

Abstract
Generation of novel protein functions is a major goal in biotechnology and also a rigorous test for our understanding of the relationship between protein structure and function. Early examples of protein engineering focused on design and directed evolution within the constraints of the original protein architecture, exemplified by the highly successful fields of antibody and enzyme engineering. Recent studies show that protein engineering strategies which step away from these natural architectures, i.e. by manipulating the organization of domains and modules thus mimicking nonhomologous recombination, is highly effective in producing complex and sophisticated functions both in terms of molecular recognition and regulation.
Analysis of genomes shows that protein evolution progresses via point mutations, duplication and recombination of genes under selective pressure. Technologies that simulate these mechanisms can be effective strategies to generate novel protein functions and architectures for diverse applications, a major goal in the field of protein biotechnology. To date, a majority of protein engineering efforts has utilized methodologies that recapitulate the processes of gene duplication and subsequent sequence divergence. Specific and random mutations are introduced by knowledge-based design and/or more random means such as error prone PCR into a single, essentially invariant scaffold, and mutants exhibiting a desired function are identified through screening and selection. [1–9]. This type of functional evolution generally is incremental and requires a starting scaffold that is already predisposed to the desired type of function.
Nonhomologous genetic recombination either by genome rearrangement and alternative splicing can produce new combinations of gene fragments and thereby drastically different polypeptide sequences. Bioinformatic analyses have suggested that domain recombination is a major driving force for leaps in protein function [10–14]. This view was also supported by simulations showing that evolution via nonhomologous recombination of protein segments is many orders of magnitude more effective than point mutation in acquiring significant new function [15]. Herein a domain is defined as an evolutionary and structurally separable unit within a protein. Individual domains of eukaryotic, multi-domain proteins are often encoded in an exon. A domain in isolation may or may not be autonomously folded into a well-defined structure. In contrast, a module, another term often used in the context of protein evolution, is defined as a functionally minimal unit that is transferable from one protein context to another [16]. A module may contain multiple domains, or it may not even contain a domain, as in the case of a short peptide segment containing a binding site for another module. There have been increasing number of successes in generating new protein functions by methodologies that recapitulate rearrangement and combinations of domains and modules, demonstrating they are indeed powerful means to generate large functional changes and expand the repertoire of synthetic proteins. This review focuses on recent design and engineering studies that are based on a structural and mechanistic understanding of how combinations of domains and modules define protein functions.
Enhancement of molecular recognition functions through domain/module recombination
The design and engineering of specific and high-affinity recognition functions has been a major goal in protein engineering with clear applications in therapeutics and diagnostics. The main approach in this field has been to use a single, stable scaffold corresponding to a single module, such as the Fab and Fv segments of immunoglobulins and also non-antibody scaffolds [4,7]. Mutations are introduced in a small portion that is expected to form a contiguous "patch" within such a scaffold (e.g. the complementarity-determining regions of the immunoglobulins) to produce a repertoire while maintaining the overall domain architecture and tertiary structure, and variants are then identified using methods such as phage display (Fig. 1a) [17,18]. These strategies mimic evolution through point mutations and homologous recombinations. Recent studies described below suggest that binding proteins with multiple recognition patches each residing on separate domain offer distinct advantages over the traditional, single-patch binding proteins.
Figure 1.

Generation of new molecular recognition functions by combining domains. (a–c) Schematic drawing of different types of binding protein architectures. (a) "Single-patch" recognition by a conventional binding protein. (b) Multivalent (multi-epitope) recognition by multiple modules linked with flexible linkers. (c) Synergistic recognition of a contiguous target surface by a single binding site constructed at the interface of two domains. The target (or antigen) is colored in gray. Recognition surfaces of the binding proteins are marked in yellow. (d) Crystal structures of two affinity clamps (PDB ID: 2QBW and 3CH8) showing the ability of directed domain-interface evolution to produce diverse interface configurations. The schemes below the structural models show simplified views of the interactions between the 8-residue target peptide and the affinity clamps. The molecular graphics was generated using PyMOL (www.pymol.org).
Domain combination for avidity
Enhancement of affinity and specificity through multivalent interactions, or avidity, is widely exploited at molecular and cellular levels [19]. Here I classify avidity into homotropic and heterotropic. The former refers to enhancement due to multiple copies of identical interactions and the latter due to multiple distinct interactions.
