

Abstract
Mammalian cells can choose either nonhomologous end joining (NHEJ) or homologous recombination (HR) for repair of chromosome breaks. Of these two pathways, HR alone requires extensive DNA synthesis and thus abundant synthesis precursors (dNTPs). We address here if this differing requirement for dNTPs helps determine how cells choose a repair pathway. Cellular dNTP pools are regulated primarily by changes in ribonucleotide reductase activity. We show that an inhibitor of ribonucleotide reductase (hydroxyurea) hypersensitizes NHEJ-deficient cells, but not wild type or HR-deficient cells, to chromosome breaks introduced by ionizing radiation. Hydroxyurea additionally reduces the frequency of irradiated cells with a marker for an early step in HR, Rad51 foci, consistent with reduced initiation of HR under these conditions. Conversely, promotion of ribonucleotide reductase activity protects NHEJ-deficient cells from ionizing radiation. Importantly, promotion of ribonucleotide reductase activity also increases usage of HR in cells proficient in both NHEJ and HR at a targeted chromosome break. Activity of ribonucleotide reductase is thus an important factor in determining how mammalian cells repair broken chromosomes. This may explain in part why G1/G0 cells, which have reduced ribonucleotide reductase activity, rely more on NHEJ for DSB repair.
Keywords: Chromosome breaks, pathway choice, ribonucleotide reductase
2. Introduction
Efficient and accurate repair of DNA double strand breaks (DSBs) is essential for cell survival. Eukaryotic cells employ two major pathways for DSB repair: nonhomologous endjoining (NHEJ) and homologous recombination (HR) (reviewed in [1]). NHEJ religates broken ends with minimal or no requirement for DNA synthesis, and is active during the whole cell cycle [2–6]. In contrast, HR has extensive requirements for DNA synthesis and is primarily employed for DSB repair in S and G2 phases [3–6]. A key step in HR, and the point where cells commit to repair by HR over repair by NHEJ [6,7], involves resection of 100s to 1000s of nucleotides from 5′-ends to produce long single stranded 3′-overhangs. These 3′-overhangs then invade the sister chromatid or homologous chromosome and serve as primers for re-synthesis of the previously degraded sequence around the break (reviewed in [8]). HR’s need for extensive DNA synthesis suggests it will be much more dependent than NHEJ on the presence of sufficient dNTPs.
Availability of dNTPs is primarily reliant on the de novo pathway and specifically activity of ribonucleotide reductase (RNR). RNR activity has been additionally linked to the cellular capacity to survive DNA damage [9,10]. RNR possesses a large subunit (R1) and one of two possible small subunits (R2 or p53R2) [11]. RNR activity is regulated over the cell cycle by limiting transcription of the primary version of the small subunit, R2, to S and G2 [12], as well as destruction of this protein in M phase [13]. As a consequence, RNR activity rises in early S, and falls after G2 – a fluctuation that correlates well with the extent cells perform HR.
Cells nevertheless retain some ability to generate nucleotide pools de novo in G1 by using a complex of R1 and the alternative version of the small subunit, p53R2 [14]. P53R2 is expressed at low levels throughout the cell cycle [15], but expression can be further augmented after DNA damage through a p53-dependent mechanism [16]. p53R2 protein is additionally stabilized after DNA damage through an ataxia telangiectasia mutated (ATM) dependent mechanism [17]. This up-regulation and stabilization after DNA damage is consistent with a specific role for p53R2 in providing nucleotides for DNA repair [14].
Here we address whether manipulation of cellular capacity to generate dNTPs de novo by RNR has an impact on whether cells repair chromosome breaks by HR or by NHEJ. We show that treatment with hydroxyurea (HU), which inhibits RNR activity, suppresses HR; conversely, stimulation of nucleotide synthesis promotes HR. We conclude that de novo nucleotide production is an important determinant of repair pathway choice.
3. Materials and Methods
3.1. Cell culture and colony formation assays
All cell lines were grown at 37°C and 5% CO2 in DMEM (Gibco) supplemented with 10% fetal bovine serum (Gibco), 1x MEM non-essential amino acids (Gibco), penicillin, and streptomycin. The Brca2−/− cell line (V-C8) and the matching parental line (V79) was the kind gift of Dr. M.Z. Zdzienicka. Cells were synchronized essentially as previously described [4], except that cells were grown in presence of 5 mM N-acetyl-cysteine (Sigma) and 10 mM HEPES (Gibco) for synchronization in G1/G0. For enrichment in G1/G0, 8×104 cells/cm2 were plated out and grown to confluency during 3 days. For enrichment in S phase, G1/G0 cells were replated at a density of 4×104/cm2, incubated for 16 hours in the presence of aphidicolin (1 μg/ml, Sigma), and released by medium change. 3 hours after release (3.5 hours for HR-deficient cell line irs1SF), the majority of the cells reached mid-S phase. Synchronization was verified by analysis of cell cycle profiles by flow cytometry (Dako Cyan ADP) after propidium iodide staining (Roche). Only experiments with at least 80% pure populations were analyzed (e.g. supplemental Figure 1).
