

Abstract
The multifunctional papillomavirus E2 protein serves important roles in transcriptional activation and genome maintenance and cooperates with the viral E1 helicase for the initiation of viral DNA replication. The bovine papillomavirus genome contains seventeen E2 binding sites, largely concentrated within the long control region, and a single E1 binding site at the origin of viral replication. Using chromatin immunoprecipitation (ChIP) followed by restriction enzyme digestion and PCR, we show that BPV E1 was present only in the region of an active origin of replication and that BPV E2 remained attached to definable segments of the viral genome at specific stages of the cell cycle.
Keywords: Bovine Papillomavirus, Papillomavirus E2 protein, RED-ChIP, BPV-1 genome
Introduction
Papillomaviruses (PV) are responsible for a variety of benign and malignant epithelial lesions including carcinoma of the cervix (Zur Hausen, 2002). Papillomavirus genomes consist of approximately 8 kilobases of double stranded DNA that, upon infection, are stably maintained as low copy episomes in the basal layer of epithelial cells until induced by cellular differentiation to initiate programmed viral genome amplification. Bovine papillomavirus type 1 (BPV-1) has been historically utilized as a model for the study of PV genome replication. BPV-1 genomic or sub-genomic DNAs that are transfected into murine C127 and NIH3T3 cell lines can replicate autonomously, displaying a stable copy number and expressing the essential early viral proteins (Law et al., 1981). The murine C127 cell line is also transformed by BPV-1 without integration of viral DNA into the host genome. The mechanism of cell transformation by BPV-1 is still not fully understood, however both the BPV-1 E5 and E6 proteins efficiently perform this task. (Bergman et al., 1988; Schiller, Vass, and Lowy, 1984).
The major function of E2 is regulation of BPV-1 transcription. The N-terminal two-thirds of E2 encodes a potent transcriptional activation domain (TAD) that stimulates expression from E2 binding site containing reporters in mammalian and yeast cells (Haugen et al., 1987; Morrissey, Barsoum, and Androphy, 1989; Spalholz, Yang, and Howley, 1985; Stanway et al., 1989). E2, through its C-terminal domain, binds to a specific palindromic sequence found in all PVs (Androphy, Lowy, and Schiller, 1987; McBride, Schlegel, and Howley, 1988; Sedman and Stenlund., 1995). There are 17 potential E2 binding sites of variable affinities within the BPV-1 genome (Li et al., 1989), the majority of which are clustered in the long control region (LCR), which includes the major promoter elements and the viral origin (Figure 1). Several E2 binding sites appear adjacent to promoters within the E1 and E2 genes.
Figure 1.
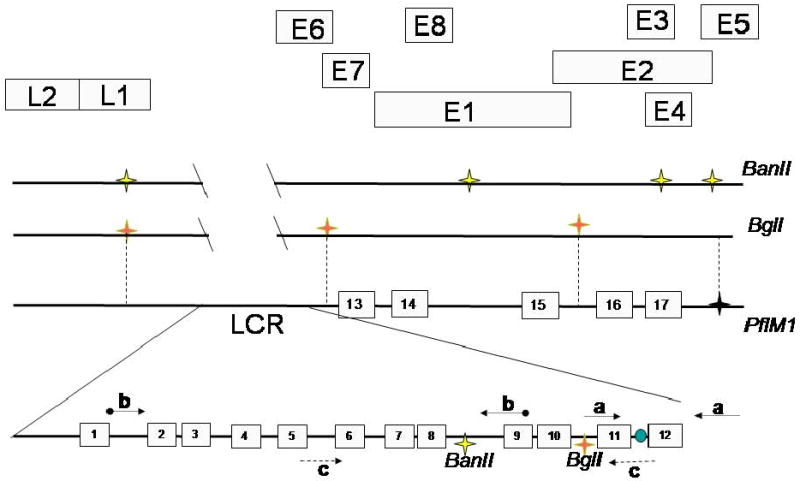
Schematic of the BPV-1 genome showing E1 and E2 binding sites. E2 binding sites are shown as numbered boxes 1-17. Symbols on the upper line represent BanII restriction sites outside the viral LCR. The viral origin is depicted between E2 sites 11 and 12. The LCR sense and antisense primers which flank the BglI and BanII cut sites are represented by dashed arrows; the primers for the origin are shown as simple arrows, with the primers for the upstream region of the LCR which flank only the BanII cut site represented by ball and arrow. The letters A, B and C represent primer pairs listed in Materials and methods.
In addition to its function in transcriptional activation, the E2 protein is necessary for both transient replication and persistence of replicated viral episomes. BPV and HPV genomes have been shown to associate with cellular chromatin during mitosis. This is thought to retain viral episomes following dissolution of the nuclear envelope in order to ensure equal partitioning of newly replicated PV into emerging daughter cells (Ilves, Kivi, and Ustav, 1999; Lehman and Botchan, 1998). The E2 C-terminal DNA binding domain identifies the viral genome, while cellular factors that interact with the N-terminal transactivation domain (TAD) facilitate genomic tethering to mitotic chromosomes (Abroi et al., 2004). The cis-acting sites involved in mitotic segregation of the viral genome were defined by deletional studies and overlap the cluster of E2 binding sites in the 5′ BPV-1 LCR. This region has been labeled as the minichromosome maintenance element (MME) (Abroi et al., 2004; Lehman and Botchan, 1998; Pirsoo et al., 1996). The trans factors that connect E2 to mitotic chromosomes have been intensively studied.
Our lab reported that the interaction of E2 with ChlR1, a DNA helicase involved in establishment of proper chromatid cohesion, is necessary for maintenance of viral episomes (Parish et al., 2006). While it was proposed that the bromodomain protein Brd4 tethers E2 proteins to mitotic chromosomes more recent investigations indicate that Brd4 is also necessary for the transcriptional functions of E2 (Ilves et al., 2006; McPhillips et al., 2006; Schweiger et al., 2007; Wu et al., 2006; You et al., 2004).
