2) is abrogated by treatment with Dox (compare lanes 2 and 3). In contrast, cells treated with ATMi showed only a modest reduction in Chk1 phosphorylation suggestive of a minor role for ATM in phosphorylating this protein.
Fig. 7.

ATR- and DNA-PK-dependent Chk2 phosphorylation in MMS and 4-AN treated cells. (A) Extracts were prepared from ATRkd cells 20 h after mock-treatment, or treatment with MMS (0.5 mM for 1 h) + 4-AN (10 μM continuously) alone (-), MMS + 4-AN combined with Dox (Dox) (1 μg/ml; 48 h pretreatment and throughout the MMS + 4-AN exposure), MMS + 4-AN with ATMi (10 μM continuously) (ATMi), MMS + 4-AN with caffeine (2 mM continuously). Samples were immunoblotted with antibody directed against phospho-T68 Chk2 (upper panel), bulk Chk2 (middle panel) and tubulin (lower panel). (B) Samples were prepared as described in panel A and immunoblotted with antibody directed against phospho-S345 Chk1 (upper panel), bulk Chk1 (middle panel) and tubulin (lower panel). (C) Extracts were prepared from AT cells (-ATM) or AT-comp (+ATM) cells 20 h after mock-treatment, or treatment with MMS (0.1 mM for 1 h) + 4-AN (10 μM continuously) or MMS and 4-AN with DNA-PK inhibitor NU7026 (PKi) (10 μM continuously). Samples were immunoblotted as described in panel A.
DNA-dependent protein kinase (DNA-PK) has also been reported to affect the phosphorylation state of Chk2 following IR [35]. To determine whether DNA-PK had any role after treatment with MMS and 4-AN, we analyzed the level of Chk2 phosphorylation in AT cells treated with the DNA-PK inhibitor NU7026 (PKi). As shown in Fig. 7C, the level of Chk2 phosphorylation observed in AT cells was reduced with PKi treatment (compare lanes 3 and 5), providing evidence that DNA-PK is involved in the response to MMS and 4-AN.
The results of this study identify the importance of ATM in the cellular response to inhibition of PARP during MMS-mediated base damage. ATM plays an important role in prolonging the S phase delay observed in MMS and 4-AN-treated cells and our observations suggest that ATM signaling through Chk2 kinase is involved. However, the impact of caffeine on cell cycle profiles and of ATR and DNA-PK on Chk2 phosphorylation in MMS and 4-AN treated cells suggests there is cross-talk between multiple signaling pathways in this DNA damage response.
4. Discussion
4.1. Inactivation of PARP and replication-dependent DSB formation
The activity of PARP-1, the major isoform in the PARP family involved in DNA damage responses, is required for efficient cellular BER (reviewed in [36]). PARP becomes activated following binding to SSB intermediates formed during BER and as a result of this activation and autoribosylation, its affinity for DNA is reduced allowing it to dissociate from its DNA binding sites and facilitating access of repair proteins [8]. When cells are subjected to DNA methylation along with PARP inhibition, PARP-1 remains bound to strand break intermediates of repair. Prolonged DNA binding of inactivated PARP is potentially disruptive to the completion of repair since it is likely to impair PARP-1-mediated recruitment of BER factors as well as chromatin modification.
The persistence of SSBs and stabilization of PARP binding at DNA lesions in the presence of 4-AN may impair the progression of the replication machinery through the genome. A similar situation occurring after inhibition of DNA topoisomerase I is known to negatively impact DNA replication. Camptothecin stabilizes the topoisomerase I/DNA covalent complex, resulting in persistent SSBs (reviewed in [37]), replication fork stalling, and DSB formation due fork collapse.
We propose that PARP inhibition in the presence of methylated base damage also results in formation of replication-dependent DSBs. Both SSBs and stabilized PARP-1 are potential impediments to DNA replication, however, exposure to the combination of MMS and 4-AN during S phase is critical for generating the observed replication defect [12]. We have demonstrated that MMS combined with 4-AN results in an ATR- and Chk1-dependent S phase delay [11,12]. It is well known that S phase delay following replication fork stalling is due to ATR- and Chk1-mediated signaling (reviewed in [13]). As shown in Fig. 2, ATM is important for sustaining the MMS and 4-AN-induced S phase delay. Combined treatment with MMS and 4-AN also results in increased levels of γ-H2A.X [11] and phosphorylation of Chk2 (Fig. 5). Both of these phosphorylation events can be mediated by the kinase activity of ATM and have been directly associated with the DSB response. However, we note that phosphorylation of ATM, Chk2 and H2A.X has been associated with other forms of cellular stress [38-40]. With this possibility in mind, preliminary experiments using Pulsed Field Gel Electrophoresis of genomic DNA have been conducted and suggest that DSBs are formed following MMS and 4-AN treatment (Michelle Heacock, personal communication). Such evidence of DSB damage occurring in the background of an S phase delay and ATR signaling events are compelling evidence of replication fork collapse.