Xrs6 was complemented by generating a subclone that stably integrated a cDNA containing the C. griseus gene encoding Ku80 (the kind gift of Dr. D.B. Roth) and that was grown in presence of 400 μg/ml Geneticin (Invitrogen). In order to measure repair by homologous recombination, pDR-GFP [18] (the kind gift of Dr. M. Jasin) was stably integrated into the Chinese hamster ovary (CHO) cell line K1, resulting in the cell line K-DR, which was grown in presence of 10 μg/ml puromycin (Sigma). The murine gene encoding p53R2 was inserted into pcDNA6/myc-His A (Invitrogen) and stably integrated into the CHO cell lines K1 and Xrs6, resulting in lines K+P and X+P. p53R2 overexpressing lines were grown in presence of 10 μg/ml blasticidin (Invitrogen). Expression of myc-tagged p53R2 was verified by western blot with the monoclonal mouse antibody 9B11 (Cell Signaling). An actin-specific polyclonal rabbit antibody (A2066; Sigma) was used for the loading control.
For colony formation assays, synchronized cells were plated out in presence or absence of 0.2 mM hydroxyurea or 1 μg/ml aphidicolin (both Sigma). After one hour, cells were irradiated with 1 Gy in a Gammacell 40 irradiator (137Cs). 7 hours after irradiation cells were washed with 1x phosphate buffered saline (PBS) and provided with fresh medium without drug. The number of colonies was assessed 7 days later by Comassie staining (50% methanol, 5% Comassie) and counted using ImageJ (NIH) as software. All experiments were repeated at least 3 times, and the mean and standard error of the mean for each experiment were calculated with Prism 4.0c (Graphpad).
3.2. Repair substrate assay
1×106 exponentially growing wild type CHO cells containing the recombination substrate DR-GFP, K-DR, were transfected with 2 μg plasmid DNA (Amaxa, Kit T (VCA-1002), program H-014): empty vector alone (pcDNA6/LacZ-myc) in combination with expression vectors for p53R2 or p53R2-Y138V, a catalytic mutant, respectively. In parallel reactions, expression vectors for I-SceI and p53R2 or p53R2-Y138V, respectively, were delivered by electroporation. GFP expression was analyzed by flow cytometry 48 hours later. All experiments were repeated 3 times and means as well as standard errors of the mean were calculated with Prism 4.0c (Graphpad).
3.3. Immunofluorescence
G1/G0 or S phase enriched cells were seeded onto collagen-coated coverslips (Becton-Dickinson) in medium containing 0.2 mM hydroxyurea, 1 μg/ml aphidicolin, or no drug. Cells were irradiated one hour later with 8 Gy. Cells were then fixed with 4% buffered paraformaldehyde in PBS at indicated times; no apoptotic cells could be detected up to 10 hours after irradiation (Supplemental Figure 2). Fixed cells were then permeabilized with 0.2% Triton X-100 for 3 minutes and analyzed with primary antibodies (Santa Cruz; catalogue numbers and dilutions noted for each antibody in parenthesis) against R2 (sc-10848; 1:200), Rad51 (sc-8349; 1:150), and Cyclin A (sc-751; 1:100) and secondary antibodies Alexa Fluor 488 conjugated donkey anti-goat (Molecular Probes; 1:1000) or Cy3 conjugated donkey anti-rabbit (Jackson ImmunoResearch; 1:1000). After three washing steps with PBS, whereof the first contained DAPI (5 μg/ml), coverslips were mounted onto glass slides with Fluorescent Mounting Medium (DakoCytomation). Specimens were analyzed in an AxioScope II (Zeiss) using a 40x objective and Openlab software. At least 100 cells were analyzed for each condition and experiments were repeated at least three times. Cells with ≥3 Rad51 foci were considered to be focus-positive. The mean and standard error of the mean was calculated with Prism 4.0c as software.
4. RESULTS
4.1. Nucleotide synthesis influences survival after damage
We first tested whether inhibition of nucleotide synthesis impairs cell survival after ionizing radiation (IR) in a pattern consistent with HR’s requirement for extensive DNA synthesis. For this analysis we varied the ability of cells to use the two pathways by making use of well-established Chinese hamster cell lines with mutations in various genes essential for efficient HR or NHEJ.