Exploiting homotropic avidity requires relatively small costs in engineering effort. This type of multivalent approach can be used with any molecular recognition module. For example, the immune system exploits this principle by using IgM, which contains a total of ten identical antigen-binding sites that increases the effective affinity particularly to homo-oligomeric antigens. An oligomeric version of synthetic binding protein based on the fibronectin type III domain (FN3), a small antibody-like domain, showed significantly higher affinity to a cell surface antigen [20].
Heterotropic avidity can be created by concatenating multiple domains with flexible linkers where each domain recognizes different epitopes on a target (Fig. 1b). Prominent natural examples of such "beads-on-a-string" proteins for molecular recognition are nucleic acid-binding proteins using tandem repeats of the zinc-fingers (ZFs), ~23-residue modular domains each folding into a compact structure. Structural studies have revealed that each ZF recognizes three nucleotide base pairs and that tandem repeats of the zinc-finger modules can bind to long stretches of nucleotide sequences to achieve high levels of specificity. By exploiting this modular design, several groups have developed synthetic zinc-finger proteins [21]. It is now possible to construct nucleases and transcription factors with exquisite specificity using poly-ZF proteins [22].
Molecules for recognizing proteins conceptually equivalent to multi-ZF proteins, termed "avimers", have been developed using small, disulfide-linked protein modules called A domains as a building block [23]. The A domains are small (~35 residues) autonomously folded domains primarily stabilized by disulfide bonds and calcium binding. Similar to ZFs, A domains occur as multiple repeats in human receptor proteins, and a target is contacted by multiple A domains with each domain binding to a distinct epitope. By mimicking these natural receptors, these avimers were generated using phage display methods by step-wise procedures that mimic early ZF engineering [21], where a single A domain binding to a target is initially selected, then an adjacent A domain is optimized in the context of the first selected domain and so on. Individual A domains in an avimer recognize distinct epitopes of a target, which leads to high specificity and affinity (sub-nM Kd) using two or three A domain modules. This work has established a powerful strategy to generate high levels of molecular recognition function.
Recognition sites at the interface of combined domains
Structural analysis of multi-domain proteins, particularly enzymes, suggests that dramatic changes in function have emerged through joining protein domains and adjusting the newly created interface between them [10,24]. For example, the classical NAD-binding Rossmann fold domain is joined to distinct substrate recognition domains to form a series of active, NAD(P)-dependent enzymes [11]. A recent comprehensive study of the haloacid dehalogenase superfamily convincingly show that its subfamilies with distinct substrate specificity have emerged as a result of multiple, independent evolutionary events that inserted a domain (termed "cap") into the core catalytic domain at different positions [25]. The emergence of distinct caps correlate with the acquisition of new catalytic capacities. In this type of domain combination, there are extensive interactions between the two linked domains, suggesting that the inter-domain interface has been evolutionarily tuned by a series of point mutations. These observations support an evolutionary path for generating sophisticated functions by constructing a single active site at the interface between two newly combined, evolutionally unrelated domains (Fig. 1c).
Huang et al. demonstrated that highly specific and complex protein functions indeed can be generated at a newly created domain interface [26]. Their goal was to produce highly specific and tight binding proteins to short, unstructured peptides, a class of difficult targets for molecular recognition using the conventional, single-patch strategy. A fusion protein of the erbin PDZ domain (a low-affinity peptide-binding domain) and a functionally inert second domain that serves as a diversity-presenting scaffold (the FN3 domain) was made and subsequently the interface between the two domains was optimized by combinatorial library selection of FN3 loops using phage display. This process, termed "directed domain-interface evolution", dramatically enhanced both affinity and specificity to a target peptide (~500 fold to single nM Kd and ~6,000 fold, respectively), levels unattainable by optimizing the binding interface of a single PDZ domain [27]. The x-ray crystal structure of the evolved fusion protein confirmed that the design principles were successful. The two fused domains formed a clamshell architecture inside which the target peptide was bound (Fig. 1d). The optimized FN3 loops interact with both the target peptide and the PDZ domain extensively, thus significantly enlarging the binding surface for the peptide. These binding proteins, termed "affinity clamps", have a single-nanomolar dissociation constant and outperform a monoclonal antibody in immunochemical applications, demonstrating their practical utility.