E1 and E2 are the only viral proteins required for transient replication of the genome (Mohr et al., 1990; Sedman and Stenlund, 1995; Ustav and Stenlund, 1991). The E1 protein is an ATP dependent replicative helicase that binds DNA with low specificity (Sedman and Stenlund, 1995; Ustav et al., 1993). In a cell free system, E1 can initiate DNA replication in the absence of the viral E2 protein (Grossel et al., 1996; Lusky, Hurwitz, and Seo., 1993) The role of the E2 binding sites in replication in vivo has been characterized by transfection of engineered plasmids with mutant LCR constructs along with E1 and E2 expression plasmids, and in cell lines stably expressing E1 and E2 (Liang and Botchan, 1990; Lusky, Hurwitz, and Seo, 1993; Lusky, Hurwitz, and Seo, 1994; Mohr et al., 1990; Sedman and Stenlund, 1995; Ustav et al., 1993; Ustav and Stenlund, 1991). In vitro and in vivo data demonstrated that a functional origin for transient replication consisted of E2 binding site 12 (BS12) flanking an E1 specific binding sequence (Lusky, Hurwitz, and Seo, 1993; Sedman and Stenlund., 1995; Ustav et al., 1993). The prevailing model is that E2 directly binds and targets E1 to the origin (Mohr et al., 1990). In vitro and structural data indicate that E2 must dissociate from E1 to allow for assembly of E1 double hexamers and initiation of DNA replication (Abbate, Berger, and Botchan, 2004; Lusky, Hurwitz, and Seo., 1994)(Sedman and Stenlund., 1996; Sedman and Stenlund., 1998; Yang et al., 1993). The experiments presented herein provide physical evidence to support this model as we detected the presence of both E1 and E2 at the origin in vivo during G1/S.
In this report we used a modified chromatin immunoprecipitation (ChIP) assay to define the segments of the BPV-1 episome that interact with E2 and E1 during cell cycle in monolayer cultures of transformed mouse cells. Published data would suggest that E2 is released from the E1 complex that assembles on the origin during G1/S phase; however the question remains what happens to E2 between G1/S and mitosis? Our ChIP protocol was modified to include restriction enzyme digestion, following immunoprecipitation to release segments of the viral genome that were not cross-linked to the E2 complexes. Bound regions of the DNA were differentiated using specific primers and amplification by polymerase chain reaction (PCR). Since sonication stochastically disrupts DNA, this modified protocol allows for finer resolution of binding site occupancy. With this assay we show binding of E2 to specific sites in different phases of the cell cycle and place E1 at the origin at initiation of viral replication.
Results
To investigate E2 binding site occupancy in vivo, we used the C127 cell lines ID13 and A3 that stably replicate and maintain BPV-1. The A3 genome contains three serine-to-alanine mutations in the hinge region of the E2 protein at amino acids 290, 298 and 301. These mutations stabilize the E2 protein and result in increased average genome copy number (Lehman and Botchan, 1998; Lehman, King, and Botchan, 1997; McBride and Howley, 1991). To initiate the ChIP studies, it was first necessary to test the efficacy of the available antibodies to the BPV-1 proteins. A rabbit antibody to E2 (II-1) was used in immunoprecipitations from ID13 and A3 cells along with the parental C127 cell line. Detection by on an immunoblot was achieved with the monoclonal E2 antibody B201 (Figure 2 top panel). It is interesting to note that the C-terminal repressor form of E2 (E2R) (Hubbert et al., 1988; Lambert, Spalholz, and Howley, 1987) was not detected in the A3 cell lysates. The BPV-1 E1 protein is expressed at low levels in cells that stably replicate viral episomes. Prior to this study detection of E1 has been largely restricted to overexpression systems and natural expression has been rarely visualized (Sun et al., 1990). Here the BPV-1 E1 protein was detectable in A3 and ID13 cells using an antiserum raised to its C-terminal 20 amino acids (Figure 2, middle panel). An additional immunoblot was performed in an attempt to detect E1 in synchronized A3 cells. E1 was present at detectable levels in asynchronous cells and cells blocked at G1/S, but not in mitotic A3 cells (Figure 3, bottom panel).
Figure 2.
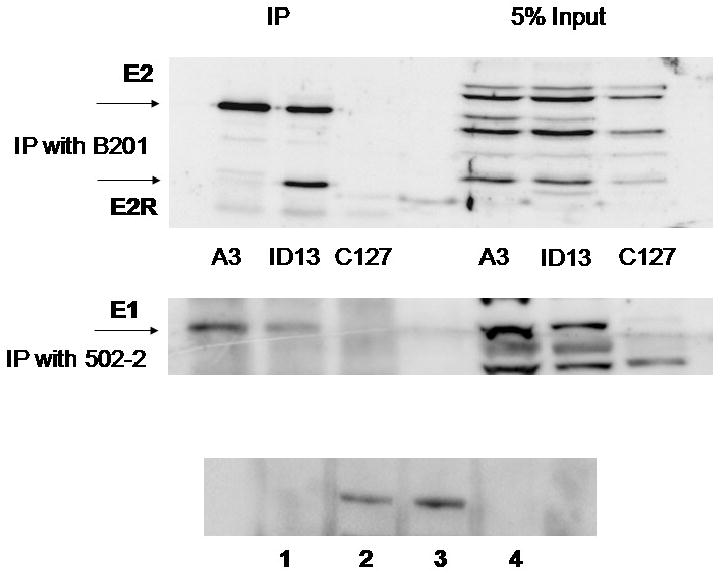
A3 cells, ID13 cells and the parental cell line C127, were lysed and immunoprecipitated with monoclonal antibody to BPV-1 E2 (B201). The proteins were immunoblotted using the rabbit antibody II-1 and visualized with the Thermo-scientific dura ECL detection kit (top panel). A3, ID13 and C127 cells were blocked overnight in 5 mM thymidine and BPV-1 E1 was immunoprecipitated using the 502-2 rabbit antibody. E1 was then detected with 502-2 (center panel). A3 cells were blocked in thymidine (lane 2), uncycled (lane 3) or blocked overnight with nocodazole (lane 4) and immunoprecipitated and blotted with the 502-2 rabbit antibody. Lane 1 shows thymidine treated C127 cells (bottom panel).
