

Abstract
Glypican-3 (Gpc3) is a heparan sulfate proteoglycan (HSPG) expressed widely during vertebrate development. Loss-of-function mutations cause Simpson Golabi Behmel Syndrome (SGBS), a rare and complex congenital overgrowth syndrome with a number of associated developmental abnormalities including congenital heart disease. We found that Gpc3-deficient mice display a high incidence of congenital cardiac malformations like ventricular septal defects, common atrioventricular canal and double outlet right ventricle. In addition we observed coronary artery fistulas, which have not been previously reported in SGBS. Coronary artery fistulas are noteworthy because little is known about the molecular basis of this abnormality. Formation of the coronary vascular plexus in Gpc3-deficient embryos was delayed compared to wild-type, and consistent with GPC3 functioning as a co-receptor for fibroblast growth factor-9 (FGF9), we found a reduction in sonic hedgehog (Shh) mRNA expression and signaling in embryonic mutant hearts. Interestingly, we found an asymmetric reduction in SHH signaling in cardiac myocytes, as compared with perivascular cells, resulting in excessive coronary artery formation in the Gpc3-deficient animals. We hypothesize that the excessive development of coronary arteries over veins enables the formation of coronary artery fistulas. This work has broad significance to understanding the genetic basis of coronary development and potentially to molecular mechanisms relevant to revascularization following ischemic injury to the heart.
Keywords: proteoglycans, heparan sulfate, FGF, SHH, vasculature, coronary, heart
Introduction
HSPGs are expressed widely on both the surface of cells and within the extracellular matrix where they bind and modify the functions of a diverse array of ligands. Mutations in the genes encoding both enzymes in the heparan sulfate biosynthetic pathway, as well as those encoding specific core proteins that bear heparan sulfate, have been shown to result in significant developmental abnormalities due to defects in growth factor signaling (Perrimon and Bernfield, 2000). Although heparan sulfate itself is ubiquitous on cell surfaces this post-synthetic modification occurs predominantly on the proteins encoded by members of two gene families, syndecans (Saunders et al., 1989) and glypicans (David et al., 1990).
There are six members of the vertebrate glypican gene family (Paine-Saunders et al., 1999; Veugelers et al., 1999). These glypicans share a common mechanism of attachment to the cell surface through glycophosphatidylinositol linkages, and homologies within the core protein primary sequences suggests the existence of subfamilies of structurally related glypicans (Paine-Saunders et al., 1999; Veugelers et al., 1999). Multiple studies have provided evidence for the involvement of various glypicans in the modulation of cell signaling, including specifically several studies both in vitro and in vivo suggested a function for glypican-3 (Gpc3) in the BMP (Paine-Saunders et al., 2000), Hedgehog (Capurro et al., 2008), Wnt (De Cat et al., 2003; Song et al., 2005), and FGF (Grisaru et al., 2001) signaling pathways
Loss-of-function mutations in GPC3 causes an X-linked disorder in humans known as Simpson Golabi Behmel syndrome (SGBS), a disorder associated with both pre- and postnatal overgrowth, a predisposition to certain childhood cancers and a complex assortment of developmental birth defects including cardiac malformation. In the most comprehensive review of cardiac anomalies in patients with a clinical diagnosis of SGBS, Lin et al. described 101 patients (89 from the literature and 12 newly described patients) (Lin et al., 1999), 36 of whom had an abnormality of cardiac structure or function and 26 who had an anatomic heart defect. Of patients less than 3 years of age, death was associated with a cardiac abnormality in 23%. When only patients with a documented GPC3 gene mutation were considered, 46% had a cardiac abnormality.
Gpc3 knockout mice have not previously been evaluated for cardiac defects, but we suspected them given a consistent observation of as much as a 50% perinatal lethality in Gpc3-deficient animals (Paine-Saunders et al., 2000). To determine the potential effect of Gpc3 loss-of-function on cardiac development, we collected newborn (P0) mouse pups and performed a detailed histological evaluation of their cardiac anatomy. As expected, we found a high incidence of defects as seen in human patients with SGBS with ventricular septal defects (VSD) most common. Other structural defects included double outlet right ventricle and common atrioventricular canal. To our surprise, we found an especially high frequency of coronary artery fistulas, a defect rarely reported in association with mutations of other cardiac developmental genes and for which the molecular basis is poorly understood. We therefore evaluated the consequence of the loss of Gpc3 function to the process of coronary vascular development. Loss of Gpc3 function causes a delay in coronary vascular plexus formation and a reduction in downstream Shh signaling, consistent with a loss of Gpc3 function as a co-receptor for FGF9 signaling in the developing heart. Loss of Gpc3 function is associated with a selective reduction of SHH signaling in the cardiomyocytes, with relative preservation of signaling in the perivascular cells. This correlates with an excessive development of coronary arteries relative to veins that we propose is the pathogenic mechanism for the formation of coronary artery fistulas.
Methods
Animals
These experiments made use of the tm108 targeted null allele of Gpc3 as has been previously described (Paine-Saunders et al., 2000) and bred onto a pure background of C57Bl6. Because Gpc3 is an X-linked allele, we restricted our analysis to male animals either wild-type or hemizygous null for Gpc3 (Gpc3/Y). Genotypes of embryos were determined by PCR for the wild-type and Gpc3-mutant alleles, and sex was determined by PCR for Sry, both as previously described (Paine-Saunders et al., 2000).
Cardiac Histopathology
Neonatal pups were collected each morning within hours of birth to prevent cannibalization of animals that have hemodynamically significant congenital heart disease. The pups were euthanized. The thorax was fixed in 10% neutral buffered formalin and then transferred to 70% ethanol. Fixed hearts were dissected free and embedded in paraffin. The hearts were entirely sectioned in the frontal plane at 6 μm thickness. The sections were stained with hematoxylin and eosin, and two individuals independently inspected each heart under a 5× microscope objective for defects.