Their subsequent work mapped the specificity profile of a highly specific affinity clamp using a phage display peptide library. These results revealed that "affinity clamping" can expand the peptide recognition epitope significantly beyond that of the starting PDZ domain [28]. The crystal structure of this affinity clamp showed a deep binding groove for a larger portion of the target peptide constructed at the domain interface involving extensive contacts between the two domains, rationalizing the exquisite specificity of the affinity clamp (Fig. 1d). A comparison of two crystal structures revealed a considerable difference in the relative orientation of the two domains, explaining different functional properties between the two and also implies that diverse functions can be generated by controlling the mode of interactions between the two domains. These studies experimentally establish evolutionary paths that dramatically enhance the function of single-domain modules.
Generation of new regulatory functions by combination and rearrangement of domains and modules
The ability to control protein's molecular recognition function is the basis for engineering higher-level functionalities such as regulatory circuits and signal sensing. Eukaryotic regulatory proteins are often constructed with discrete modules, each responsible for specific molecular recognition or catalysis [16]. Such regulatory proteins utilize a limited number of module families rather than diverse individual proteins. Comparisons of oncogenes and proto-oncogenes show that alterations in modular architecture through nonhomologous combination and rearrangement can trigger dramatic changes in phenotype, such as carcinogenesis. Examples include BCR-Abl (fusion of dimeric BCR protein to Abl kinase) and v-Src (elimination of a phospho-Tyr site for intramolecular SH2 interaction), both of which result in kinase disregulation [29,30]. These observations in natural proteins suggest the effectiveness of nonhomologous recombination and rearrangement of protein modules in constructing diverse regulatory pathways.
Protein modules and their respective targets are autonomous and portable, offering "mix and match" simplicity in designing this type of proteins, which can be exploited to create synthetic regulatory proteins, a major goal in the field of synthetic biology. For instance, hybrid scaffold/adaptor proteins have successfully designed that redirect the flow of cellular signals by simple mass action [31–33]. Recently, a synthetic signal transduction pathway in plant was constructed using a component of a bacterial histidine kinase pathway, demonstrating a high level of portability of such modular components [34]. Recent studies described below demonstrate that more complex regulatory mechanisms can be designed by covalently linking modules and creating and controlling interactions between them.
Allosteric regulation using modular components
Many modular proteins act as regulated switches. Modular proteins can control the activity of an "output" module at the molecular level by two distinct modes of allostery, steric and conformational [31]. Steric allostery physically masks the active site of an output module by employing another module (Fig. 2a). This masking can be relieved by the binding of an effector to the inhibitory input module. Conformational allostery is similar to the traditional model of allostery [35], where an output module samples active and inactive conformations and the equilibrium between the two states is modulated by other modules connected to the output module (Fig. 2b). Conformational allostery requires an output module whose active site is malleable, such as protein kinase modules [29].
Figure 2.
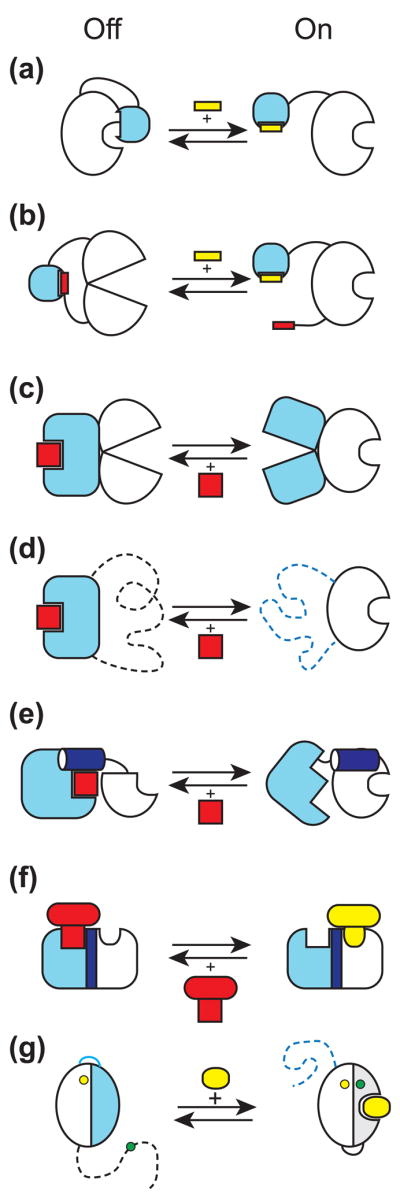
Molecular mechanisms used to create synthetic regulatory circuits and sensors by module/domain recombination and rearrangement. The input module is shown in light blue and the output module is in white. The output module is in the "on" state on the right side and "off" on the left. An activator is shown in yellow and an inhibitor in red. (a) Steric allostery. The input module physically occludes the active site of the output module, which can be relieved by a ligand (yellow) for the input module. (b) Conformational allostery. Interactions involving the input module and its intramolecular ligand distort the conformation of the output module. A ligand in trans removes the inhibition. (c) Conformational coupling. Two modules are connected in such a way that only one of them is in the active