Figure 3.
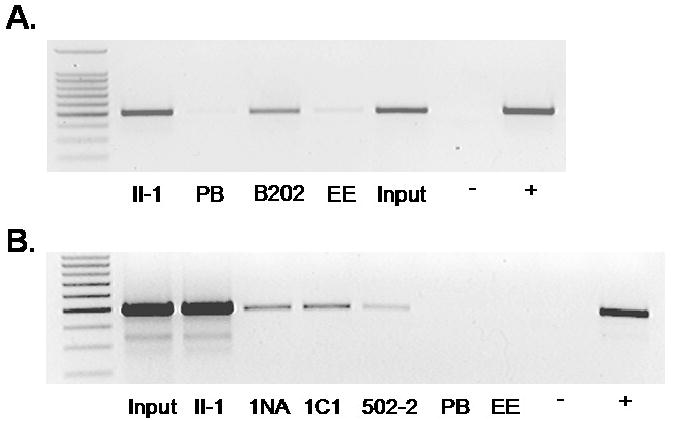
A3 cells were harvested for ChIP assay. PCR was performed for the BPV LCR on samples immunoprecipitated with the anti-BPV E2 rabbit antibody II-1, a pre-immune rabbit sera (PB), the mouse monoclonal antibody to BPV E2 (B202) or the mouse monoclonal antibody to the epitope tag EE (c 452) 0.1% input was included for reference, along with negative and positive controls for the PCR (A). ChIP samples using anti-BPV1 rabbit E2 antibody II-1, anti-BPV-1 E1 rabbit antibodies 1NA, 1C1, 502-2, pre-immune serum and EE antibody as negative controls. PCR negative control and a BPV-1 genome positive control lanes were included. Lane 1 in both figures is a 100 bp molecular weight marker (B).
Utilizing these antibodies, we attempted to determine E2 site occupancy in asynchronous A3 cells in a segment of the PV DNA LCR (nucleotides 7522-67, primer set C Figure 1) that spans the viral origin and the surrounding E2 binding sites. Pre-immune rabbit serum and the EE monoclonal antibody were included as negative controls. Following ChIP with both the rabbit E2 antisera containing the II-1 antibody and the B202 monoclonal E2 antibody DNA products were detectable with 25 cycles of standard PCR (Figure 3A). Using asynchronous A3 cells, ChIP was performed using anti-BPV-1 E1 antibodies developed in this lab. Three rabbit antibodies were used to detect E1 on the viral genome, utilizing rabbit pre-immune serum and EE antibody as negative controls and the E2 II-1 antibody as a positive control. The 1NA antibody (raised to the N-terminal 24 kilodaltons of BPV-1 E1 expressed in E. coli), and two C-terminal antibodies, 1C1 and 502-2 (raised to the C-terminal 36 kDa and a C-terminal 20 amino acid peptide of E1, respectively), co-precipitated the BPV-1 origin in synchronized cells following 27 cycles of standard PCR (Figure 3B). This places both E1 and E2 in the region of the origin of replication and demonstrates the specificity of the ChIP by utilizing antibodies to several different epitopes of both proteins to detect them in association with the viral genome.
To determine whether E2 and E1 were associated with the BPV-1 episome during G1/S and in mitosis, A3 cells were effectively synchronized in G1/S using 5 mM thymidine (16 hours) and blocked in mitosis using 100 ng/ml nocodazole (12 hours), with well defined peaks visualized using flow cytometry analysis (Figure 4A). Sub-confluent asynchronous cells were distributed throughout the cell cycle with the majority in G1. ChIPs performed on these synchronized cells illustrated the ability to detect E2 associated with viral DNA (Figure 4B). However, E1 only associated with viral genomes in asynchronous and G1/S cells and not in nocodazole arrested cells. E1 was not detectable in an immunoblot of nocodazole blocked A3 (Figure 2) consistent with data that E1 is degraded following S phase (Malcles et al., 2002).
Figure 4.
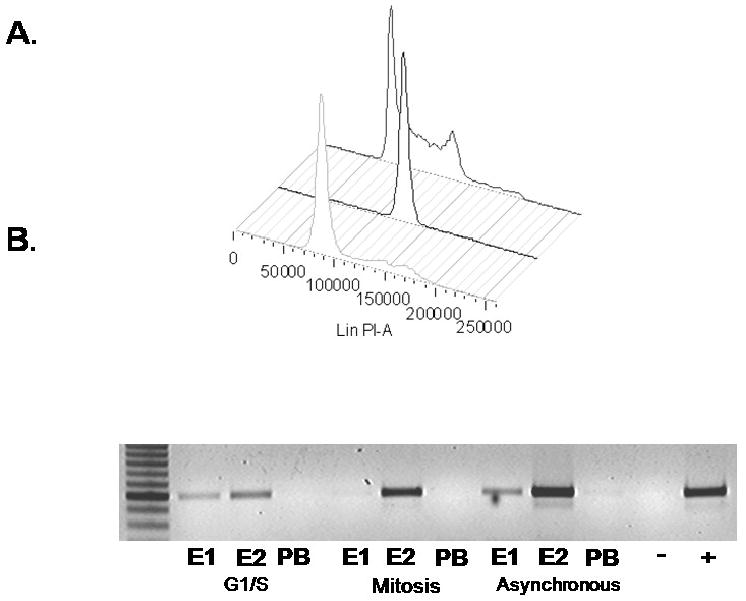
A3 cells were synchronized using either 5mM thymidine to block in G1/S (forefront), 100 ng/ml nocodazole to block in mitosis (center plot), or were left untreated (rear plot), for 12 to 16 hours then harvested and stained with propidium iodide for FACS (A). Cycled cells were harvested and ChIP assays performed using E1 antibody 502-2, the E2 antibody II-1 or rabbit pre-immune serum (B).