We manipulated the cellular ability to synthesize dNTPs by treating cells with hydroxyurea (HU), which specifically blocks production of nucleotides de novo through inhibition of the enzyme ribonucleotide reductase (RNR) [11,16]. In order to focus on the impact of HU on repair of radiation-induced breaks we targeted G1/G0 enriched cells, so that HU alone could not introduce damage (e.g. during replication). We also limited both the amounts of HU used (200μM; 5–10 fold less than used in most protocols) as well as the contact time to the hour immediately prior to irradiation and the additional 7 hours following. Cells were then washed extensively and returned to normal growth conditions. The frequency of cells that survive this treatment was determined by assessing their capacity to form colonies relative to untreated controls (Table 1). To further summarize this data and focus on the impact of genotype, we then additionally compared surviving fractions for each deficient cell line to its parental line (Figure 1).
Table 1.
Sensitivity to ionizing radiation after pretreatment with hydroxyurea and aphidicolin at different stages of the cell cycle.
| A (G1) | AA8 | irs1SF | V79 | V-C8 | K1 | Xrs6 | XR-1 | Xrs6 compl |
|---|---|---|---|---|---|---|---|---|
| HU (200μM) | 0.93 (0.06) | 0.94 (0.10) | 0.83 (0.04) | 1.16 (0.06) | 0.93 (0.04) | 0.75 (0.13) | 0.57 (0.11) | 0.88 (0.08) |
| APH (1μg/ml) | 0.99 (0.06) | 0.85 (0.15) | 0.76 (0.04) | 0.85 (0.04) | 0.93 (0.04) | 0.83 (0.15) | 0.60 (0.09) | 0.91 (0.05) |
| IR (1Gy) | 0.64 (0.05) | 0.51 (0.04) | 0.85 (0.04) | 0.66 (0.04) | 0.74 (0.05) | 0.027 (0.008) | 0.081 (0.010) | 0.71 (0.06) |
| HU&IR | 0.54 (0.02) | 0.53 (0.06) | 0.66 (0.04) | 0.71 (0.02) | 0.63 (0.04) | 0.0051 (0.0015) | 0.0137 (0.0058) | 0.57 (0.03) |
| APH&IR | 0.35 (0.04) | 0.37 (0.01) | 0.53 (0.03) | 0.70 (0.04) | 0.53 (0.04) | 0.0031 (0.0008) | 0.0116 (0.0084) | 0.61 (0.02) |
| B (S) | AA8 | irs1SF | K1 | Xrs6 |
|---|---|---|---|---|
| HU (200μM) | 0.36 (0.04) | 0.38 (0.06) | 0.54 (0.05) | 0.28 (0.06) |
| IR (1Gy) | 0.76 (0.04) | 0.46 (0.05) | 0.85 (0.02) | 0.0364 (0.0041) |
| HU&IR | 0.12 (0.02) | 0.12 (0.02) | 0.35 (0.02) | 0.0019 (0.0007) |
Fig. 1.
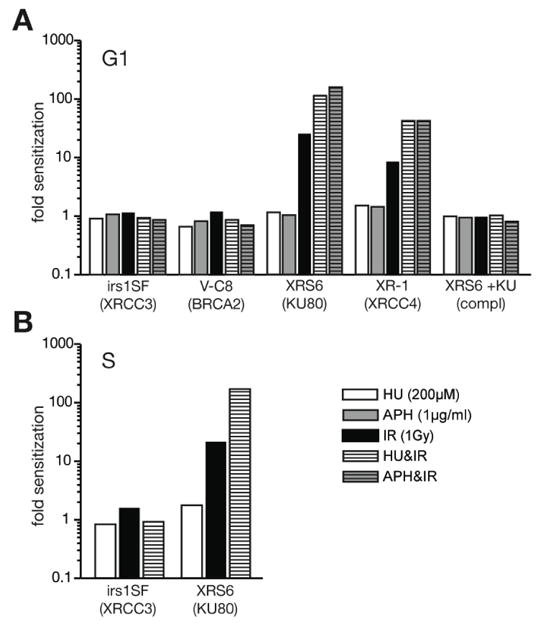
Sensitivity to ionizing radiation. Results from colony survival assays obtained with mutant cell lines were compared to their corresponding parental line (irs1SF to AA8; V-C8 to V79; Xrs6, XR-1, and Xrs6 complemented to K1). Bars thus represent the impact of the mutation on sensitivity to irradiation and/or drug treatment for (A) G1/G0 or (B) S phase enriched cells.
G1/G0 enriched cells deficient in HR (irs1SF and V-C8) [19,20] were not significantly sensitive to 1 Gray (Gy) of IR (Table 1A, Figure 1A), relative to matched wild type controls (AA8 and V79, respectively). This result is consistent with prior studies indicating that in G1/G0, IR-induced breaks are primarily repaired by NHEJ [3–5]. However, we were able to detect significant radiosensitivity in G1/G0-enriched V-C8 cells at higher doses (5 Gy; data not shown). Critically, our data indicated neither wild type nor HR-deficient cells were made significantly more radiosensitive at either dose when also treated with HU (Table 1A; Figure 1A). We conclude that blocking the ability to generate dNTPs de novo does not significantly impact how well NHEJ proficient, G1/G0 enriched cells repair IR-generated DSBs, consistent with our predictions.