4.2. Dose dependence of methylation damage and PARP inhibitor
Previous studies have linked PARP inhibition and activation of ATM. For example, Haince et al. reported that PARP inhibition diminished the ATM response to N-methyl-N′-nitro-N-nitrosoguanidine (MNNG)-mediated DNA damage and suggested that poly(ADP-ribosyl)ation plays a direct role in ATM activation [41]. Bryant and Helleday demonstrated that 4-AN treatment alone, and therefore inhibition of PARP during repair of endogenous base damage, resulted in activation of ATM [42]. Additionally, PARP-1 has been shown to interact with and to poly(ADP-ribosyl)ate ATM, and inhibition of PARP-1 has been shown decrease ATM kinase activity towards several substrates, suggestive of a direct role for PARP in the ATM-mediated response [43,44]. It should be noted that in some experiments cell treatments were conducted at highly toxic concentrations. In one study, the concentration of MNNG was by itself sufficient to reduce cell viability to less than 10% [41]. Another study utilized 4-AN at a concentration 10-fold higher than in our experiments [42] and possibly leading to a similar DSB formation and response as treating with lower doses of 4-AN combined with a base-damaging agent. Here, we used MMS and 4-AN concentrations and exposure times that when tested as single agents exhibited low toxicity. Furthermore, neither MMS nor 4-AN individually under our conditions had detectable effects on the cell cycle (data not shown) [11], or on Chk1 and Chk2 phosphorylation (Fig. 6). By combining low doses of MMS and 4-AN in this way, we are able to demonstrate the impact of inhibition of PARP activity during BER on the checkpoint response.
4.3. ATM response to DNA base damage and PARP inhibition
Our results indicate that the combination of methylated base damage and PARP inhibition activates DSB-induced signaling pathways. Repair of DSBs is achieved through homologous recombination (HR) or non-homologous end joining. Consistent with the enhanced MMS sensitivity of ATM-mutant cells treated with 4-AN (Fig. 1B), cells lacking either Brca1 or Brca2 also exhibit sensitivity to PARP inhibitors [45,46]. Brca1 and Brca2 are considered to be important components of repair by HR since cells mutated in these genes exhibit deficiencies in this repair pathway [47,48]. Presumably, the increased sensitivity of BRCA1 and -2 mutants to PARP inhibitors reflects the impaired HR of DSBs resulting from replication fork collapse. In contrast to our experimental system, where PARP inhibition required MMS-mediated base damage to produce these effects, BRCA1 and -2 mutants were sensitive to PARP inhibitors alone. It is likely that, in the absence of efficient HR, the levels of DSBs resulting from PARP inhibition combined with endogenous base damage are detrimental to cell survival.
ATM-mediated signaling is also associated with HR. ATM is thought to activate an exonuclease activity of the MRN (Mre11-Rad50-Nbs1) complex involved in the strand resection step of recombination [49]. HR as a result of damage caused by inhibition of PARP is also absent when cells lack ATM activity [42]. This apparent role for ATM, considered together with the increased sensitivity to MMS when PARP and ATM activity are inhibited (Fig. 1A and 1C), suggest that the cell utilizes HR for repair of DSB damage resulting from PARP inhibition during BER.
Our results clearly demonstrated the importance of ATM in responding to DSBs resulting from inhibition of PARP during BER. Although ATM was important for sustaining an S phase delay (Fig. 2, left panel), Chk2 appeared to play a similar role that was independent of ATM (Fig. 3B). Phosphorylation of Chk2 in MMS and 4-AN-treated cells was still observed, albeit to a lesser degree, in cells lacking ATM (Fig. 6A). We have shown here that ATR and DNA-PK also contribute to the phosphorylation of Chk2 (Fig. 7B and C). Such functional redundancy is not surprising. Both ATR and DNA-PK had been shown to phosphorylate Chk2 in response to IR [35,50]. DNA-PK also was shown to phosphorylate Chk2 in response to high doses of PARP inhibitors [42]. Overlapping roles for ATM and DNA-PK in the phosphorylation of H2A.X in response to IR have been demonstrated [29,51]. We are currently investigating the role DNA-PK plays a role in checkpoint signaling in response to PARP inhibition during BER.