Non-Radioactive In Situ Hybridization
Slides containing zinc fixed, paraffin-embedded hearts or torsos from embryonic day 12.5-14.5 (E12.5-14.5) were deparaffinized and hydrated. Sections were fixed in 4% paraformaldehyde in PBS for 25 min, rinsed once in PBS, then digested with 10 μg/ml proteinase K for 10 min at 37°C. The digestion was stopped by incubating for 10 min in 0.2% glycine in PBS. Slides were then washed twice in PBS, acetylated in 0.1M triethanolamine (pH 8.0) and 0.25% acetic anhydride for 10 min, rinsed once more in PBS, before incubating at 65°C for 4 hours in prehybridization buffer, containing 50% formamide, 1.3× SSC, 5mM EDTA, 50 μg/ml yeast tRNA, 0.2% Tween 20 and 0.5% CHAPS. Hybridization was carried out using Digoxigenin-labeled antisense probes at 65°C for 16-20 hours in a humidity chamber. Slides were rinsed and treated with 20 μg/ml RNase A for 30 min at 37°C, washed twice with 2× SSC, 0.1% CHAPS for 10 min at 65°C, then twice with 0.2× SSC, 0.1% CHAPS for 10 min at 65°C, then twice in KTBT buffer containing 50 mM Tris pH 7.5, 150mM NaCl, 10mM KCl and 1% Triton X-100 for 5 min. Sections were blocked for 2 hours at 4°C in KTBT with 0.1% Blocking Reagent (Roche) and 20% sheep serum (Sigma) and then incubated with alkaline phosphatase labeled anti-digoxigenin (Roche) for 16-20 hours, washed with KTBT five times 10 min, then washed with NTMT containing 40 mM Tris pH 9.5, 100 mM NaCl, 40 mM MgCl and 0.2% Tween 20 twice for 5 min. Development was with BCIP/ NBT (Vector Labs) and slides were counterstained with Nuclear Fast Red (Vector Labs).
Whole-mount In Situ Hybridization
E13.5 hearts were fixed in 4% paraformaldehyde and processed according to the protocol described by Colvin et al. (2001) (Colvin et al., 2001) with the following modifications: a 0.2% glycine wash was included after the proteinase K digestion, the hybridization solution did not contain any heparin and we included 0.1% Blocking Reagent (Roche) in the antibody blocking buffer.
Whole-mount Immunohistochemistry
E13.5 hearts were fixed in 4% paraformaldehyde and processed according to the protocol described by White et al. (2007) (White et al., 2007) with the following modifications: a 0.1% Blocking Reagent (Roche) was added to the blocking buffer, the secondary antibody and subsequent steps were from an ABC elite kit (Vector Labs) and the hearts were incubated in the ABC reagent overnight at 4°C.
Results
Loss of Gpc3 function results in structural heart defects in mice including coronary artery fistulas
Gpc3 is widely expressed in the cardiomyocytes of the embryonic heart, and human patients with SGBS commonly have congenital heart defects (Lin et al., 1999) thus demonstrating the importance of Gpc3 function to cardiac development. To assess the effect of the loss of Gpc3 function in cardiac development, a detailed histological examination was performed of thirty hearts from newborn (P0) mice either wild-type (N=14) or deficient for Gpc3 (N=16). While the wild-type hearts were all normal, there was a high incidence of heart defects in the Gpc3-deficient animals (69%) representing a variety of abnormalities with the most common being VSD (5/16 hearts; 4 perimembranous, 1 inlet). Defects of the outflow tract and atrioventricular canal development were also found and one mutant animal had a double outlet right ventricle. Whereas the pulmonary artery and aorta normally arise from the right and left ventricles, respectively (Fig. 1A, B), in the double outlet right ventricle the pulmonic valve (Fig. 1C, arrowhead) was ventral and leftward to the aortic valve, which also arose from the right ventricle (Fig. 1D, arrowhead). The VSD was in the subaortic position, and there was fibrous discontinuity between the aortic and mitral valves (not shown). In contrast to the separate tricuspid and mitral valves, as seen in a wild-type littermate (Fig. 1E), two mutant animals had a common atrioventricular canal with leaflets of the common valve bridging the two ventricles (Fig. 1F). Figure 1G shows an example of a perimembranous VSD and a summary of the cardiac defects and their frequency in Gpc3-deficient animals are shown (Fig. 1H).
Figure 1. Congenital heart defects in Gpc3-deficient hearts.
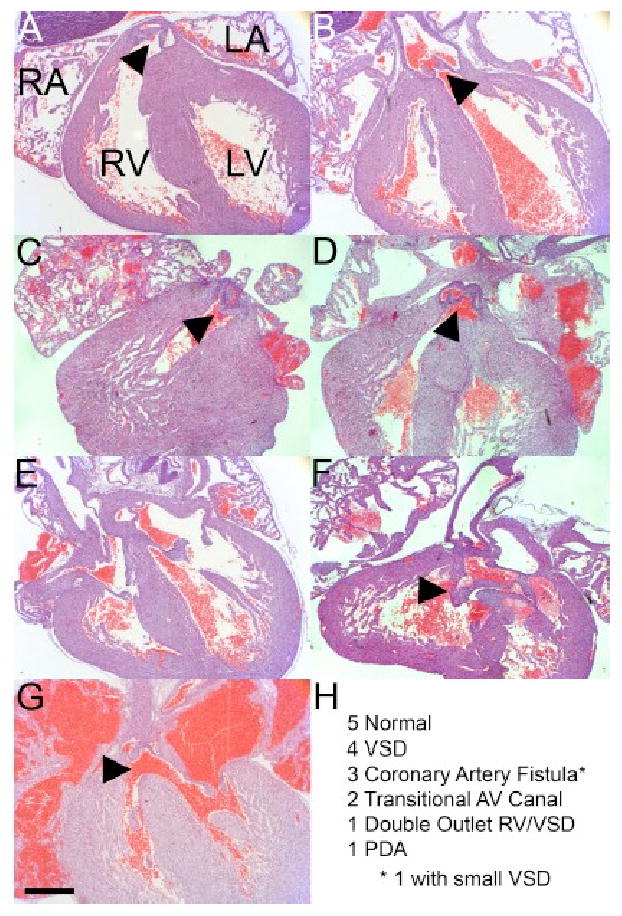
(A, B) The normal origins of the pulmonary artery and aorta (arrowheads) from the right and left ventricles, respectively, are shown. (C, D) Double outlet right ventricle in a Gpc3/Y mouse. Both great arteries originate from the right ventricle. The pulmonic valve (C, arrowhead) is ventral and leftward to the aortic valve (D, arrowhead). (E) Separate tricuspid and mitral valves are shown in this wild-type mouse. (F) Common atrioventricular canal in a Gpc3/Y mouse. The common atrioventricular valve (arrowhead) is shown. (G) Perimembranous VSD in a Gpc3/Y mouse (arrowhead). (H) Summary of observed cardiac phenotypes in Gpc3/Y mice. Of the 5 VSDs, 4 were small perimembranous and 1 was a small inlet. * One heart with a coronary artery fistula had a small perimembranous VSD. The double outlet right ventricle was associated with a subaortic VSD. We observed 1 patent ductus arteriosus (PDA). All 14 wild-type littermate control hearts were structurally normal. Bar, 500 μm.