E2 binding site occupancy was assessed in cycled A3 cells. The majority of E2 binding sites in the BPV-1 genome are located in the LCR, with some as few as 17 bases apart, as illustrated in Figure 1. A restriction enzyme digest was added to the ChIP protocol to discriminate between these LCR segments. Three sets of primers were designed to distinguish ChIP products in adjacent regions of the LCR as depicted in Figure 1. Set (A) amplifies a product spanning nucleotides 7867–297, includes the viral origin and is 3′ to both the BanII and BglI sites. Primer set (B) encompasses nucleotides 7205–7685 and amplifies DNA 5′ to the BPV-1 origin and the BglI restriction enzyme recognition sequence. Primer set C will amplify nucleotides 7522–67 and spans both the BanII and BglI restriction sites used in this assay (Figures 3A, B, and 4B). Inclusion of restriction digest by BglI prior to PCR facilitated the examination of the LCR at the origin of replication by releasing adjacent genomic DNA and allowing for specific detection of either the origin (primer set A) or the E2 binding cluster (primer set B) by PCR. Inclusion of digestion with BanII further demonstrates specificity of binding to the upstream E2 cluster by preferentially eliminating this PCR product following a ChIP of this region of the genome. Digestion with BanII at nucleotide 7653 or BglI at 7832 will interfere with amplification using primer set C, and the absence of a PCR product indicates the digestion is virtually complete. The LCR fragment amplified by primer set C was immunoprecipitated by E2 and E1 antibodies in undigested samples (Figure 5C) and, as predicted, digestion with either BanII or BglI prior to PCR resulted in absence of this product when RED-ChIP was applied to thymidine blocked A3 cells and binding to the BPV-1 origin was subsequently examined by PCR.
Figure 5.
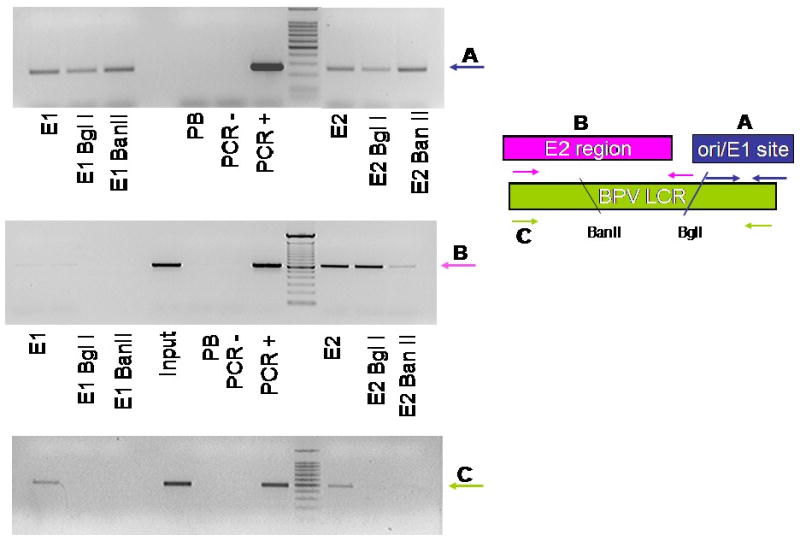
A3 cells were synchronized and harvested for ChIP assay. Lysates were digested with either Bgl I or Ban II then immunoprecipitated with 502-2, II-1 or pre-immune sera. (A) PCR reactions were performed for the BPV origin of replication; (B) the region of the LCR immediately upstream of the origin, or (C) spanning the origin and upstream regions. The figure on the right is a schematic representation of primer usage with A, B and C representing primer pairs and amplified regions as described in Materials and methods.
We used RED-ChIP to determine whether E2 is associated with sequences in the MME upstream of the viral origin. Figure 5B shows results of immunoprecipitation and PCR with primer set B (7205-7685) from thymidine blocked A3 cells. This region of the DNA contains a cluster of E2 binding sites (sites 1-8) that overlap the MME. E2 antibodies immunoprecipitated this region using both standard ChIP and RED-ChIP (BglI). PCR in the BanII digested lane was attenuated due to the location of the BanII restriction site between the set B primers, indicating that E2 is present at the MME in cells arrested at G1/S. E1 did not bind this region of the viral genome using either standard ChIP conditions or in digested ChIP samples.
To establish whether E1 and E2 are present at the origin of replication, the RED-ChIP assay was used again in A3 cells blocked at G1/S. Nucleotides 7867-297 encompass BPV-1 binding sites 11 and 12 as well as the BPV-1 origin of replication and are 3′ to both the BanII and BglI restriction sequences. This region of the LCR is amplified with primer set A. In cells blocked in G1/S both E1 and E2 bound to the genome at the origin of replication and immunoprecipitated with the corresponding 502-2 and II-1 antibodies (Figure 5A). The results from ChIP and RED-ChIP utilizing these three sets of PCR primers indicate that E2 binds to distinct regions in the viral LCR, in the upstream E2 cluster and at the origin of replication at the onset of S phase. E1, however, could only be detected in the immediate vicinity of the viral origin.
To acquire a more complete picture of the association of E2 with other binding sites distributed over the entire genome, additional RED-ChIP assays were performed on A3 cells blocked at G1/S (thymidine), and at the onset of mitosis (nocodazole), or in un-cycled cells (asynchronous). Standard ChIP assays with PCR primer set C and the II-1 E2 antibody detected binding of the protein to the LCR (Figure 6 lane 1 LCR in panels A, B and C), as would be expected from previous results (see Figure 3). Samples were also digested with the restriction enzymes PflM1 (nucleotide 3682) and BglI (RED-ChIP (PflM1, BglI) Figure 6, lane 2). The result of this digestion is the formation of a segment of viral DNA encompassing nucleotides 3682-6531 that contains no predicted E2 binding sequences (See Figure 1). Subsequently, a RED-ChIP (PflM1, BglI) with the II-1 E2 antibody would be expected to yield no PCR product in thymidine and/or nocodazole blocked cells using primers to detect this region of the viral genome (nucleotides 5638-6039) in the L1 coding sequence (primer set L1). The L1 primer set was used for PCR on the same samples in which E2 binding to the LCR was detected. There was no sequence specific binding of E2 in digested or undigested samples, consistent with the absence of E2 binding sites in the L1 coding region (Figure 6, lane 2, L1, A, B and C). Near complete digestion by BglI is demonstrated by a lack of product in lane 2 in all LCR samples (Fig. 6 lane 2, LCR, A, B and C). The viral sequences flanked by the BglI restriction site at 2818 and the PflM1 site at 3682 contain E2 binding sites, 16 and 17 (Figure 1). We attempted to detect binding of E2 to this region in ChIP assays using primers for nucleotides 2953-3085; however, E2 binding was not observed in undigested or digested samples (Figure 6, lanes 1 and 2, BS 16 & 17). Therefore, interaction of E2 in vivo with sites outside the LCR in any phase of the cell cycle was not detected (compare thymidine, asynchronous and nocodazole groups, Figure 6). The results of this PCR assay were quantified using ImageJ software and are shown beneath each panel in Figure 6. Background was subtracted (pre-immune serum lane) and data are relative to input for each sample.