In contrast, HU treatment significantly increased the radiation sensitivity of G1/G0 enriched cells deficient in NHEJ (Xrs6 and XR-1 [21,22]; Table 1A, Figure 1A). Survival of NHEJ-deficient cells was impaired after exposure to 1 Gy of IR, but survival was an additional ~5 fold lower if also treated with HU (Table 1A, p<0.05, Student’s T-test). The behavior of Xrs6 cells complemented with a Ku80 cDNA was indistinguishable from the parental line (K1), confirming that these effects can be attributed to deficiency in the NHEJ component Ku80 (Table 1A, Figure 1A). HU treatment thus radiosensitizes NHEJ-deficient cells, but has little to no impact on radiosensitivity of wild type cell lines or an HR-deficient cell line. We also observed similar effects with the polymerase α/δ inhibitor aphidicolin (APH; Table 1A, Figure 1A), arguing for a general requirement for processive DNA synthesis if NHEJ deficient, G1/G0 cells are to survive IR.
We also analyzed the impact of HU treatment after irradiation during S phase. Even though cells are undergoing DNA replication at the time of treatment (Table 1B, Figure 1B), HU treatment alone still has only mild effects on colony formation of wild type or HR deficient cells. As previously reported, HR-deficient cells are sensitive to irradiation during S phase [3,4]. However, further treatment with HU had an only modest additive impact on survival (Table 1B), similar to that observed in wild type cells (Figure 1B).
Survival of NHEJ-deficient cells is slightly reduced after treatment with HU alone during S phase (reduced by a factor of 2, relative to wild type cells; Table 1B). Nevertheless, HU reduced survival by an additional 20 fold relative to that observed by IR alone in these cells (Xrs6; Figure 1B). This effect is considerably greater than what would be expected if effects of the two treatments were additive, as is the result in wild type cells. The ability to generate dNTPs de novo may thus be critical for radioresistance in NHEJ deficient cells during S phase as well as during G1.
4.2. Hydroxyurea treatment inhibits early steps of HR
We suggest radioresistance in NHEJ deficient cells is 1) best attributed to successful repair by HR, even in G1, and 2) that HR’s ability to repair DSBs is compromised by blocking de novo nucleotide production. To more directly address this issue we followed formation of Rad51 foci, an early marker for attempted DSB repair by HR, by immunofluorescence. Once again, we note HU alone is sufficient to induce damage as well as Rad51 foci in S phase cells (e.g. supplemental Figure 3) [23,24] and therefore focused again on G1/G0 enriched cells. To exclude any contribution from possible contaminating S/G2 cells we also co-stained cells with an antibody to a marker for S/G2 cells. We used the RNR subunit R2 as an S/G2 marker after first confirming R2-positive cells correlated well with cells positive for Cyclin A, another marker for cells in S/G2 (Figure 2A).
Fig. 2.
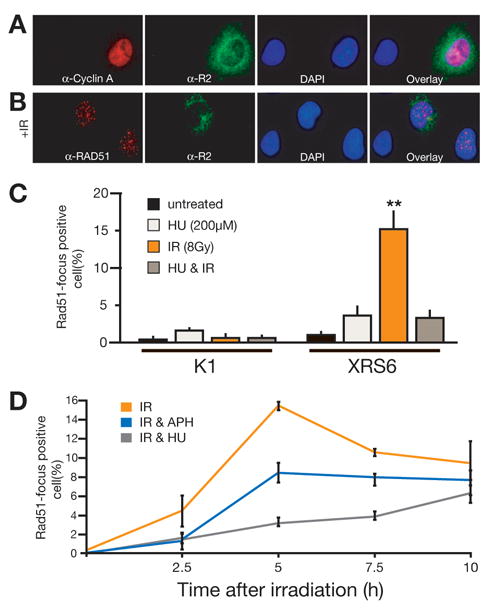
Analysis of Rad51 focus formation by immunofluorescence. (A) Representative immunofluorescence data for cells co-stained for Cyclin A and R2; shown are S phase enriched Xrs6 cells. (B) Representative immunofluorescence data for cells co-stained for Rad51 and the R2 subunit of ribonucleotide reductase, a marker for S phase cells. Shown are Xrs6 cells after irradiation with 8 Gy. (C) Mean frequency of Rad51 focus formation in R2-negative, G1/G0 cells from three independent experiments. Shown is the mean +/− SEM. **Ionizing radiation significantly stimulated formation of Rad51 foci in Xrs6 cells; p=0.0014. (D) Quantification of Rad51 focus formation in R2-negative, G1 phase Xrs6 cells at different time points after irradiation and in dependence of pretreatment with hydroxyurea (200μM) or aphidicolin (1 μg/ml). Shown is the mean +/−SEM from three independent experiments.