Unexpectedly, we diagnosed coronary artery fistulas in 3 of 16 Gpc3-deficient animals (Fig. 1H). In contrast to the normal proximal left anterior descending (LAD) and circumflex (CIRC) arteries in a wild-type littermate (Fig. 2A), some Gpc3-deficient animals had a dilated LAD (Fig. 2B, arrowhead) feeding a coronary artery fistula (Fig. 2B) that coursed through the interventricular septum and drained into the right ventricle (Fig. 2C, arrowhead). Congenital heart defects have frequently been described in mouse models, but coronary artery fistulas have rarely been reported (Li et al., 2002; Ratajska et al., 2005). We therefore concluded that Gpc3 plays an important role in coronary vascular development. This novel foothold into the molecular basis of coronary artery fistulas motivated us to investigate the developmental and signaling mechanisms related to Gpc3 function in coronary development.
Figure 2. Gpc3-deficient hearts display coronary artery fistulas.

The proximal left anterior descending (LAD) and circumflex (CIRC) arteries are shown in a wild-type mouse (A). (B, C) A coronary artery fistula from a Gpc3/Y mouse. The proximal portion of the fistula shows a dilated LAD (arrowhead, B) feeding a coronary artery fistula (CAF) that courses through the interventricular septum ultimately draining into the right ventricle (arrowhead, C). Although the connection to the right ventricle appears small in the fixed specimen, the shunt volume in the animal must have been large enough to cause significant coronary artery dilatation. Bar, 500 μm.
Loss of Gpc3 expression causes delayed development of the coronary vascular plexus
We evaluated the timing and progression of the development of the coronary vascular plexus by whole-mount immunohistochemical staining of E13.5 hearts with an antibody against platelet endothelial cell adhesion molecule-1 (PECAM-1), a vascular marker. Blood vessels normally emerge from the atrioventricular groove at approximately E11.5, expand towards the apices of the left and right ventricle through E12.5 and eventually cover the entire ventricular surface by E13.5 (Lavine et al., 2006). Various heparin-dependent growth factors, including FGF and SHH, are critical to the initial formation of the plexus, and loss of function mutations in these signaling pathways cause a delay in its development (Lavine et al., 2006). Compared to wild-type littermate controls, Gpc3-deficient hearts show a nearly identical delay in the development of the coronary vascular plexus (Fig. 3A, B). To quantify the delay, the percentage of the ventricles covered by blood vessels was measured in collections of wild-type and Gpc3-deficient animals from a total of five different litters. Although variation existed between individual litters in the overall magnitude of delay in coronary vascular plexus formation, a consistent delay was observed between the mutant and wild-type embryos within a litter. The Gpc3-deficient vascular plexus covered an average of 46.6-84.1% of the heart, compared to 91.5-97.2% in the wild-type littermates. The results from a representative litter are shown. In this litter 97.2% ± 1.7% of the 5 wild-type hearts were covered with vessels at E13.5, compared to 81.4% ± 5.0% (p<0.0001) for the 5 Gpc3-deficient hearts (Fig. 3C).
Figure 3. Gpc3-deficient hearts exhibit a delayed development of the coronary vascular plexus.
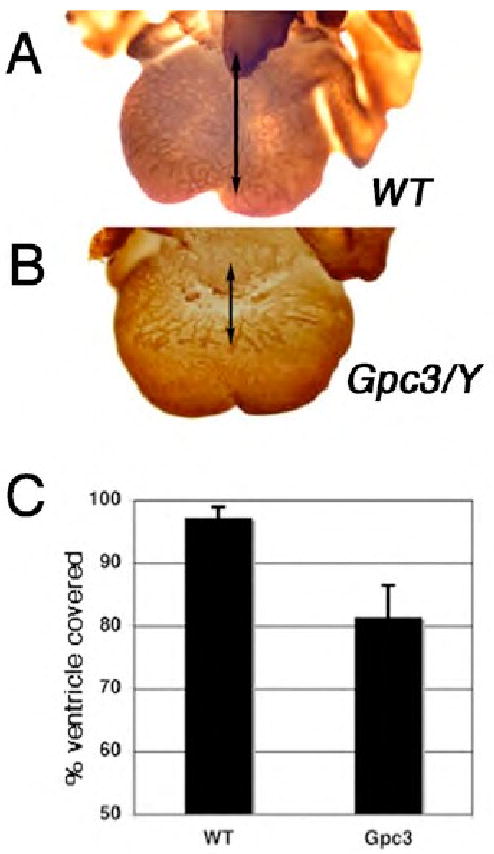
Whole mount PECAM staining of E13.5 day hearts demonstrated that when compared to the vascular plexus of wild-type control hearts (A), Gpc3-deficient hearts (B) display delayed development of the coronary vascular plexus. Arrows indicate the extent of the vascular plexus progression down the ventricle. (C) Quantification of the percentages of the ventricle covered by blood vessels is shown from a single representative litter containing 5 wild-type (97.2% ± 1.7%) and 5 Gpc3-deficient animals (81.4% ± 5.0%) p<0.0001.
Gpc3-deficient hearts express reduced levels of Sonic Hedgehog (Shh) mRNA
Several heparan sulfate-dependent growth factors have been shown to be critical for normal coronary vascular development and as such could be responsible for the observed phenotype in Gpc3-deficent animals. In particular, FGF9 has been shown to be critical to coronary artery development. Like all FGFs, FGF9 requires a heparan sulfate proteoglycan to form an active ternary signaling complex (Lavine et al., 2006). FGF9 functions indirectly in coronary artery development through the actions of Sonic Hedgehog (SHH), secreted by the epicardium as a consequence of FGF9 signaling. Mice deficient for Fgf9 displayed a reduction in Shh mRNA expression and a reduction in SHH signaling as revealed by a reduction in the mRNA expression of Patched1 (Ptc1), a component of the SHH signaling complex and a direct target gene for SHH. The reduction in SHH signal strength is responsible for the delay in coronary vascular plexus formation (Lavine et al., 2006).
The delay in coronary vascular plexus formation in Fgf9 null animals is nearly identical to the phenotype we observed in Gpc3-deficient animals. Therefore, to characterize the potential relationship between Gpc3 and FGF9/SHH signaling axis in the embryonic heart, we evaluated the level of expression of Shh mRNA levels by whole mount in situ hybridization in wild-type and Gpc3-deficient animals. Shh mRNA expression was diminished in hearts that lacked expression of Gpc3 compared to wild-type controls (Fig. 4), similar to Fgf9-deficient animals, and consistent with a function of GPC3 as a co-receptor for FGF9 signaling.
Figure 4. Shh mRNA levels are decreased in Gpc3-deficient hearts.