In the study presented here, we provide an approach that combines a sub-lethal level of a DNA methylating agent with a non-toxic concentration of a PARP inhibitor to inhibit cell growth. Similar clinical approaches are under consideration. For example, PARP inhibitors combined with the chemotherapeutic methylating agent temozolomide have been used in experimental systems to treat different tumors including glioblastoma, and colon and lung carcinoma (reviewed in [6,20]) and clinical trials using PARP inhibitors and temozolomide are underway [19]. Understanding how this approach impacts the cell at the molecular level, as demonstrated here, should prove important for fully exploiting its potential in treating cancer.
Acknowledgments
We thank Rebecca Childress and Jennifer Zeng for technical assistance. This research was supported by Research Project Number ES050159 in the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences. Dr. Kedar's contribution was funded in whole with federal funds from NIH/NIEHS, under delivery order HHSN273200700046U to Constella/SRA, LLC.
Footnotes
Conflict of Interest statement: The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zharkov DO. Base excision DNA repair. Cell Mol Life Sci. 2008 doi: 10.1007/s00018-008-7543-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Horton JK, Joyce-Gray DF, Pachkowski BF, Swenberg JA, Wilson SH. Hypersensitivity of DNA polymerase β null mouse fibroblasts reflects accumulation of cytotoxic repair intermediates from site-specific alkyl DNA lesions. DNA Repair (Amst) 2003;2:27–48. doi: 10.1016/s1568-7864(02)00184-2. [DOI] [PubMed] [Google Scholar]
- 3.Sobol RW, Prasad R, Evenski A, Baker A, Yang XP, Horton JK, Wilson SH. The lyase activity of the DNA repair protein β-polymerase protects from DNA-damage-induced cytotoxicity. Nature. 2000;405:807–810. doi: 10.1038/35015598. [DOI] [PubMed] [Google Scholar]
- 4.Sobol RW, Horton JK, Kühn R, Gu H, Singhal RK, Prasad R, Rajewsky K, Wilson SH. Requirement of mammalian DNA polymerase-β in base-excision repair. Nature. 1996;379:183–186. doi: 10.1038/379183a0. [DOI] [PubMed] [Google Scholar]
- 5.Lavrik OI, Prasad R, Sobol RW, Horton JK, Ackerman EJ, Wilson SH. Photoaffinity labeling of mouse fibroblast enzymes by a base excision repair intermediate. Evidence for the role of poly(ADP-ribose) polymerase-1 in DNA repair. J Biol Chem. 2001;276:25541–25548. doi: 10.1074/jbc.M102125200. [DOI] [PubMed] [Google Scholar]
- 6.Haince JF, Rouleau M, Hendzel MJ, Masson JY, Poirier GG. Targeting poly(ADP-ribosyl)ation: a promising approach in cancer therapy. Trends Mol Med. 2005;11:456–463. doi: 10.1016/j.molmed.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 7.Horton JK, Baker A, Vande Berg BJ, Sobol RW, Wilson SH. Involvement of DNA polymerase β in protection against the cytotoxicity of oxidative DNA damage. DNA Repair (Amst) 2002;1:317–333. doi: 10.1016/s1568-7864(02)00008-3. [DOI] [PubMed] [Google Scholar]
- 8.Satoh MS, Lindahl T. Role of poly(ADP-ribose) formation in DNA repair. Nature. 1992;356:356–358. doi: 10.1038/356356a0. [DOI] [PubMed] [Google Scholar]
- 9.Satoh MS, Poirier GG, Lindahl T. NAD(+)-dependent repair of damaged DNA by human cell extracts. J Biol Chem. 1993;268:5480–5487. [PubMed] [Google Scholar]
- 10.Satoh MS, Poirier GG, Lindahl T. Dual function for poly(ADP-ribose) synthesis in response to DNA strand breakage. Biochemistry. 1994;33:7099–7106. doi: 10.1021/bi00189a012. [DOI] [PubMed] [Google Scholar]
- 11.Horton JK, Stefanick DF, Kedar PS, Wilson SH. ATR signaling mediates an S-phase checkpoint after inhibition of poly(ADP-ribose) polymerase activity. DNA Repair (Amst) 2007;6:742–750. doi: 10.1016/j.dnarep.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horton JK, Stefanick DF, Naron JM, Kedar PS, Wilson SH. Poly(ADP-ribose) polymerase activity prevents signaling pathways for cell cycle arrest after DNA methylating agent exposure. J Biol Chem. 2005;280:15773–15785. doi: 10.1074/jbc.M413841200. [DOI] [PubMed] [Google Scholar]