Figure 6.
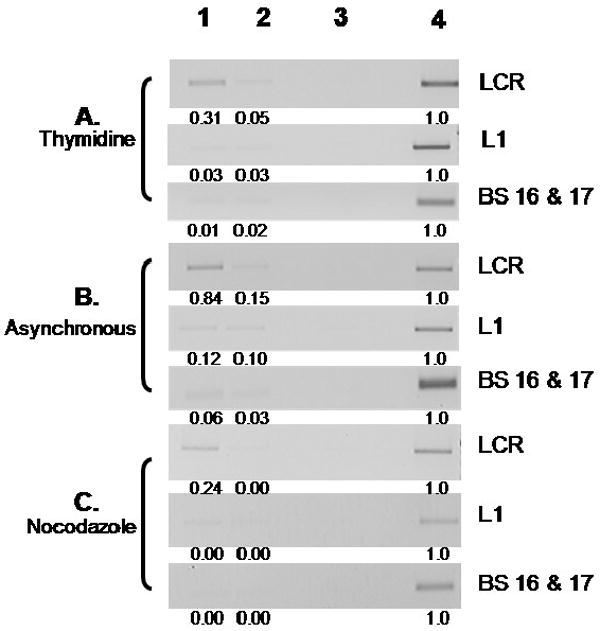
A3 cells were synchronized and E2 was immunoprecipitated without digestion (lane1) or digested with BglI and PflM1 (lane 2). Lane 3 is undigested ChIP sample using rabbit pre-immune serum, and Input DNA diluted 1:10 is also shown (lane 4). PCR was performed using either primers for the LCR of BPV, for nucleotides 5638-6039 in the L1 region, or for nucleotides 2952-3085 flanking E2 binding sites 16 and 17. The bands obtained from PCR were then quantified relative to input DNA levels using ImageJ software. Negative values are shown as 0.00.
Discussion
While extensive work has been undertaken to characterize the E2 binding sites within the genome of the bovine papillomavirus, the majority of these studies utilized either purified proteins or in vivo techniques where E1, E2 and portions of the viral LCR were transfected into cell lines (Chiang et al., 1992; Ilves, Kivi, and Ustav, 1999; Liang and Botchan., 1990; Lusky, Hurwitz, and Seo., 1994; Mohr et al., 1990; Pirsoo et al., 1996). These model systems utilized E1 and E2 at levels higher than endogenously expressed and do not account for the influence of the cell cycle on viral protein levels, expression, modification or nuclear localization. In our ChIP assays we utilized the C127 A3 cell line that maintains the full BPV-1 genome with serine to alanine mutations in the E2 protein at amino acids 290, 298 and 301. Levels of E2 and E1 are predicted to be higher in this cell type since in wild type BPV-1, phosphorylation, ubiquitination and degradation of E2 seem to rely heavily on phosphorylation at serine 301 (McBride and Howley, 1991). While mutation of this serine alone resulted in elevated levels of E2 and increased viral DNA copy number, the genome appears to otherwise replicate and be maintained in a manner consistent with previous findings (Lehman, King, and Botchan, 1997; Penrose and McBride, 2000). This phenotype may be enhanced by the absence of BPV-1 E2R normally expressed by wild type BPV-1, however as there has been no role shown for E2R in stable genome maintenance, the loss of this protein would have a negligible effect on the results of ChIP in mitotic cells (Figure 2)(Lambert, Monk, and Howley, 1990). The antibodies developed in our lab are not able to differentiate between E2 and E2R and the lack of E2R simplifies the interpretation of the data-only full length E2 is found bound to these consensus sequences.
Traditional ChIP has been used to distinguish between closely aligned transcription factor binding sites within cellular genomic DNA utilizing only sonication to disrupt the chromatin. Sonication results in variably sized fragments with no consistent restriction points. Most often, sonication alone was unable to disrupt the genome between the E2 binding sites of the BPV-1 LCR. The combination of traditional ChIP with restriction enzyme digestion (RED-ChIP) allowed us to detect specific binding of BPV-1 E2 with its predicted sites in the BPV-1 LCR, and helped demonstrate a lack of binding at predicted sites outside the LCR in a monolayer cell culture system. The BPV-1 E2 binding sites have been determined by sequence similarity and detection of E2 bound to DNA in vitro using EMSA (Li et al., 1989). The affinity of several of these binding sites is remarkably low, and they are not conserved among HPV types, leading us to question their functionality. While binding of E2 to it sequences outside the viral LCR does not appear to be required for growth or maintenance of BPV-1 in monolayer culture, it is possible that these are utilized for amplification of the viral genome in differentiated epithelial cells. It is also interesting that the inability to detect the E2R protein in A3 cells correlates with the absence of E2 at the putative E2R promoter. Why the A3 mutant of E2 did not bind to this site in vivo is uncertain. It may be that binding to these sites is below the level of detection of our assay conditions.