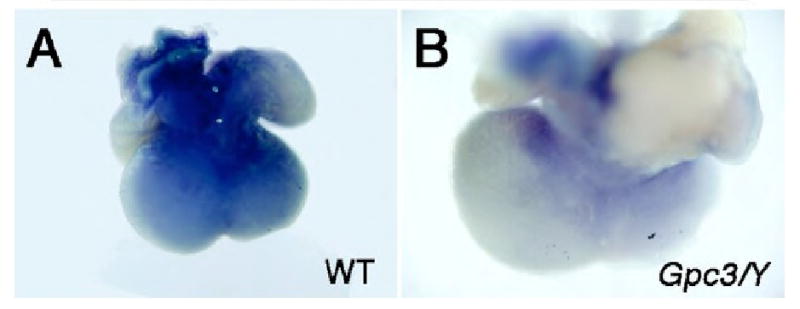
Whole-mount in situ hybridization at embryonic day 12.5 demonstrates that mRNA levels of Shh are significantly decreased in Gpc3 deficient hearts (B) compared to wild-type controls (A).
Loss of Gpc3 function selectively reduces SHH signaling by cardiomyocytes with relatively preserved signaling within the perivascular cells
A reduction in Shh mRNA expression does not necessarily confirm a reduction in SHH protein levels, or more specifically the level or strength of SHH signaling. The reduction in SHH mRNA expression in Fgf9-deficient animals causes a reduction in Ptc1 mRNA expression, a component of as well as a direct target of the SHH signaling pathway, and therefore directly confirms a reduction in SHH signaling. We found a similar reduction in Ptc1 mRNA expression in Gpc3-deficient hearts by whole mount in situ hybridization (data not shown), but unlike Fgf9-deficient animals the reduction in Ptc1 expression was cell-type specific. Although SHH signaling, as assessed by Ptc1 mRNA in situ expression at E13.5, is markedly reduced in the cardiomyocytes of Gpc3-deficient hearts, signaling is relatively preserved in the perivascular cells (Fig. 5B and C). The observation that SHH signaling is asymmetrically altered in Gpc3-deficient hearts, while surprising, is consistent with and may well explain the complex phenotype of coronary artery fistulas in Gpc3-deficient animals as described below.
Figure 5. Ptc1 mRNA expression is reduced in cardiomyocytes, but preserved in the perivascular cells of Gpc3-deficient animals.
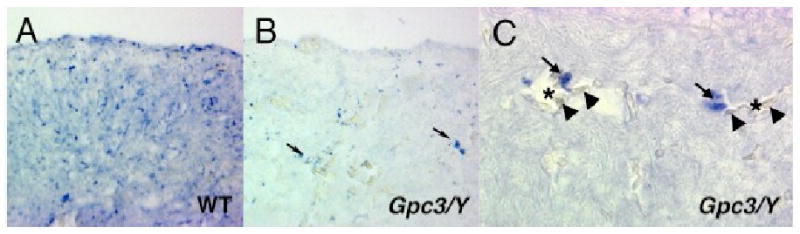
In situ hybridization detection of Ptc1 mRNA in tissue sections of embryonic day 13.5 wild-type (A) and Gpc3-deficient hearts (B), reveals that although there is an overall reduction in Ptc1 mRNA expression in Gpc3-deficient animals, there is relatively preserved expression by perivascular cells responsible for the formation of coronary arteries (arrows). Higher magnification view of Gpc3-deficient heart showing expression of Ptc1 cytoplasmic mRNA in cells with perivascular localization (arrows). Red cells (arrowheads) in the vascular lumen (asterisks) are as marked.
Loss of Gpc3 alters the relative abundance of coronary arteries and veins
The impairment of the FGF/SHH signaling axis in the embryonic hearts of Gpc3-deficient animals can explain the delayed formation of the coronary vascular plexus but does not by itself explain the pathogenesis of coronary artery fistulas. Somehow, the fistulas must arise from an inability of arterial progenitors to connect to the venous system, which could result from a defect in any number of processes like cell-type specification or cell-cell communication. Coronary arteries are well established to develop from vessels deep within the myocardium, while the veins arise and are localized superficially in the subepicardium (Lavine et al., 2008b; Ratajska et al., 2003), and Shh signaling in perivascular cells and cardiomyocytes, respectively, specify the development of these coronary arteries and veins in their distinct histological locations (Lavine et al., 2008b). The differential effects of the loss of Gpc3 function on SHH signaling in cardiac myocytes in contrast with perivascular cells, as demonstrated above, would be expected to lead to an asymmetrical development of the coronary arteries and veins. To address this issue, we examined and counted intramyocardial and subepicardial vessels in representative 400× fields of view from sections at the bases of PECAM-stained, E13.5 Gpc3-deficient and wild-type hearts. Representative sections clearly showed an increased number of PECAM-stained cells in the myocardium of Gpc3-deficient hearts compared to wild-type (Fig. 6A, B). There were approximately five times as many intramyocardial (aterial) vessels in the mutant as in the wild-type, whereas the number of subepicardical (venous) vessels tended to be less (Fig. 6C). Therefore, loss of Gpc3 function causes a disproportionate increase in the number of coronary arteries relative to veins. Given an excess of arteries, those that do not connect to a vein might default in some animals to a cardiac chamber to form a fistula.
Figure 6. Loss of Gpc3 expression results in an increase in the number of intramyocardial vessels.

Whole-mount PECAM-stained hearts were sectioned and then counterstained with hematoxylin. The coronary veins are found in the subepicardial layer noted in (A) and (B) by arrows, while coronary arteries are found in the intramyocardial layer demarked by the arrowhead in (B). As shown quantitatively (C), there is a significant increase in the number of intramyocardial vessels in Gpc3 deficient hearts compared to wild-type hearts. Sections were photographed at 400×.
Discussion
A high incidence of congenital heart defects is found in SGBS in association with the loss of function of the heparan sulfate proteoglycan (HSPG) known as Gpc3. HSPGs in general, and glypicans in particular, are widely expressed and function as critical modulators of a diverse array of signaling pathways during development. These include FGFs, Hedgehogs, Wnts and BMPs (Filmus et al., 2008) pathways that have all been implicated in the pathogenesis of congenital heart malformation (Andelfinger, 2008). FGF and Hedgehog signaling, in particular, function in the development of the cardiac outflow tract and atrioventricular canal, and the absence of Gpc3 expression in the developing heart and associated tissues likely contribute to the heart defects observed in our mouse model (Goddeeris et al., 2008; Goddeeris et al., 2007; Hutson et al., 2006; Park et al., 2008). Although coronary vascular abnormalities have not been specifically described in SGBS, there has been a case report describing clinically significant abnormalities of the carotid artery (Penisson-Besnier et al., 2008) suggesting the importance of studying Gpc3 to understanding the biology of vascular development. The unanticipated discovery of coronary artery fistulas in Gpc3-deficient mice offers novel insight into the molecular basis of coronary development.