- 13.Paulsen RD, Cimprich KA. The ATR pathway: fine-tuning the fork. DNA Repair (Amst) 2007;6:953–966. doi: 10.1016/j.dnarep.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 14.Woodhouse BC, Dianova II, Parsons JL, Dianov GL. Poly(ADP-ribose) polymerase-1 modulates DNA repair capacity and prevents formation of DNA double strand breaks. DNA Repair (Amst) 2008;7:932–940. doi: 10.1016/j.dnarep.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 15.Shiloh Y. The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci. 2006;31:402–410. doi: 10.1016/j.tibs.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 16.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 17.Brem R, Fernet M, Chapot B, Hall J. The methyl methanesulfonate induced S-phase delay in XRCC1-deficient cells requires ATM and ATR. DNA Repair (Amst) 2008;7:849–857. doi: 10.1016/j.dnarep.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 18.Horton JK, Watson M, Stefanick DF, Shaughnessy DT, Taylor JA, Wilson SH. XRCC1 and DNA polymerase β in cellular protection against cytotoxic DNA single-strand breaks. Cell research. 2008;18:48–63. doi: 10.1038/cr.2008.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plummer R, Jones C, Middleton M, Wilson R, Evans J, Olsen A, Curtin N, Boddy A, McHugh P, Newell D, Harris A, Johnson P, Steinfeldt H, Dewji R, Wang D, Robson L, Calvert H. Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res. 2008;14:7917–7923. doi: 10.1158/1078-0432.CCR-08-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plummer ER, Calvert H. Targeting poly(ADP-ribose) polymerase: a two-armed strategy for cancer therapy. Clin Cancer Res. 2007;13:6252–6256. doi: 10.1158/1078-0432.CCR-07-0617. [DOI] [PubMed] [Google Scholar]
- 21.Cliby WA, Roberts CJ, Cimprich KA, Stringer CM, Lamb JR, Schreiber SL, Friend SH. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 1998;17:159–169. doi: 10.1093/emboj/17.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ziv Y, Bar-Shira A, Pecker I, Russell P, Jorgensen TJ, Tsarfati I, Shiloh Y. Recombinant ATM protein complements the cellular A-T phenotype. Oncogene. 1997;15:159–167. doi: 10.1038/sj.onc.1201319. [DOI] [PubMed] [Google Scholar]
- 23.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9159. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 24.Butler WB. Preparing nuclei from cells in monolayer cultures suitable for counting and for following synchronized cells through the cell cycle. Anal Biochem. 1984;141:70–73. doi: 10.1016/0003-2697(84)90426-3. [DOI] [PubMed] [Google Scholar]
- 25.Arienti KL, Brunmark A, Axe FU, McClure K, Lee A, Blevitt J, Neff DK, Huang L, Crawford S, Pandit CR, Karlsson L, Breitenbucher JG. Checkpoint kinase inhibitors: SAR and radioprotective properties of a series of 2-arylbenzimidazoles. Journal of medicinal chemistry. 2005;48:1873–1885. doi: 10.1021/jm0495935. [DOI] [PubMed] [Google Scholar]
- 26.Biade S, Sobol RW, Wilson SH, Matsumoto Y. Impairment of proliferating cell nuclear antigen-dependent apurinic/apyrimidinic site repair on linear DNA. J Biol Chem. 1998;273:898–902. doi: 10.1074/jbc.273.2.898. [DOI] [PubMed] [Google Scholar]
- 27.Kedar PS, Kim SJ, Robertson A, Hou E, Prasad R, Horton JK, Wilson SH. Direct interaction between mammalian DNA polymerase β and proliferating cell nuclear antigen. J Biol Chem. 2002;277:31115–31123. doi: 10.1074/jbc.M201497200. [DOI] [PubMed] [Google Scholar]
- 28.Barfknecht TR, Little JB. Hypersensitivity of ataxia telangiectasia skin fibroblasts to DNA alkylating agents. Mutat Res. 1982;94:369–382. doi: 10.1016/0027-5107(82)90299-8. [DOI] [PubMed] [Google Scholar]
- 29.Wang H, Wang M, Wang H, Böcker W, Iliakis G. Complex H2AX phosphorylation patterns by multiple kinases including ATM and DNA-PK in human cells exposed to ionizing radiation and treated with kinase inhibitors. J Cell Physiol. 2005;202:492–502. doi: 10.1002/jcp.20141. [DOI] [PubMed] [Google Scholar]