conformation at a given time. A ligand binding to the active state of the input module inhibits the output module. (d) Mutually exclusive folding. The dashed lines indicate unfolded segments. This is an extreme case of conformational coupling depicted in panel c. (e) Segmental overlap creating mutually exclusive folding of a shared helix (shown in dark blue) and thus conformational allostery. A ligand for the input module stabilizes docking of the shared helix to the rest of the input module and thus inactivates the output module. (f) Segmental overlap creating steric allostery. Two modules are fused in such a way that a ligand to the input module (red) physically occludes binding of a ligand to the output module (yellow). (g) Alternative folding frames for creating a sensor. The blue and gray portions denote equivalent segment of the starting protein that has been duplicated. The blue segment is mutated so that it no longer binds to a ligand but it interacts with the rest of the protein more strongly than the gray segment thereby favoring the inactive state (the left side of the arrows). A ligand shifts the equilibrium to the right, which changes the distance between fluorophores (the yellow and green circles).
The schemes (a) and (b) are made after Dueber et al. [31], (e) after Strickland et al. [44] and (g) after Stratton et al. [48].
Building upon their pioneering work on the design of synthetic switches by modular recombination [36], Dueber et al. constructed switches that exhibit nonlinear responses [37]. These switches utilized multiple copies (up to five) of low-affinity SH3-peptide interactions as negative regulators of the WASP protein. In contrast to the original switch, which showed a linear response with respect to the input (SH3 ligand), the newly designed switches showed strong positive cooperativity with a Hill coefficient as high as 3.9. This study demonstrates that homotropic cooperativity akin to that exhibited by oligomeric proteins (e.g. hemoglobin) can be designed by modular recombination.
Regulation by designed mutual exclusivity between modules
Proteins that exhibit conformational allostery are usually multi-domain proteins in which an active site is located at a domain interface. This architecture enables conformational coupling between the active site and an effector site (or among active sites) through domain reorientation. One can construct synthetic allostery by connecting two modules in such as way that their respective functional states are coupled. It is most straightforward to set up such coupling in a mutually exclusive manner. Then, the functional state of the output module can be controlled perturbed by altering the conformational state of the input module by a signal (e.g. ligand binding) (Fig. 2c). Insertion of one protein within another, rather than end-to-end fusion, offers higher probability of successful coupling. This topic has been reviewed recently [38,39]. A bottleneck in constructing this type of conformational coupling is the identification of a proper insertion site, which usually requires screening of a large number of constructs. A facile method based on a transposon to systematically generate random insertion mutations has been developed [40]. From ~50 β-lactamase mutants in which cytochrom b562 was inserted, a variant was identified that confer >100 fold change in antibiotic resistance in response to heme binding. The extreme case of this type of design is mutually exclusive folding, where a protein with a long distance between the N- and C-termini is inserted in a loop of another protein so that folding of the two proteins are negatively coupled (Fig. 2d) [41]. Detailed biophysical and computational studies were performed on the effects of the length of linkers connecting two modules [42], demonstrating that linkage plays an essential role in the context of mutually exclusive folding.
Light-triggered conformational changes of natural light-sensing domains (a subfamily of the light-oxygen-voltage or LOV domains) have been exploited as an input domain for constructing light-sensitive proteins [43]. The LOV domains are a subfamily of the Per–Arnt–Sim (PAS) domain family that frequently occurs fused to another protein through N- or C-terminal helix. Light activation detaches and unfolds the C-terminal Jα helix. A light-sensitive histidine kinase exhibiting 1,000-fold change in activity has been generated by replacing an oxygen-sensing PAS domain of a histidine kinase with a light-sensitive LOV domain while maintaining the Jα helix-mediated linkage [43].