While the A3 cell line performed relatively well when synchronized, there were minor limitations imposed on this study by the irregularity of cycling. Serum starvation did not maintain cells in G1. Thymidine blocks cells at the initiation of replication by inhibiting the synthesis of deoxycytidine and initiating the DNA repair response. This stalls the progress of replication forks and delays the transition into S phase (Bjursell and Reichard, 1973; Xeros, 1962) and here was employed to analyze cells at the border of G1/S to investigate E1 and E2 at the onset of replication. A double thymidine block is the more common approach, however this resulted in a significant amount of cell death. As shown in Figure 3A, a single thymidine block was successful at stalling the majority of cells at G1/S and was the preferable method. Cells released from a single thymidine block did not progress into S phase as a synchronous population and therefore it was not possible to examine E2 binding as cells exit S phase and enter G2/M. We thus resorted to the use of nocodazole to examine E2 binding to the genome of cells blocked in mitosis. Nocodazole acts as a microtubule depolymerizing agent and prevents assembly of the mitotic spindle required for genome segregation, effectively accumulating cells in prometaphase. Nocodazole does not interfere with BPV-1 E2 mediated segregation of the viral episome, which does not require mitotic spindle formation (Dao et al., 2006), nor should it interfere with Brd4 mediated attachment to chromatin, as Brd4 has been shown to localize to condensed chromatin with only minor staining visible on mitotic spindles (Abbate, Voitenleitner, and Botchan, 2006; You et al., 2004). Although Brd4 is a potential candidate for the cellular protein required for viral tethering neither our lab (data not shown) nor Ives et al were able to place Brd4 on the viral genome in mitotic cells using chromatin immunoprecipitation (Ilves et al., 2006). ChIP has been shown to be relatively sensitive; however, it is possible that the binding of Brd4 to the genome was below the level of detection of our assay or that Brd4 epitopes were unavailable in a chromatin-associated complex.
Our data reveal that E2 remains associated with the BPV-1 LCR throughout the cell cycle, and localized to the origin and MME during S phase. It has been reported that central LCR E2 binding sites, predominantly sites 5-8, are the cis element required for BPV-1 genome tethering (Pirsoo et al., 1996). Our results confirm that the E2 protein is specifically bound at the MME region of the viral genome during mitosis.
BPV-1 E2 binding sites 11 and 12 flank the viral origin. It has been reported that the binding of E1 to the viral origin excludes E2 from BS12 (Lusky, Hurwitz, and Seo., 1994). Our data do not distinguish specific binding of E2 to either BS12 or the nearby BS11 during assembly of the replication complex (Figure 5). We could not demonstrate that E1 is present on the origin while E2 is bound to BS12. The use of sequential ChIP of the origin would be necessary to show concurrent association of both viral proteins. Attempts at this technique were not effective due to high background. Additional restriction enzyme digestion would also be required to differentiate between binding of E2 to sites 11 and 12, both of which have been shown to be capable of a role in initiating replication in vitro (Sanders and Stenlund, 2000).
The herpesvirus protein Epstein Barr Nuclear Antigen 1 (EBNA-1) has been compared to papillomavirus E2. Both are multifunctional with roles in replication, transcription and viral genome maintenance. The DNA binding and dimerization domain of EBNA-1 folds in a beta-barrel structure similar to E2 and binds to the cellular protein Brd4 during mitosis (Lin et al., 2008; Lindner and Sugden, 2007). The genome of EBV is more than 20 times the size of the BPV-1 genome and in contrast to the PV genome which may replicate several times per S phase, replicates only once per cell cycle, being subject to the restrictions of DNA licensing factors (Lindner and Sugden, 2007). ChIP assays have placed a number of replication proteins at the EBV origin (Chaudhuri et al., 2001). Our lab is currently using this protocol to compare replicons and determine the cellular factors present during PV replication. The use of ChIP combined with the restriction enzyme digest (RED-ChIP) is a useful tool for finer mapping of origins and promoters in regions of the genome where these proposed sites can be separated by a restriction enzyme. Utilizing this technique it should be possible to analyze the occupancy of E2 binding sites throughout the viral replicative program.
Materials and Methods
Cell culture
The C127 A3 cells were a generous gift from M. Botchan. All cells were cultured at 37°C in Dulbecco's Modified Eagle Medium (Gibco) with 10% fetal bovine serum, penicillin/streptomycin (100 U/ml) in 5% CO2. 1.4 × 106 cells were seeded in a 10 cm dish and 24 hours later were synchronized using 5 mM thymidine (Sigma) for 12 to 16 hours (G1/S) or 100 ng/ml nocodazole for 12 hours (M).
Chromatin immunoprecipitation (ChIP)
ChIP was performed using a protocol from Upstate Cell Signaling. For restriction enzyme digestion coupled ChIP (RED-ChIP), cells were treated with 0.33% formaldehyde and ChIP was performed according to the manufacturer's instructions. Following immunoprecipitation and washing, samples were resuspended in 100 μl of the appropriate restriction enzyme digest buffer, 1% bovine serum albumin (BSA) and either 30 units of BanII (New England Biolabs), 70 units high concentrate BglI (Promega), 70 units of BglI and 8 units PflM1 (New England Biolabs), or no enzyme. Following overnight digestion, samples were washed twice with TE buffer and processed using the standard ChIP protocol.
PCR Primers
| Primer | Sequence | Nucleotides | |
| Origin | Forward | tcaaaatgcagcattatattttaagct | 7867-297 |
| Primer Set A | Reverse | tggaacaccttgccaaagtcttc | |
| Upstream E2 | Forward | acacccggtacacatcctgt | 7205-7685 |
| Binding Region | Reverse | ctgtctgtagtttaaggcg | |
| Primer Set B | |||
| BPV-1 LCR | Forward | aaagtttccattgcgtctgg | 7522-67 |
| Primer Set C | Reverse | gctttttctagttagctggctatttt | |
| BS 16 and 17 | Forward | ggtggtagaggtggagtttgatg | 2953-3085 |
| Reverse | agtagtagagcccagttccgtcag | ||
| L1 | Forward | atctccctccaacccctgtaag | 5638-6039 |
| Reverse | gcctgtttgtttcctgtcatctg | ||
FACS
Briefly, thymidine blocked A3 cells were washed with PBS then trypsinized and washed once in DMEM with FBS, twice in PBS, then centrifuged at 1000 rpm. The cell pellet was resuspended in 200 μl ice cold PBS and gently agitated while 800 μl ice cold 100% ethanol was added drop-wise and left at 4°C overnight. Following fixation, cells were centrifuged at 1500 rpm for 5 minutes and fixation buffer removed. Samples were resuspended in 1 ml of propidium iodide (PI) staining solution (glucose 1 g/L, RNase A 100 U/ml, propidium iodide 0.05 mg/ml in PBS buffer (PBS, glucose 1g/L)). Prior to FACS analysis (Becton Dickinson), samples were incubated in the dark at room temperature for 30 minutes and filtered through gauze. Data were analyzed using FlowJo.