We assessed Rad51 focus formation in G1/G0 enriched K1 and Xrs6 cells after irradiation with 8 Gy (Figure 2B, C, and D). Rad51 focus-positive cells were apparent in Xrs6 cells but not K1 cells, indicating HR is employed for DSB repair in G1 cells only when NHEJ is not available (Figure 2C) [25]. Importantly, HU treatment reduced radiation-dependent Rad51 foci 4.6 fold in Xrs6 cells. This latter result is consistent with the attribution of HU’s ability to radiosensitize these cells to reduced attempts at HR. Moreover, Rad51 focus formation is an early step in HR, and precedes synthesis-dependent steps [26]. Inhibition of Rad51 focus formation under these conditions suggests capacity to provide nucleotides for repair might help control whether cells commit to HR. Also consistent with this suggestion, HU treatment (and presumably HU’s ability to inhibit de novo nucleotide formation) was more effective in suppressing Rad51 foci than was inhibition of DNA synthesis per se (through aphidicolin treatment; Figure 2D).
4.3. Overexpression of p53R2 promotes efficiency of HR
The results obtained after HU treatment are in line with our hypothesis that inhibition of RNR, and thus a reduced capacity to provide dNTPs for repair, prevents use of HR. However, it remains formally possible that the effects of HU treatment described above are independent of HU’s ability to inhibit RNR. Moreover, we also wanted to test whether increased RNR activity had the opposite effect. We therefore stably overexpressed a myc-tagged version of p53R2 in both KU80-deficient cells (Xrs6) and the matched wild type parental line (K1; Figure 3A). We confirmed this was sufficient to significantly impact whole cell dNTP pools during G1/G0, though not in S phase (supplemental Figure 4), presumably because the primary small subunit (R2) is present at high levels in S phase but not G1.
Fig. 3.
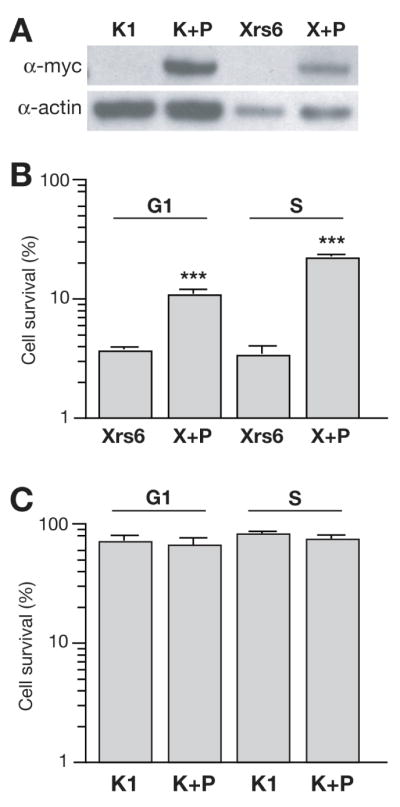
Effect of p53R2 overexpression on cell survival. (A) Characterization of p53R2 overexpressing CHO cell lines. Cell lysates of wild type (K1) and NHEJ-deficient (Xrs6) cells as well as myc-p53R2 overexpressing subclones of these cell lines (K+P, X+P) were probed for myc or actin in parallel Western blots. (B) Mean of three experiments comparing the frequency of survival for the NHEJ-deficient cell lines (Xrs6) after irradiation with 1 Gy relative to un-irradiated controls. Shown is mean +/− SEM. ***p53R2 overexpression made NHEJ-deficient cells significantly radioresistant; p<0.0001. (C) Mean of three experiments comparing the frequency of survival for the wild type CHO cell lines (K1) after irradiation with 1 Gy relative to un-irradiated controls. Shown is mean +/− SEM.
We irradiated cells that were enriched in either G1/G0 or S phase, and analyzed colony formation as before (Figure 3B, and C). Importantly, overexpression of p53R2 protected NHEJ deficient cells (X+P) from IR (Figure 3B). Survival improved 2.9-fold after irradiation in G1 and 6.6-fold in S phase, respectively (p<0.0001, Student’s T-test). The protective effect of p53R2 overexpression was thus most evident in S phase, despite our observation that p53R2 overexpression did little to impact S phase whole-cell dNTP pools. Finally, p53R2 had little impact on the radiosensitivity of wild type (K1) cells regardless of when in the cell cycle these cells were irradiated (Figure 3C). We suggest this is because proficiency in both pathways in wild type cells means differences in RNR activity does not significantly determine if cells successfully repair double strand breaks, only which pathway (HR or NHEJ) they use to repair them.