A light-controlled transcription factor was engineered by designing a sequence overlap between two proteins so as to create mutually exclusive folding of an α-helix between them (Fig. 2e) [44]. In this fusion construct, the C-terminal Jα helix of another LOV domain (LOV2) and the N-terminal helix of Trp repressor were designed to be partially overlapped, i.e. encoded by the same stretch of polypeptide segment, so that the Trp repressor is functional only when light triggers the detachment and unfolding of the LOV Jα helix. One of 12 fusion proteins constructed in this manner showed light-dependent activity, validating this rational design strategy.
Light-induced conformational change was also used to regulate the enzymatic activity of dihydroforate reductase [45]. In this work, the insertion position was designed based on statistic coupling analysis of family members, which indicated this position as a hot spot linking a distant surface with the enzyme active site. Small light-dependent changes in enzyme activity were observed.
Another light-dependent synthetic signaling system was using a modular design concept similar to rewiring of adaptor proteins [46]. The light-dependent interaction between a plant photosensory domain (phytochrome B) and its binding domain (Pif3) was exploited as the basis for controlling the interaction between the WASP protein and its activator Cdc42. Red illumination induces complex formation of the Cdc42-phytochrome B fusion and the WASP-Pif3 fusion and consequently activates WASP. The activation can be reversed by far-red illumination. This simple but elegant modular strategy should be applicable to many other systems with minimal modification.
Protein interaction switches have been generated by designing a hybrid sequence that encodes overlapping and mutually exclusive binding sites for two unrelated interactions [47]. This strategy is similar to the concept of mutually exclusive folding described above, but the design is based on steric allostery instead (Fig. 2e). Sallee et al. successfully designed three types of such hybrid sequences, (a) hybrid of two modular domains, (b) hybrid of one modular domain and a short peptide motif, and (c) hybrid of two peptide motifs. Biophysical characterization of a type (a) switch constructed from the syntrophin PDZ and the WASP p21-binding domain supported that the switch functions by the steric allostery mechanism.
Designing sensors by creating an alternative folding frame
The mutually exclusive folding principle has been expanded to construct a biosensor using only a single folded domain, rather than two domains, as the fundamental building block [48]. Biosensor engineering usually couple ligand-induced conformational change with readout, and consequently it is difficult to design a sensor from a protein that does not exhibit a large conformational change upon ligand binding. The "alternative folding frame" strategy does not rely on this prerequisite. Instead, it creates a protein containing duplicated segments so that only one of the two fragments is incorporated in the folded protein at a time (Fig. 2f). Hence there is no net change in the total amount of folded protein regardless of which fragment is incorporated. Mutations are introduced to the duplicated segment in such a way that ligand binding alters the equilibrium between the two folding frames, thus creating large ligand-dependent conformational rearrangement suitable for readout. Stratton et al. created a calcium sensor by designing alternative folding frames in calbinding D9k, a calcium-binding domain that does not undergo a large conformational change upon calcium binding. Biophysical analysis supports the designed mode of action. The response half time of this sensor was 5–25 seconds, reflecting relatively slow kinetics due to a large conformational rearrangement. Although this strategy still requires that the duplicate segments do not cause the protein to aggregate and that the folding/unfolding kinetics are reasonably fast, it makes it possible to produce sensors from simple ligand-binding proteins.
Simultaneous emergence of new recognition and allostery
Liang et al. discovered that a synthetic allosteric enzyme constructed by recombining β-lactamase and maltose-binding protein acquired an additional mode of allostery in response to zinc binding, even though the two parent proteins are not known to bind zinc [49]. Thus, the recombination event created a new zinc-binding site that preferentially stabilizes the off state of β-lactamase. These results nicely illustrate that module/domain recombination can be an effective route to new function in both molecular recognition and regulation. They also give a cautionary note on interpreting in vivo data on synthetic switches engineered by module recombination, because an unidentified cellular compound might act as a new allosteric effector.
Future Perspectives
The studies discussed above demonstrate the effectiveness and potential of module/domain-level protein engineering in creating sophisticated functions. To date, design efforts have been focused on generating either new regulatory mechanisms or new molecular recognition functions. Clearly, these are not mutually exclusive goals [49], and synergistic uses of both would greatly expand our capacity to build new protein functionalities. As such, it is highly likely that this area of research and development will see significant growth in the near future.