Antibodies
Rabbit anti-BPV-1 E1 antibodies INA, 1C1, 502-2 (Santucci et al., 1990), rabbit anti-BPV-1 E2 antibody II-1 (Androphy, Lowy, and Schiller, 1987), monoclonal antibodies to E2 (B201 and B202) and anti-Brd4 rabbit antibody AP8051a (Abgent) were used for ChIP experiments.
Acknowledgments
We thank Joanna Parish for many helpful suggestions. This work was supported by NIH R01CA58376 to EJA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbate EA, Berger JM, Botchan MR. The X-ray structure of the papillomavirus helicase in complex with its molecular matchmaker E2. Genes and Development. 2004;18:1981–1996. doi: 10.1101/gad.1220104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbate EA, Voitenleitner C, Botchan MR. Structure of the Papillomavirus DNA-Tethering Complex E2:Brd4 and a Peptide that Ablates HPV Chromosomal Association. Molecular Cell. 2006;24:877–889. doi: 10.1016/j.molcel.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Abroi A, Ilves I, Kivi S, Ustav M. Analysis of Chromatin Attachment and Partitioning Functions of Bovine Papillomavirus Type 1 E2 Protein. Journal of Virology. 2004;78(4):2100–2113. doi: 10.1128/JVI.78.4.2100-2113.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Androphy EJ, Lowy DR, Schiller JT. Bovine papillomavirus E2 trans-activating gene product binds to specific sites in papillomavirus DNA. Nature. 1987;325:70–73. doi: 10.1038/325070a0. [DOI] [PubMed] [Google Scholar]
- Bergman P, Ustav M, Sedman J, Moreno-Lopez J, Vennstrom B, Pettersson U. The E5 gene of bovine papillomavirus type 1 is sufficient for complete oncogenic transformation of mouse fibroblasts. Oncogene. 1988;2(5):453–459. [PubMed] [Google Scholar]
- Bjursell G, Reichard P. Effects of Thymidine on Deoxyribonucleoside Triphosphate Pools and Deoxyribonucleic Acid Synthesis in Chinese Hamster Ovary Cells. The Journal of Biological Chemistry. 1973;248(11):3904–3909. [PubMed] [Google Scholar]
- Chaudhuri B, Xu H, Todorov I, Dutta A, Yates JL. Human DNA replication initiation factors, ORC and MCM, associate with oriP of Epstein-Barr virus. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(18):10085–10089. doi: 10.1073/pnas.181347998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang CM, Ustav M, Stenlund A, Ho TF, Broker TR, Chow LT. Viral E1 and E2 proteins support homologous and heterologous papillomaviral origins. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(13):5799–5803. doi: 10.1073/pnas.89.13.5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao LD, Duffy A, Tine BAV, Wu SY, Chiang CM, Broker TR, Chow LT. Dynamic localization of the human papillomavirus type 11 origin binding protein E2 through mitosis while in association with the spindle apparatus. Journal of Virology. 2006;80(10):4792–4800. doi: 10.1128/JVI.80.10.4792-4800.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossel MJ, Barsoum J, Prakash SS, Androphy EJ. The BPV-1 E2 DNA-contact helix cysteine is required for transcriptional activation but not replication in mammalian cells. Virology. 1996;217(1):301–310. doi: 10.1006/viro.1996.0117. [DOI] [PubMed] [Google Scholar]
- Haugen TH, Cripe TP, Ginder GD, Karin M, Turek LP. Trans-activation of an upstream early gene promoter on bovine papilloma virus-1 by a product of the viral E2 gene. The EMBO Journal. 1987;6(1):145–152. doi: 10.1002/j.1460-2075.1987.tb04732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbert NL, Schiller JT, Lowy DR, Androphy EJ. Bovine papilloma virus-transformed cells contain multiple E2 proteins. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(16):5864–5868. doi: 10.1073/pnas.85.16.5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilves I, Kivi S, Ustav M. Long-term episomal maintenance of bovine papillomavirus type 1 plasmids is determined by attachment to host chromosomes, which is mediated by the viral E2 protein and its binding sites. Journal of Virology. 1999;73:4404–4412. doi: 10.1128/jvi.73.5.4404-4412.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilves I, Maemets K, Silla T, Kanikson K, Ustav M. Brd4 Is Involved in Multiple Processes of the Bovine Papillomavirus Type 1 Life Cycle. Journal of Virology. 2006;80(7):3660–3665. doi: 10.1128/JVI.80.7.3660-3665.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert PF, Monk BC, Howley PM. Phenotypic Analysis of Bovine Papillomavirus Type 1 E2 Repressor Mutants. Journal of Virology. 1990;64(2):950–956. doi: 10.1128/jvi.64.2.950-956.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert PF, Spalholz BA, Howley PM. A transcriptional repressor encoded by BPV-1 shares a common carboxy-terminal domain with the E2 transactivator. Cell. 1987;50(1):69–78. doi: 10.1016/0092-8674(87)90663-5. [DOI] [PubMed] [Google Scholar]
- Law MF, Lowy DR, Dvoretzky I, Howley PM. Mouse cells transformed by bovine papillomavirus contain only extrachromosomal viral DNA sequences. Proceedings of the National Academy of Sciences of the United States of America. 1981;78(5):2727–2731. doi: 10.1073/pnas.78.5.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman CW, Botchan MR. Segregation of viral plasmids depends on tethering to chromosomes and is regulated by phosphorylation [see comment] Proceedings of the National Academy of Sciences of the United States of America. 1998;95:4338–4343. doi: 10.1073/pnas.95.8.4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman CW, King DS, Botchan MR. A papillomavirus E2 phosphorylation mutant exhibits normal transient replication and transcription but is defective in transformation and plasmid retention. Journal of Virology. 1997;71:3652–3665. doi: 10.1128/jvi.71.5.3652-3665.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Knight J, Bream G, Stenlund A, Botchan M. Specific recognition nucleotides and their DNA context determine the affinity of E2 protein for 17 binding sites in the BPV-1 genome. Genes and Development. 1989;3:510–526. doi: 10.1101/gad.3.4.510. [DOI] [PubMed] [Google Scholar]