- 30.Zhou BB, Chaturvedi P, Spring K, Scott SP, Johanson RA, Mishra R, Mattern MR, Winkler JD, Khanna KK. Caffeine abolishes the mammalian G(2)/M DNA damage checkpoint by inhibiting ataxia-telangiectasia-mutated kinase activity. J Biol Chem. 2000;275:10342–10348. doi: 10.1074/jbc.275.14.10342. [DOI] [PubMed] [Google Scholar]
- 31.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, Abraham RT. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999;59:4375–4382. [PubMed] [Google Scholar]
- 33.Kaufmann WK, Heffernan TP, Beaulieu LM, Doherty S, Frank AR, Zhou Y, Bryant MF, Zhou T, Luche DD, Nikolaishvili-Feinberg N, Simpson DA, Cordeiro-Stone M. Caffeine and human DNA metabolism: the magic and the mystery. Mutat Res. 2003;532:85–102. doi: 10.1016/j.mrfmmm.2003.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pommier Y, Sordet O, Rao VA, Zhang H, Kohn KW. Targeting chk2 kinase: molecular interaction maps and therapeutic rationale. Current pharmaceutical design. 2005;11:2855–2872. doi: 10.2174/1381612054546716. [DOI] [PubMed] [Google Scholar]
- 35.Li J, Stern DF. Regulation of CHK2 by DNA-dependent protein kinase. J Biol Chem. 2005;280:12041–12050. doi: 10.1074/jbc.M412445200. [DOI] [PubMed] [Google Scholar]
- 36.Petermann E, Keil C, Oei SL. Importance of poly(ADP-ribose) polymerases in the regulation of DNA-dependent processes. Cell Mol Life Sci. 2005;62:731–738. doi: 10.1007/s00018-004-4504-2. [DOI] [PubMed] [Google Scholar]
- 37.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 38.Bolderson E, Scorah J, Helleday T, Smythe C, Meuth M. ATM is required for the cellular response to thymidine induced replication fork stress. Human molecular genetics. 2004;13:2937–2945. doi: 10.1093/hmg/ddh316. [DOI] [PubMed] [Google Scholar]
- 39.Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. γH2AX and cancer. Nat Rev Cancer. 2008;8:957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 41.Haince JF, Kozlov S, Dawson VL, Dawson TM, Hendzel MJ, Lavin MF, Poirier GG. Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to DNA-damaging agents. J Biol Chem. 2007;282:16441–16453. doi: 10.1074/jbc.M608406200. [DOI] [PubMed] [Google Scholar]
- 42.Bryant HE, Helleday T. Inhibition of poly (ADP-ribose) polymerase activates ATM which is required for subsequent homologous recombination repair. Nucleic Acids Res. 2006;34:1685–1691. doi: 10.1093/nar/gkl108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aguilar-Quesada R, Muñoz-Gámez JA, Martín-Oliva D, Peralta A, Valenzuela MT, Matínez-Romero R, Quiles-Pérez R, Menissier-de Murcia J, de Murcia G, de Almodóvar MR, Oliver FJ. Interaction between ATM and PARP-1 in response to DNA damage and sensitization of ATM deficient cells through PARP inhibition. BMC Mol Biol. 2007;8:29. doi: 10.1186/1471-2199-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haince JF, McDonald D, Rodrigue A, Déry U, Masson JY, Hendzel MJ, Poirier GG. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J Biol Chem. 2008;283:1197–1208. doi: 10.1074/jbc.M706734200. [DOI] [PubMed] [Google Scholar]
- 45.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 46.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 47.Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- 48.Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7:263–272. doi: 10.1016/s1097-2765(01)00174-5. [DOI] [PubMed] [Google Scholar]
- 49.Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 50.Wang XQ, Redpath JL, Fan ST, Stanbridge EJ. ATR dependent activation of Chk2. J Cell Physiol. 2006;208:613–619. doi: 10.1002/jcp.20700. [DOI] [PubMed] [Google Scholar]
- 51.Stiff T, O'Driscoll M, Rief N, Iwabuchi K, Löbrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004;64:2390–2396. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]