Acknowledgments
I would like to thank M. Biancalana, R. Gilbreth and S. Yan for critical reading of the manuscript. SK was supported by National Institutes of Health grants.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pluckthun A, Mayo SL. The design of evolution and the evolution of design. Curr Opin Struct Biol. 2007;17:451–453. [Google Scholar]
- 2.Kortemme T, Baker D. Computational design of protein-protein interactions. Curr Opin Chem Biol. 2004;8:91–97. doi: 10.1016/j.cbpa.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 3.Winter G, Griffiths AD, Hawkins RE, Hoogenboom HR. Making antibodies by phage display technology. Annu Rev Immunol. 1994;12:433–455. doi: 10.1146/annurev.iy.12.040194.002245. [DOI] [PubMed] [Google Scholar]
- 4.Binz HK, Amstutz P, Pluckthun A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat Biotechnol. 2005;23:1257–1268. doi: 10.1038/nbt1127. [DOI] [PubMed] [Google Scholar]
- 5.Looger LL, Dwyer MA, Smith JJ, Hellinga HW. Computational design of receptor and sensor proteins with novel functions. Nature. 2003;423:185–190. doi: 10.1038/nature01556. [DOI] [PubMed] [Google Scholar]
- 6.Yoshikuni Y, Ferrin TE, Keasling JD. Designed divergent evolution of enzyme function. Nature. 2006;440:1078–1082. doi: 10.1038/nature04607. [DOI] [PubMed] [Google Scholar]
- 7.Sidhu SS, Fellouse FA. Synthetic therapeutic antibodies. Nat Chem Biol. 2006;2:682–688. doi: 10.1038/nchembio843. [DOI] [PubMed] [Google Scholar]
- 8.Bershtein S, Tawfik DS. Advances in laboratory evolution of enzymes. Curr Opin Chem Biol. 2008;12:151–158. doi: 10.1016/j.cbpa.2008.01.027. [DOI] [PubMed] [Google Scholar]
- 9.Persson H, Lantto J, Ohlin M. A focused antibody library for improved hapten recognition. J Mol Biol. 2006;357:607–620. doi: 10.1016/j.jmb.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 10.Bashton M, Chothia C. The generation of new protein functions by the combination of domains. Structure. 2007;15:85–99. doi: 10.1016/j.str.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 11.Rossmann MG, Moras D, Olsen KW. Chemical and biological evolution of nucleotide-binding protein. Nature. 1974;250:194–199. doi: 10.1038/250194a0. [DOI] [PubMed] [Google Scholar]
- 12.Gilbert W. Why genes in pieces? Nature. 1978;271:501. doi: 10.1038/271501a0. [DOI] [PubMed] [Google Scholar]
- 13.Blake CCF. Do genes-in-pieces imply proteins-in-pieces? Nature. 1978;273:267. [Google Scholar]
- 14.Todd AE, Orengo CA, Thornton JM. Sequence and structural differences between enzyme and nonenzyme homologs. Structure. 2002;10:1435–1451. doi: 10.1016/s0969-2126(02)00861-4. [DOI] [PubMed] [Google Scholar]
- 15.Bogarad LD, Deem MW. A hierarchical approach to protein molecular evolution. Proc Natl Acad Sci U S A. 1999;96:2591–2595. doi: 10.1073/pnas.96.6.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pawson T, Nash P. Assembly of cell regulatory systems through protein interaction domains. Science. 2003;300:445–452. doi: 10.1126/science.1083653. [DOI] [PubMed] [Google Scholar]
- 17.Fellouse FA, Esaki K, Birtalan S, Raptis D, Cancasci VJ, Koide A, Jhurani P, Vasser M, Wiesmann C, Kossiakoff AA, et al. High-throughput Generation of Synthetic Antibodies from Highly Functional Minimalist Phage-displayed Libraries. J Mol Biol. 2007;373:924–940. doi: 10.1016/j.jmb.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 18.Sidhu SS, Koide S. Phage display for engineering and analyzing protein interaction interfaces. Curr Opin Struct Biol. 2007;17:481–487. doi: 10.1016/j.sbi.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 19.Bromley SK, Burack WR, Johnson KG, Somersalo K, Sims TN, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML. The immunological synapse. Annu Rev Immunol. 2001;19:375–396. doi: 10.1146/annurev.immunol.19.1.375. [DOI] [PubMed] [Google Scholar]
- 20.Duan J, Wu J, Valencia CA, Liu R. Fibronectin type III domain based monobody with high avidity. Biochemistry. 2007;46:12656–12664. doi: 10.1021/bi701215e. [DOI] [PubMed] [Google Scholar]
- 21.Choo Y, Klug A. Designing DNA-binding proteins on the surface of filamentous phage. Curr Opin Biotechnol. 1995;6:431–436. doi: 10.1016/0958-1669(95)80073-5. [DOI] [PubMed] [Google Scholar]