Although the phenotypes observed on this C57Bl6 inbred strain display incomplete penetrance and variable expressivity, this observation is neither surprising nor is it a contradiction to the conclusion that Gpc3 is critical to these aspects of cardiac development. Variability in both penetrance and expressivity are well established observations in association with other genetic mutations resulting in cardiac developmental defects in inbred strains, and speaks to the important role of epigenetic influences and stochastic events in the phenotypic presentation of cardiac defects (Black, 2006). Similarly, we would anticipate, as in the case of other phenotypes previously observed in association with loss of Gpc3 function (Cano-Gauci et al., 1999; Chiao et al., 2002; Paine-Saunders et al., 2000), that there is strain-specific variability reflective of the important role of genetic modifiers. However, exploration of these various factors was not the focus of the present study.
Development of the vasculature typically begins in most tissues with the formation of a primary capillary plexus via the process of vasculogenesis, which is then remodeled through a process of angiogenesis (Flamme et al., 1997; Risau, 1997). During the remodeling process, the various caliber arterial and venous vessels emerge permitting blood to flow from the arteries to the arterioles, capillaries, venules and finally veins. The coronary vascular system follows a similar sequence of development, which is tightly regulated by a cascade of growth factor signals. The epicardium has been identified as a critical modulator of this development because removal of the epicardium results in the loss of signaling molecules critical to the initiation of this cascade and therefore defects in coronary development (Morabito et al., 2002; Reese et al., 2002; Wada et al., 2003). FGF9 released from the epicardium signals through FGF receptors on the cardiomyocytes to result in the release of a wave of Shh expression by the epicardium (Lavine et al., 2006). This in turn modulates via Vegf the wave of growth and development of the coronary vascular plexus as it originates in the atrial-ventricular groove and extends toward the ventricular apices (Lavine et al., 2006). Loss of either Fgf9 expression by the epicardium or conditional loss of both Fgfr1 and Fgfr2 receptor expression by the cardiomyocytes results in a delay in formation of the coronary vascular plexus identical to what we observed for Gpc3 mutants (Lavine et al., 2006).
One of the many functions of HSPGs is as an essential co-receptor for FGF signal transduction (Rapraeger et al., 1991; Yayon et al., 1991). Although heparan sulfate (HS) carbohydrate chains of HSPGs bind FGFs with low affinity, this low affinity interaction promotes the formation of a higher affinity complex between FGFs and their tyrosine kinase signaling receptors FGFR1-FGFR4 (Nugent and Edelman, 1992; Roghani et al., 1994). While the precise stoichiometry and mechanism of this functional interaction remains an issue of active investigation (Goodger et al., 2008), it is well established that HS binds to both FGFs (Ornitz et al., 1995) and the FGF receptors (Kan et al., 1993) thus increasing the affinity of FGFs for their signaling receptors and are therefore essential for receptor dimerization and subsequent signaling (Herr et al., 1997; Pantoliano et al., 1994; Springer et al., 1994; Venkataraman et al., 1996).
Glypican family members are capable of augmenting FGF signaling both in in vitro and in vivo systems (Aikawa et al., 2008; Bashkin et al., 1992; Bonneh-Barkay et al., 1997; Brunner et al., 1991; Lai et al., 2008; Steinfeld et al., 1996; Yan and Lin, 2007); however, little is known about the normal functional relationships between specific glypicans and specific FGF ligand/receptor pairs during development. Gpc3 is co-expressed with the Fgfr1 and Fgfr2 receptors of the cardiomyocytes required for the reception and subsequent relay of the FGF9 signals responsible for the control of coronary vascular development, and loss of function of Gpc3 in these same cells mimics the loss of Fgf9 function with respect to both the induction of Shh mRNA expression and the resulting progression of development of the coronary vascular plexus. These findings are most readily explained by GPC3 providing an essential co-receptor function for FGF9 in these cells, although we can not exclude the possibility that loss of Gpc3 expression interferes with other heparin-dependent cellular process that independently down regulates Shh mRNA expression in the heart.
While loss of Gpc3 function by the cardiomyocytes mimics the phenotype of loss of Fgf9 function with respect to the initiation and progression of the vascular plexus, we also found an imbalance between the formation of coronary arteries and veins that is not seen in association with either the loss of Fgf9 function or a generalized reduction in Shh mRNA induction alone. Anatomical studies using corrosion casting of late gestation hearts have unambiguously demonstrated that coronary arteries and veins can be identified by their development within distinct regions of the heart (Ratajska et al., 2003). Using the conditional disruption of SHH signal reception and specific molecular markers distinguishing coronary arteries and veins, the Ornitz laboratory have furthermore confirmed that coronary arteries develop deep within the myocardial wall in response to SHH signal reception within the perivascular cells, while coronary veins form more superficially in the subepicardial region in response to SHH signaling to the cardiomyocytes (Lavine et al., 2008b). Our results clearly show that loss of Gpc3 function results in a disproportionate accumulation of coronary arteries as compared with veins, an alteration that we speculate is responsible ultimately for the routing of these excess coronary arteries to the cardiac chamber, and thus the formation of the coronary artery fistulas we observed in Gpc3-deficient animals.
The imbalance between artery and vein formation in Gpc3-deficient animals would imply that although there is an overall reduction in Shh mRNA expression, SHH signaling is disproportionately reduced in the cardiomyocytes of Gpc3-deficient mice as compared with perivascular cells, a result confirmed by the examination of Ptc1 mRNA expression in Gpc3-deficient hearts. We do not know at the moment the molecular details by which the loss of Gpc3 function might lead to differential SHH signaling between these two populations of cells in the heart. Although the Filmus Lab has recently provided evidence for alternative functions of GPC3 including a direct inhibitory activity of GPC3 on SHH signaling in other tissues, a similar activity in the heart would not explain our results. We found that loss of Gpc3 function causes a reduction in Shh mRNA expression and a reduction in SHH signaling in cardiomyocytes. Gpc3 is widely expressed by cardiomyocytes, and loss of a SHH-inhibitory function in these cells would be expected to result in an increase in SHH signaling in those cells, rather than a reduction, and therefore an accelerated rate of vascular plexus formation and a relative excess in vein formation over arteries. Preserved and/or disproportionate SHH signaling in perivascular cells in the setting of a reduction in the production of SHH by the epicardium could be a reflection of enhanced diffusion of SHH in the absence of GPC3 on the surface of the cardiomyocytes. In addition and/or alternatively, loss of Gpc3 function might lead to the unopposed promotion of SHH signaling in perivascular cells by a different HSPG that actually promotes SHH signaling, perhaps another member of the glypican gene family. The Drosophila glypican Dally-like for example, has been shown to actually promote Hedgehog signaling in flies (Beckett et al., 2008; Gallet et al., 2008), and the vertebrate ortholog of this glypican, Gpc6, is also expressed in the developing heart (data not shown). Further studies will be necessary to distinguish between these alternative hypotheses.