To determine if repair pathway choice in wild type cells is influenced by RNR activity, we assessed repair more directly with the well-characterized recombination reporter construct, DR-GFP. DR-GFP contains a recognition site for the homing endonuclease I-SceI within a defective GFP gene (Figure 4A) [18]. Expression of I-SceI generates a double strand break that can be repaired by a downstream homologous donor sequence, and this version of HR repair can be quantitatively assessed by GFP expression (Figure 4 and supplemental Figure 5). We stably integrated DR-GFP in wild type (K1) cells, thereby constructing the cell line K-DR, and induced recombination by transient expression of I-SceI. RNR activity was varied by additionally co-transfecting empty vector, or vectors that promote expression of either wild type p53R2 or a catalytically inactive p53R2 point mutant (p53R2-Y138V; [27] and references therein).
Fig. 4.
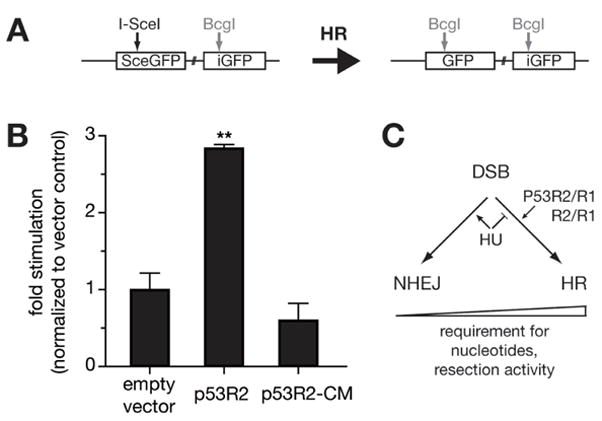
Repair of a targeted double strand break by homologous recombination. (A) DR-GFP; GFP is expressed only after homology directed repair. (B) Effect of p53R2 overexpression on recombination after 48 hours. The cell line K-DR was assessed for GFP expression after transfection with empty vector alone or with I-SceI expression. The influence of simultaneous expression of wild type p53R2 or a catalytic mutant of p53R2 (p53R2-CM), respectively, was analyzed and is represented as fold stimulation of GFP expression. **p53R2 overexpression stimulated GFP expression significantly; p=0.001. (C) Possible impact of nucleotide pool availability on repair pathway choice.
We observe that the marker for HR, GFP expression, accumulates 3 fold more efficiently in clones that transiently overexpress wild type p53R2 than in the parental DR-GFP containing line (Figure 4B). Contrastingly, overexpression of the catalytic mutant p53R2-Y138V did not stimulate GFP expression. Since K-DR cells are proficient in both NHEJ and HR we conclude overexpression of p53R2, and thus increased capacity for de novo dNTP synthesis, promotes the use of HR over NHEJ for repair of DSBs.
5. DISCUSSION
In this work we identify RNR activity, and thus the capacity for generation of nucleotides de novo, as an important determinant for whether cells can use HR to survive chromosome breaks made by ionizing radiation. By comparison, manipulation of the de novo pathway (either inhibition or promotion) had no significant impact on whether cells could similarly use NHEJ to repair these breaks. NHEJ’s ability to sustain efficient repair regardless of the activity of the de novo pathway is thus a strong rationale for using a repair pathway suggested to be intrinsically more error prone than the alternative (HR).
It is notable that manipulation of RNR activity, and p53R2-dependent RNR activity in particular, has an impact on DSB repair by HR that might be considered to exceed the expected impact of these manipulations on whole cell dNTP pools. We demonstrated HU has a clear effect on radiation sensitivity in G1, even though the effects of HU on whole cell pools in G1 cells are probably modest (unpublished data and [15,28,29]). Similarly, overexpression of p53R2 protects S phase cells from ionizing radiation, yet we observe no obvious impact on whole cell dNTP pools (Supplemental Figure 4). These observations parallel prior work indicating replication is arrested in mammalian cells by inhibition of RNR, even when dNTP pools are only subtly reduced [28,30]. As with replication, there are many reasons why changes in RNR activity might have an impact on HR that appears to exceed its impact on whole cell pools (see e.g. [29], and references therein). In this regard, reports that DNA damage both stabilizes and relocalizes p53R2 to the nucleus [14,17], is consistent with the possibility that p53R2 could contribute to a privileged or locally restricted pool that promotes efficient HR. However, we note that damage-dependent relocalization is not universally observed [31].
We observed very different effects of HU on Rad51 foci depending on cell-cycle phase. Consistent with our hypothesis, HU inhibits formation of radiation-induced Rad51 foci in G1 (Figure 2): in contrast, HU is sufficient for formation of Rad51 foci in S phase cells (e.g. supplementary Figure 3). In S phase cells, HU treatment results in fork stalling but, as noted above, dNTP pools are only subtly reduced (~2 fold; [28,30]). Therefore, S phase pools in mammalian cells even after HU treatment are still much higher than would be present normally in G1/G0, and this could explain how HR in HU-treated S phase cells remains active.