- 22.Blancafort P, Segal DJ, Barbas CF., 3rd Designing transcription factor architectures for drug discovery. Mol Pharmacol. 2004;66:1361–1371. doi: 10.1124/mol.104.002758. [DOI] [PubMed] [Google Scholar]
- 23••.Silverman J, Liu Q, Bakker A, To W, Duguay A, Alba BM, Smith R, Rivas A, Li P, Le H, et al. Multivalent avimer proteins evolved by exon shuffling of a family of human receptor domains. Nat Biotechnol. 2005;23:1556–1561. doi: 10.1038/nbt1166. This paper describes the design principle of avimers, generating of very high affinity molecules to multiple targets, and in vivo efficacy of an anti-interleukin 6 avimer. [DOI] [PubMed] [Google Scholar]
- 24.Vogel C, Bashton M, Kerrison ND, Chothia C, Teichmann SA. Structure, function and evolution of multidomain proteins. Curr Opin Struct Biol. 2004;14:208–216. doi: 10.1016/j.sbi.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 25•.Burroughs AM, Allen KN, Dunaway-Mariano D, Aravind L. Evolutionary genomics of the HAD superfamily: understanding the structural adaptations and catalytic diversity in a superfamily of phosphoesterases and allied enzymes. J Mol Biol. 2006;361:1003–1034. doi: 10.1016/j.jmb.2006.06.049. A particularly comprehensive comparison of structures of an enzyme superfamily that supports nonhomologous recombination (in this case insertion) as a dominant driving force for functional diversification. [DOI] [PubMed] [Google Scholar]
- 26••.Huang J, Koide A, Makabe K, Koide S. Design of protein function leaps by directed domain interface evolution. Proc Natl Acad Sci U S A. 2008;105:6578–6583. doi: 10.1073/pnas.0801097105. This work establishes the affinity clamp concept, generates high-affinity affinity peptide-binding proteins and shows their utility as antibody alternatives. A high-resolution crystal structure of a clamp-peptide complex validates the design. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrer M, Maiolo J, Kratz P, Jackowski J, Murphy D, Delagrave S, Inglese J. Directed evolution of PDZ variants to generate high-affinity detection reagents. Protein Eng Des Sel. 2005;18:165–173. doi: 10.1093/protein/gzi018. [DOI] [PubMed] [Google Scholar]
- 28.Huang J, Makabe K, Biancalana M, Koide A, Koide S. Structural basis for exquisite specificity of affinity clamps, synthetic binding proteins generated through directed domain-interface evolution. J Mol Biol. 2009 doi: 10.1016/j.jmb.2009.07.067. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- 30.Hantschel O, Superti-Furga G. Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat Rev Mol Cell Biol. 2004;5:33–44. doi: 10.1038/nrm1280. [DOI] [PubMed] [Google Scholar]
- 31.Dueber JE, Yeh BJ, Bhattacharyya RP, Lim WA. Rewiring cell signaling: the logic and plasticity of eukaryotic protein circuitry. Curr Opin Struct Biol. 2004;14:690–699. doi: 10.1016/j.sbi.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 32.Bhattacharyya RP, Remenyi A, Yeh BJ, Lim WA. Domains, motifs, and scaffolds: the role of modular interactions in the evolution and wiring of cell signaling circuits. Annu Rev Biochem. 2006;75:655–680. doi: 10.1146/annurev.biochem.75.103004.142710. [DOI] [PubMed] [Google Scholar]
- 33.Howard PL, Chia MC, Del Rizzo S, Liu FF, Pawson T. Redirecting tyrosine kinase signaling to an apoptotic caspase pathway through chimeric adaptor proteins. Proc Natl Acad Sci U S A. 2003;100:11267–11272. doi: 10.1073/pnas.1934711100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Antunes MS, Morey KJ, Tewari-Singh N, Bowen TA, Smith JJ, Webb CT, Hellinga HW, Medford JI. Engineering key components in a synthetic eukaryotic signal transduction pathway. Mol Syst Biol. 2009;5:270. doi: 10.1038/msb.2009.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Monod J, Wyman J, Changeux JP. On the Nature of Allosteric Transitions: a Plausible Model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 36.Dueber JE, Yeh BJ, Chak K, Lim WA. Reprogramming control of an allosteric signaling switch through modular recombination. Science. 2003;301:1904–1908. doi: 10.1126/science.1085945. [DOI] [PubMed] [Google Scholar]