Our results provide the first evidence of a role for GPC3 in vascular development and specifically an important novel insight into the molecular mechanisms of coronary artery fistula formation. Furthermore, appreciating the role of GPC3 in coronary vascular development has potentially broad clinical implications beyond understanding the development of coronary artery fistulas including, specifically, a potential relationship to the prevention and treatment of ischemic heart disease. Recent attempts at developing pharmacological approaches towards the prevention and/or treatment of ischemic disease of the heart, a major source of morbidity and mortality in humans, have included the administration of either FGF or VEGF-A. While promising in animal models, these approaches have thus far shown little efficacy in humans (Henry et al., 2000; Losordo et al., 2002; Simons et al., 2002). More recently, temporal and tissue-specific disruption of SHH function in the adult heart in vivo has been found to result in a loss of the normal homeostatic maintenance of the coronary vasculature that results in ischemic injury (Lavine et al., 2008a), while activation of SHH signaling in vivo is sufficient to promote coronary neovascularization and to be protective against ischemia (Kusano et al., 2005). Together, these observations suggest that the SHH signaling pathway may be a useful target for the treatment of ischemic heart disease and that GPC3 may be one potential target for therapeutic intervention.
Acknowledgments
The authors wish to acknowledge David Ornitz for many helpful discussions and the sharing of unpublished data. S.S. is supported by NIH RO1-DK56063 and Children's Discovery Institute MD-II-2009-193. P.Y.J. is a Scholar of the Child Health Research Center of Excellence in Developmental Biology at Washington University School of Medicine (NIH K12-HD001487) and is supported by American Heart Association and March of Dimes.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aikawa T, Whipple CA, Lopez ME, Gunn J, Young A, Lander AD, Korc M. Glypican-1 modulates the angiogenic and metastatic potential of human and mouse cancer cells. J Clin Invest. 2008;118:89–99. doi: 10.1172/JCI32412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andelfinger G. Genetic factors in congenital heart malformation. Clin Genet. 2008;73:516–27. doi: 10.1111/j.1399-0004.2008.01009.x. [DOI] [PubMed] [Google Scholar]
- Bashkin P, Neufeld G, Gitay-Goren H, Vlodavsky I. Release of cell surface-associated basic fibroblast growth factor by glycosylphosphatidylinositol-specific phospholipase. C J Cell Physiol. 1992;151:126–37. doi: 10.1002/jcp.1041510117. [DOI] [PubMed] [Google Scholar]
- Beckett K, Franch-Marro X, Vincent JP. Glypican-mediated endocytosis of Hedgehog has opposite effects in flies and mice. Trends Cell Biol. 2008;18:360–3. doi: 10.1016/j.tcb.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Black BL. A single mutation causes a spectrum of cardiovascular defects: the potential role of genetic modifiers, epigenetic influences, and stochastic events in phenotypic variability. J Mol Cell Cardiol. 2006;40:201–4. doi: 10.1016/j.yjmcc.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Bonneh-Barkay D, Shlissel M, Berman B, Shaoul E, Admon A, Vlodavsky I, Carey DJ, Asundi VK, Reich-Slotky R, Ron D. Identification of glypican as a dual modulator of the biological activity of fibroblast growth factors. J Biol Chem. 1997;272:12415–21. doi: 10.1074/jbc.272.19.12415. [DOI] [PubMed] [Google Scholar]
- Brunner G, Gabrilove J, Rifkin DB, Wilson EL. Phospholipase C release of basic fibroblast growth factor from human bone marrow cultures as a biologically active complex with a phosphatidylinositol-anchored heparan sulfate proteoglycan. J Cell Biol. 1991;114:1275–1283. doi: 10.1083/jcb.114.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano-Gauci DF, Song HH, Yang H, McKerlie C, Choo B, Shi W, Pullano R, Piscione TD, Grisaru S, Soon S, Sedlackova L, Tanswell AK, Mak TW, Yeger H, Lockwood GA, Rosenblum ND, Filmus J. Glypican-3-deficient mice exhibit developmental overgrowth and some of the abnormalities typical of Simpson-Golabi- Behmel Syndrome J Cell Biol. 1999;146:255–264. doi: 10.1083/jcb.146.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capurro MI, Xu P, Shi W, Li F, Jia A, Filmus J. Glypican-3 inhibits Hedgehog signaling during development by competing with patched for Hedgehog binding. Dev Cell. 2008;14:700–11. doi: 10.1016/j.devcel.2008.03.006. [DOI] [PubMed] [Google Scholar]
- Chiao E, Fisher P, Crisponi L, Deiana M, Dragatsis I, Schlessinger D, Pilia G, Efstratiadis A. Overgrowth of a mouse model of the Simpson-Golabi-Behmel syndrome is independent of IGF signaling. Dev Biol. 2002;243:185–206. doi: 10.1006/dbio.2001.0554. [DOI] [PubMed] [Google Scholar]
- Colvin JS, White AC, Pratt SJ, Ornitz DM. Lung hypoplasia and neonatal death in Fgf9-null mice identify this gene as an essential regulator of lung mesenchyme. Development. 2001;128:2095–106. doi: 10.1242/dev.128.11.2095. [DOI] [PubMed] [Google Scholar]