The choice of DSB repair pathway is primarily coupled to cell cycle through control of resection, the first step in HR, by activity of S-CDK [6,7,32–34]. However, we note that at least in mammalian cells, NHEJ is frequently used for DSB repair during S/G2 [3–5]. HR can also be used for DSB repair in G1/G0 cells [25,35], particularly when NHEJ is blocked (Figures 1 and 2). These exceptions imply resection and thus the choice of DSB repair pathway may be regulated by factors in addition to S-CDK. We suggest activity of the RNR complex is a logical candidate for such an additional factor (Figure 4C).
Supplementary Material
Acknowledgments
We thank Dr. Beverly Mitchell and all members of the Ramsden group for discussions and input. This work was supported by US Public Health Service Grant CA-97096 to D.A.R., a Leukemia and Lymphoma Society Scholar. Support was also provided by the Novartis Foundation, the Swiss National Foundation, and the Huggenberger-Bischoff Foundation (to M.D.B.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 2.Karathanasis E, Wilson TE. Enhancement of Saccharomyces cerevisiae end-joining efficiency by cell growth stage but not by impairment of recombination. Genetics. 2002;161:1015–1027. doi: 10.1093/genetics/161.3.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, Yamaguchi-Iwai Y, Shinohara A, Takeda S. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. Embo J. 1998;17:5497–5508. doi: 10.1093/emboj/17.18.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rothkamm K, Kruger I, Thompson LH, Lobrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol. 2003;23:5706–5715. doi: 10.1128/MCB.23.16.5706-5715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hinz JM, Yamada NA, Salazar EP, Tebbs RS, Thompson LH. Influence of double-strand-break repair pathways on radiosensitivity throughout the cell cycle in CHO cells. DNA Repair (Amst) 2005;4:782–792. doi: 10.1016/j.dnarep.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 6.Aylon Y, Liefshitz B, Kupiec M. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. Embo J. 2004;23:4868–4875. doi: 10.1038/sj.emboj.7600469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth NM, Haber JE, Foiani M. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 2004;431:1011–1017. doi: 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- 9.Snyder RD. The role of deoxynucleoside triphosphate pools in the inhibition of DNA-excision repair and replication in human cells by hydroxyurea. Mutat Res. 1984;131:163–172. doi: 10.1016/0167-8817(84)90057-9. [DOI] [PubMed] [Google Scholar]
- 10.Chabes A, Georgieva B, Domkin V, Zhao X, Rothstein R, Thelander L. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell. 2003;112:391–401. doi: 10.1016/s0092-8674(03)00075-8. [DOI] [PubMed] [Google Scholar]
- 11.Nordlund P, Reichard P. Ribonucleotide reductases. Annu Rev Biochem. 2006;75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- 12.Bjorklund S, Skogman E, Thelander L. An S-phase specific release from a transcriptional block regulates the expression of mouse ribonucleotide reductase R2 subunit. Embo J. 1992;11:4953–4959. doi: 10.1002/j.1460-2075.1992.tb05602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chabes AL, Pfleger CM, Kirschner MW, Thelander L. Mouse ribonucleotide reductase R2 protein: a new target for anaphase-promoting complex-Cdh1-mediated proteolysis. Proc Natl Acad Sci U S A. 2003;100:3925–3929. doi: 10.1073/pnas.0330774100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka H, Arakawa H, Yamaguchi T, Shiraishi K, Fukuda S, Matsui K, Takei Y, Nakamura Y. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature. 2000;404:42–49. doi: 10.1038/35003506. [DOI] [PubMed] [Google Scholar]
- 15.Hakansson P, Hofer A, Thelander L. Regulation of mammalian ribonucleotide reduction and dNTP pools after DNA damage and in resting cells. J Biol Chem. 2006;281:7834–7841. doi: 10.1074/jbc.M512894200. [DOI] [PubMed] [Google Scholar]
- 16.Guittet O, Hakansson P, Voevodskaya N, Fridd S, Graslund A, Arakawa H, Nakamura Y, Thelander L. Mammalian p53R2 protein forms an active ribonucleotide reductase in vitro with the R1 protein, which is expressed both in resting cells in response to DNA damage and in proliferating cells. J Biol Chem. 2001;276:40647–40651. doi: 10.1074/jbc.M106088200. [DOI] [PubMed] [Google Scholar]