- 37••.Dueber JE, Mirsky EA, Lim WA. Engineering synthetic signaling proteins with ultrasensitive input/output control. Nat Biotechnol. 2007;25:660–662. doi: 10.1038/nbt1308. This work demonstrates the capacity of modular combination to create switches with nonlinear, highly cooperative responses. [DOI] [PubMed] [Google Scholar]
- 38.Wright CM, Heins RA, Ostermeier M. As easy as flipping a switch? Curr Opin Chem Biol. 2007;11:342–346. doi: 10.1016/j.cbpa.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 39•.Ostermeier M. Designing switchable enzymes. Curr Opin Struct Biol. 2009 doi: 10.1016/j.sbi.2009.04.007. A recent review on the design of protein switches. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Edwards WR, Busse K, Allemann RK, Jones DD. Linking the functions of unrelated proteins using a novel directed evolution domain insertion method. Nucleic Acids Res. 2008;36:e78. doi: 10.1093/nar/gkn363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Radley TL, Markowska AI, Bettinger BT, Ha JH, Loh SN. Allosteric switching by mutually exclusive folding of protein domains. J Mol Biol. 2003;332:529–536. doi: 10.1016/s0022-2836(03)00925-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cutler TA, Mills BM, Lubin DJ, Chong LT, Loh SN. Effect of interdomain linker length on an antagonistic folding-unfolding equilibrium between two protein domains. J Mol Biol. 2009;386:854–868. doi: 10.1016/j.jmb.2008.10.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moglich A, Ayers RA, Moffat K. Design and signaling mechanism of light-regulated histidine kinases. J Mol Biol. 2009;385:1433–1444. doi: 10.1016/j.jmb.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44•.Strickland D, Moffat K, Sosnick TR. Light-activated DNA binding in a designed allosteric protein. Proc Natl Acad Sci U S A. 2008;105:10709–10714. doi: 10.1073/pnas.0709610105. Design of mutual exclusivity by designing an overlap of a helix between two modules. A light-switchable transcription factor was successfully produced. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee J, Natarajan M, Nashine VC, Socolich M, Vo T, Russ WP, Benkovic SJ, Ranganathan R. Surface sites for engineering allosteric control in proteins. Science. 2008;322:438–442. doi: 10.1126/science.1159052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46••.Leung DW, Otomo C, Chory J, Rosen MK. Genetically encoded photoswitching of actin assembly through the Cdc42-WASP-Arp2/3 complex pathway. Proc Natl Acad Sci U S A. 2008;105:12797–12802. doi: 10.1073/pnas.0801232105. Unlike 39 and 40, the photoswitch of this work is based on modular design using light-dependent protein-protein interaction. In addition, this switch can be turned on and off using light of different wavelengths. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47•.Sallee NA, Yeh BJ, Lim WA. Engineering modular protein interaction switches by sequence overlap. J Am Chem Soc. 2007;129:4606–4611. doi: 10.1021/ja0672728. This describes the generation of modular switches by designing a segmental overlap between two modules that creates steric mutual exclusivity. Both modular domains and short peptides were used as building blocks. [DOI] [PubMed] [Google Scholar]
- 48•.Stratton MM, Mitrea DM, Loh SN. A Ca2+-sensing molecular switch based on alternate frame protein folding. ACS Chem Biol. 2008;3:723–732. doi: 10.1021/cb800177f. This describes a new strategy to engineer fluorescence sensors using a ligand-binding module that does not exhibit a large conformational change. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49••.Liang J, Kim JR, Boock JT, Mansell TJ, Ostermeier M. Ligand binding and allostery can emerge simultaneously. Protein Sci. 2007;16:929–937. doi: 10.1110/ps.062706007. The authors find a hybrid molecule between β-lactamase and MBP has acquired new binding function to zinc and also heterotropic allostery, demonstrating domain/module recombination as a route to new molecular recognition and regulation. [DOI] [PMC free article] [PubMed] [Google Scholar]