- David G, Lories V, Decock B, Marynen P, Cassiman JJ, Berghe HVd. Molecular cloning of a phosphatidylinositol-anchored membrane heparan sulfate proteoglycan from human lung fibroblasts. J Cell Biol. 1990;111:3165–3176. doi: 10.1083/jcb.111.6.3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cat B, Muyldermans SY, Coomans C, Degeest G, Vanderschueren B, Creemers J, Biemar F, Peers B, David G. Processing by proprotein convertases is required for glypican-3 modulation of cell survival, Wnt signaling, and gastrulation movements. J Cell Biol. 2003;163:625–35. doi: 10.1083/jcb.200302152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filmus J, Capurro M, Rast J. Glypicans. Genome Biol. 2008;9:224. doi: 10.1186/gb-2008-9-5-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamme I, Frolich T, Risau W. Molecular mechanisms of vasculogenesis and embryonic angiogenesis. J Cell Physiol. 1997;173:206–10. doi: 10.1002/(SICI)1097-4652(199711)173:2<206::AID-JCP22>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Gallet A, Staccini-Lavenant L, Therond PP. Cellular trafficking of the glypican Dally-like is required for full-strength Hedgehog signaling and wingless transcytosis. Dev Cell. 2008;14:712–25. doi: 10.1016/j.devcel.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Goddeeris MM, Rho S, Petiet A, Davenport CL, Johnson GA, Meyers EN, Klingensmith J. Intracardiac septation requires hedgehog-dependent cellular contributions from outside the heart. Development. 2008;135:1887–95. doi: 10.1242/dev.016147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddeeris MM, Schwartz R, Klingensmith J, Meyers EN. Independent requirements for Hedgehog signaling by both the anterior heart field and neural crest cells for outflow tract development. Development. 2007;134:1593–604. doi: 10.1242/dev.02824. [DOI] [PubMed] [Google Scholar]
- Goodger SJ, Robinson CJ, Murphy KJ, Gasiunas N, Harmer NJ, Blundell TL, Pye DA, Gallagher JT. Evidence that heparin saccharides promote FGF2 mitogenesis through two distinct mechanisms. J Biol Chem. 2008;283:13001–8. doi: 10.1074/jbc.M704531200. [DOI] [PubMed] [Google Scholar]
- Grisaru S, Cano-Gauci D, Tee J, Filmus J, Rosenblum ND. Glypican-3 modulates BMP- and FGF-mediated effects during renal branching morphogenesis. Dev Biol. 2001;231:31–46. doi: 10.1006/dbio.2000.0127. [DOI] [PubMed] [Google Scholar]
- Henry GD, Byrne R, Hunyh TT, Abraham V, Annex BH, Hagen PO, Donatucci CF. Intracavernosal injections of vascular endothelial growth factor protects endothelial dependent corpora cavernosal smooth muscle relaxation in the hypercholesterolemic rabbit: a preliminary study. Int J Impot Res. 2000;12:334–9. doi: 10.1038/sj.ijir.3900621. [DOI] [PubMed] [Google Scholar]
- Herr AB, Ornitz DM, Sasisekharan R, Venkataraman G, Waksman G. Heparin-induced self-association of fibroblast growth factor-2. Evidence for two oligomerization processes. Journal of Biological Chemistry. 1997;272:16382–9. doi: 10.1074/jbc.272.26.16382. [DOI] [PubMed] [Google Scholar]
- Hutson MR, Zhang P, Stadt HA, Sato AK, Li YX, Burch J, Creazzo TL, Kirby ML. Cardiac arterial pole alignment is sensitive to FGF8 signaling in the pharynx. Dev Biol. 2006;295:486–97. doi: 10.1016/j.ydbio.2006.02.052. [DOI] [PubMed] [Google Scholar]
- Kan M, Wang F, Xu J, Crabb JW, Hou J, McKeehan WL. An essential heparin-binding domain in the fibroblast growth factor receptor kinase. Science. 1993;259:1918–1921. doi: 10.1126/science.8456318. [DOI] [PubMed] [Google Scholar]
- Kusano KF, Pola R, Murayama T, Curry C, Kawamoto A, Iwakura A, Shintani S, Ii M, Asai J, Tkebuchava T, Thorne T, Takenaka H, Aikawa R, Goukassian D, von Samson P, Hamada H, Yoon YS, Silver M, Eaton E, Ma H, Heyd L, Kearney M, Munger W, Porter JA, Kishore R, Losordo DW. Sonic hedgehog myocardial gene therapy: tissue repair through transient reconstitution of embryonic signaling. Nat Med. 2005;11:1197–204. doi: 10.1038/nm1313. [DOI] [PubMed] [Google Scholar]
- Lai JP, Sandhu DS, Yu C, Han T, Moser CD, Jackson KK, Guerrero RB, Aderca I, Isomoto H, Garrity-Park MM, Zou H, Shire AM, Nagorney DM, Sanderson SO, Adjei AA, Lee JS, Thorgeirsson SS, Roberts LR. Sulfatase 2 up-regulates glypican 3, promotes fibroblast growth factor signaling, and decreases survival in hepatocellular carcinoma. Hepatology. 2008;47:1211–22. doi: 10.1002/hep.22202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavine KJ, Kovacs A, Ornitz DM. Hedgehog signaling is critical for maintenance of the adult coronary vasculature in mice. J Clin Invest. 2008a;118:2404–14. doi: 10.1172/JCI34561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavine KJ, Long F, Choi K, Smith C, Ornitz DM. Hedgehog signaling to distinct cell types differentially regulates coronary artery and vein development. Development. 2008b;135:3161–71. doi: 10.1242/dev.019919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavine KJ, White AC, Park C, Smith CS, Choi K, Long F, Hui CC, Ornitz DM. Fibroblast growth factor signals regulate a wave of Hedgehog activation that is essential for coronary vascular development. Genes Dev. 2006;20:1651–66. doi: 10.1101/gad.1411406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WE, Waldo K, Linask KL, Chen T, Wessels A, Parmacek MS, Kirby ML, Lo CW. An essential role for connexin43 gap junctions in mouse coronary artery development. Development. 2002;129:2031–42. doi: 10.1242/dev.129.8.2031. [DOI] [PubMed] [Google Scholar]
- Lin AE, Neri G, Hughes-Benzie R, Weksberg R. Cardiac anomalies in the Simpson-Golabi-Behmel syndrome. Am J Med Genet. 1999;83:378–81. [PubMed] [Google Scholar]
- Losordo DW, Vale PR, Hendel RC, Milliken CE, Fortuin FD, Cummings N, Schatz RA, Asahara T, Isner JM, Kuntz RE. Phase 1/2 placebo-controlled, double-blind, dose-escalating trial of myocardial vascular endothelial growth factor 2 gene transfer by catheter delivery in patients with chronic myocardial ischemia. Circulation. 2002;105:2012–8. doi: 10.1161/01.cir.0000015982.70785.b7. [DOI] [PubMed] [Google Scholar]
- Morabito CJ, Kattan J, Bristow J. Mechanisms of embryonic coronary artery development. Curr Opin Cardiol. 2002;17:235–41. [Google Scholar]
- Nugent MA, Edelman ER. Kinetics of basic fibroblast growth factor binding to its receptor and heparan sulfate proteoglycan: A mechanism for cooperativity. Biochemistry. 1992;31:8876–8883. doi: 10.1021/bi00152a026. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Herr AB, Nilsson M, Westman J, Svahn CM, Waksman G. FGF binding and FGF receptor activation by synthetic heparan-derived di- and trisaccharides. Science. 1995;268:432–436. doi: 10.1126/science.7536345. [DOI] [PubMed] [Google Scholar]