- 17.Chang L, Zhou B, Hu S, Guo R, Liu X, Jones SN, Yen Y. ATM-mediated serine 72 phosphorylation stabilizes ribonucleotide reductase small subunit p53R2 protein against MDM2 to DNA damage. Proc Natl Acad Sci U S A. 2008;105:18519–18524. doi: 10.1073/pnas.0803313105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu N, Lamerdin JE, Tebbs RS, Schild D, Tucker JD, Shen MR, Brookman KW, Siciliano MJ, Walter CA, Fan W, Narayana LS, Zhou ZQ, Adamson AW, Sorensen KJ, Chen DJ, Jones NJ, Thompson LH. XRCC2 and XRCC3, new human Rad51-family members, promote chromosome stability and protect against DNA cross-links and other damages. Mol Cell. 1998;1:783–793. doi: 10.1016/s1097-2765(00)80078-7. [DOI] [PubMed] [Google Scholar]
- 20.Kraakman-van der Zwet M, Overkamp WJ, van Lange RE, Essers J, van Duijn-Goedhart A, Wiggers I, Swaminathan S, van Buul PP, Errami A, Tan RT, Jaspers NG, Sharan SK, Kanaar R, Zdzienicka MZ. Brca2 (XRCC11) deficiency results in radioresistant DNA synthesis and a higher frequency of spontaneous deletions. Mol Cell Biol. 2002;22:669–679. doi: 10.1128/MCB.22.2.669-679.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeggo PA, Hafezparast M, Thompson AF, Broughton BC, Kaur GP, Zdzienicka MZ, Athwal RS. Localization of a DNA repair gene (XRCC5) involved in double-strand-break rejoining to human chromosome 2. Proc Natl Acad Sci U S A. 1992;89:6423–6427. doi: 10.1073/pnas.89.14.6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Z, Otevrel T, Gao Y, Cheng HL, Seed B, Stamato TD, Taccioli GE, Alt FW. The XRCC4 gene encodes a novel protein involved in DNA double-strand break repair and V(D)J recombination. Cell. 1995;83:1079–1089. doi: 10.1016/0092-8674(95)90135-3. [DOI] [PubMed] [Google Scholar]
- 23.Saintigny Y, Delacote F, Vares G, Petitot F, Lambert S, Averbeck D, Lopez BS. Characterization of homologous recombination induced by replication inhibition in mammalian cells. Embo J. 2001;20:3861–3870. doi: 10.1093/emboj/20.14.3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scully R, Chen J, Ochs RL, Keegan K, Hoekstra M, Feunteun J, Livingston DM. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell. 1997;90:425–435. doi: 10.1016/s0092-8674(00)80503-6. [DOI] [PubMed] [Google Scholar]
- 25.Kim JS, Krasieva TB, Kurumizaka H, Chen DJ, Taylor AM, Yokomori K. Independent and sequential recruitment of NHEJ and HR factors to DNA damage sites in mammalian cells. J Cell Biol. 2005;170:341–347. doi: 10.1083/jcb.200411083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sugawara N, Wang X, Haber JE. In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51-mediated recombination. Mol Cell. 2003;12:209–219. doi: 10.1016/s1097-2765(03)00269-7. [DOI] [PubMed] [Google Scholar]
- 27.Kolberg M, Strand KR, Graff P, Andersson KK. Structure, function, and mechanism of ribonucleotide reductases. Biochim Biophys Acta. 2004;1699:1–34. doi: 10.1016/j.bbapap.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 28.Bianchi V, Pontis E, Reichard P. Changes of deoxyribonucleoside triphosphate pools induced by hydroxyurea and their relation to DNA synthesis. J Biol Chem. 1986;261:16037–16042. [PubMed] [Google Scholar]
- 29.Koc A, Wheeler LJ, Mathews CK, Merrill GF. Hydroxyurea arrests DNA replication by a mechanism that preserves basal dNTP pools. J Biol Chem. 2004;279:223–230. doi: 10.1074/jbc.M303952200. [DOI] [PubMed] [Google Scholar]
- 30.Skoog L, Bjursell G. Nuclear and cytoplasmic pools of deoxyribonucleoside triphosphates in Chinese hamster ovary cells. J Biol Chem. 1974;249:6434–6438. [PubMed] [Google Scholar]
- 31.Pontarin G, Fijolek A, Pizzo P, Ferraro P, Rampazzo C, Pozzan T, Thelander L, Reichard PA, Bianchi V. Ribonucleotide reduction is a cytosolic process in mammalian cells independently of DNA damage. Proc Natl Acad Sci U S A. 2008;105:17801–17806. doi: 10.1073/pnas.0808198105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huertas P, Cortes-Ledesma F, Sartori AA, Aguilera A, Jackson SP. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature. 2008;455:689–692. doi: 10.1038/nature07215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Limbo O, Chahwan C, Yamada Y, de Bruin RA, Wittenberg C, Russell P. Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol Cell. 2007;28:134–146. doi: 10.1016/j.molcel.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Golding SE, Rosenberg E, Khalil A, McEwen A, Holmes M, Neill S, Povirk LF, Valerie K. Double strand break repair by homologous recombination is regulated by cell cycle-independent signaling via ATM in human glioma cells. J Biol Chem. 2004;279:15402–15410. doi: 10.1074/jbc.M314191200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.