- Paine-Saunders S, Viviano BL, Saunders S. GPC6, a novel member of the glypican gene family, encodes a product structurally related to GPC4 and is colocalized with GPC5 on human chromosome 13. Genomics. 1999;57:455–458. doi: 10.1006/geno.1999.5793. [DOI] [PubMed] [Google Scholar]
- Paine-Saunders S, Viviano BL, Zupicich J, Skarnes WC, Saunders S. glypican-3 controls cellular responses to Bmp4 in limb patterning and skeletal development. Dev Biol. 2000;225:179–87. doi: 10.1006/dbio.2000.9831. [DOI] [PubMed] [Google Scholar]
- Pantoliano MW, Horlick RA, Springer BA, Dyk DEV, Tobery T, Wetmore DR, Lear JD, Nahapetian AT, Bradley JD, Sisk WP. Multivalent ligand-receptor binding interactions in the fibroblast growth factor system produce a cooperative growth factor and heparin mechanism for receptor dimerization. Biochemistry. 1994;33:10229–10248. doi: 10.1021/bi00200a003. [DOI] [PubMed] [Google Scholar]
- Park EJ, Watanabe Y, Smyth G, Miyagawa-Tomita S, Meyers E, Klingensmith J, Camenisch T, Buckingham M, Moon AM. An FGF autocrine loop initiated in second heart field mesoderm regulates morphogenesis at the arterial pole of the heart. Development. 2008;135:3599–610. doi: 10.1242/dev.025437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penisson-Besnier I, Lebouvier T, Moizard MP, Ferre M, Barth M, Marc G, Raynaud M, Bonneau D. Carotid artery dissection in an adult with the Simpson-Golabi-Behmel syndrome. Am J Med Genet A. 2008;146A:464–7. doi: 10.1002/ajmg.a.32154. [DOI] [PubMed] [Google Scholar]
- Perrimon N, Bernfield M. Specificities of heparan sulphate proteoglycans in developmental processes. Nature. 2000;404:725–8. doi: 10.1038/35008000. [DOI] [PubMed] [Google Scholar]
- Rapraeger A, Krufka A, Olwin BB. Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science. 1991;252:1705–1708. doi: 10.1126/science.1646484. [DOI] [PubMed] [Google Scholar]
- Ratajska A, Ciszek B, Sowinska A. Embryonic development of coronary vasculature in rats: corrosion casting studies. Anat Rec A Discov Mol Cell Evol Biol. 2003;270:109–16. doi: 10.1002/ar.a.10011. [DOI] [PubMed] [Google Scholar]
- Ratajska A, Zlotorowicz R, Blazejczyk M, Wasiutynski A. Coronary artery embryogenesis in cardiac defects induced by retinoic acid in mice. Birth Defects Res A Clin Mol Teratol. 2005;73:966–79. doi: 10.1002/bdra.20200. [DOI] [PubMed] [Google Scholar]
- Reese DE, Mikawa T, Bader DM. Development of the coronary vessel system. Circ Res. 2002;91:761–8. doi: 10.1161/01.res.0000038961.53759.3c. [DOI] [PubMed] [Google Scholar]
- Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–4. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- Roghani M, Mansukhani A, Dell'Era P, Bellosta P, Basilico C, Rifkin DB, Moscatelli D. Heparin increases the affinity of basic fibroblast growth factor for its receptor but is not required for binding. Journal of Biological Chemistry. 1994;269:3976–84. [PubMed] [Google Scholar]
- Saunders S, Jalkanen M, O'Farrell S, Bernfield M. Molecular cloning of syndecan, an integral membrane proteoglycan. J Cell Biol. 1989;108:1547–1556. doi: 10.1083/jcb.108.4.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M, Annex BH, Laham RJ, Kleiman N, Henry T, Dauerman H, Udelson JE, Gervino EV, Pike M, Whitehouse MJ, Moon T, Chronos NA. Pharmacological treatment of coronary artery disease with recombinant fibroblast growth factor-2: double-blind, randomized, controlled clinical trial. Circulation. 2002;105:788–93. doi: 10.1161/hc0802.104407. [DOI] [PubMed] [Google Scholar]
- Song HH, Shi W, Xiang YY, Filmus J. The loss of glypican-3 induces alterations in Wnt signaling. J Biol Chem. 2005;280:2116–25. doi: 10.1074/jbc.M410090200. [DOI] [PubMed] [Google Scholar]
- Springer BA, Pantoliano MW, Barbera FA, Gunyuzlu PL, Thompson LD, Herblin WF, Rosenfeld SA, Book GW. Identification and concerted function of two receptor binding surfaces on basic fibroblast growth factor required for mitogenesis. Journal of Biological Chemistry. 1994;269:26879–84. [PubMed] [Google Scholar]
- Steinfeld R, Van Den Berghe H, David G. Stimulation of fibroblast growth factor receptor-1 occupancy and signaling by cell surface-associated syndecans and glypicans. J Cell Biol. 1996;133:405–416. doi: 10.1083/jcb.133.2.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataraman G, Sasisekharan V, Herr AB, Ornitz DM, Waksman G, Cooney CL, Langer R, Sasisekharan R. Preferential self-association of basic fibroblast growth factor is stabilized by heparin during receptor dimerization and activation. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:845–50. doi: 10.1073/pnas.93.2.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veugelers M, De Cat B, Ceulemans H, Bruystens AM, Coomans C, Durr J, Vermeesch J, Marynen P, David G. Glypican-6, a new member of the glypican family of cell surface heparan sulfate proteoglycans. J Biol Chem. 1999;274:26968–77. doi: 10.1074/jbc.274.38.26968. [DOI] [PubMed] [Google Scholar]
- Wada AM, Willet SG, Bader D. Coronary vessel development: a unique form of vasculogenesis. Arterioscler Thromb Vasc Biol. 2003;23:2138–45. doi: 10.1161/01.ATV.0000098645.38676.CC. [DOI] [PubMed] [Google Scholar]
- White AC, Lavine KJ, Ornitz DM. FGF9 and SHH regulate mesenchymal Vegfa expression and development of the pulmonary capillary network. Development. 2007;134:3743–52. doi: 10.1242/dev.004879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Lin X. Drosophila glypican Dally-like acts in FGF-receiving cells to modulate FGF signaling during tracheal morphogenesis. Dev Biol. 2007;312:203–16. doi: 10.1016/j.ydbio.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yayon A, Klagsbrun M, Esko JD, Leder P, Ornitz DM. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell. 1991;64:841–848. doi: 10.1016/0092-8674(91)90512-w. [DOI] [PubMed] [Google Scholar]