- Liang Y, Botchan MR. Replication of Bovine Papillomavirus Type 1 DNA Initiates within an E2-Responsive Element. Journal of Virology. 1990;64:5903–5911. doi: 10.1128/jvi.64.12.5903-5911.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A, Wang S, Nguyen T, Shire K, Frappier L. The EBNA1 protein of Epstein-Barr virus functionally interacts with Brd4. Journal of Virology. 2008;82(24):12009–12019. doi: 10.1128/JVI.01680-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner SE, Sugden B. The Plasmid Replicon of Epstein-Barr Virus: Mechanistic Insights Into Efficient, Licensed Extrachromosomal Replication in Human Cells. Plasmid. 2007;58(1):1–12. doi: 10.1016/j.plasmid.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusky M, Hurwitz J, Seo YS. Cooperative Assembly of the Bovine Papillomavirus E1 and E2 Proteins on the Replication Origin Requires and Intact E2 Binding Site. Journal of Biological Chemistry. 1993;268:15795–15803. [PubMed] [Google Scholar]
- Lusky M, Hurwitz J, Seo YS. The bovine papillomavirus E2 protein modulates the assembly of but is not stably maintained in a replication-competent multimeric E1-replication origin complex. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:8895–8899. doi: 10.1073/pnas.91.19.8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcles MH, Cueille N, Mechali F, Coux O, Bonne-Andrea C. Regulation of Bovine Papillomavirus Replicative Helicase E1 by the Ubiquitin-Proteasome Pathway. Journal of Virology. 2002;76(22):11350–11358. doi: 10.1128/JVI.76.22.11350-11358.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride AA, Howley PM. Bovine papillomavirus with a mutation in the E2 serine 301 phosphorylation site replicates at a high copy number. Journal of Virology. 1991;65(12):6528–6534. doi: 10.1128/jvi.65.12.6528-6534.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride AA, Schlegel R, Howley PM. The carboxy-terminal domain shared by the bovine papillomavirus E2 transactivator and repressor proteins contains a specific DNA binding activity. The EMBO Journal. 1988;7(2):533–539. doi: 10.1002/j.1460-2075.1988.tb02842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhillips MG, Oliveira JG, Spindler JE, Mitra R, McBride AA. Brd4 Is Required for E2-Mediated Transcriptional Activation but Not Genome Partitioning of All Papillomaviruses. Journal of Virology. 2006;80(19):9530–9543. doi: 10.1128/JVI.01105-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr IJ, Clark R, Sun S, Androphy EJ, MacPherson P, Botchan MR. Targeting the E1 replication protein to the papillomavirus origin of replication by complex formation with the E2 transactivator. Science. 1990;250:1694–1699. doi: 10.1126/science.2176744. [DOI] [PubMed] [Google Scholar]
- Morrissey LC, Barsoum J, Androphy EJ. trans Activation by the Bovine Papillomavirus E2 Protein in Saccharomyces cerevisiae. Journal of Virology. 1989;63(10):4422–4425. doi: 10.1128/jvi.63.10.4422-4425.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parish JL, Bean AM, Park RB, Androphy EJ. ChlR1 Is Required for Loading Papillomavirus E2 onto Mitotic Chromosomes and Viral Genome Maintenance. Molecular Cell. 2006;24(6):867–876. doi: 10.1016/j.molcel.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Penrose KJ, McBride AA. Proteasome-Mediated Degradation of the Papillomavirus E2-TA Protein Is Regulated by Phosphorylation and Can Modulate Viral Genome Copy Number. Journal of Virology. 2000;74(13):6031–6038. doi: 10.1128/jvi.74.13.6031-6038.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirsoo M, Ustav E, Mandel T, Stenlund A, Ustav M. Cis and trans requirements for stable episomal maintenance of the BPV-1 replicator. The EMBO Journal. 1996;15(1):1–11. [PMC free article] [PubMed] [Google Scholar]
- Sanders AM, Stenlund A. Transcription Factor-dependent Loading of the E1 Initiator Reveals Modular Assembly of the Papillomavirus Origin Melting Complex. The Journal of Biological Chemistry. 2000;275(5):3522–3534. doi: 10.1074/jbc.275.5.3522. [DOI] [PubMed] [Google Scholar]
- Santucci S, Androphy EJ, Bonne-Andrea C, Clertant P. Proteins Encoded by the Bovine Papillomavirus E1 Open Reading Frame: Expression in Heterologous Systems and in Virally Transformed Cells. Journal of Virology. 1990;64(12):602706039. doi: 10.1128/jvi.64.12.6027-6039.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller JT, Vass WC, Lowy DR. Identification of a second transforming region in bovine papillomavirus DNA. Proceedings of the National Academy of Sciences of the United States of America. 1984;81(24):7880–7884. doi: 10.1073/pnas.81.24.7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweiger M, Ottinger M, You JX, Howley PM. Brd4-Independent Transcriptional Repression Function of the Papillomavirus E2 Proteins. Journal of Virology. 2007;81(18):9612–9622. doi: 10.1128/JVI.00447-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedman J, Stenlund A. Co-operative interaction between the initiator E1 and the transcriptional activator E2 is required for replicator specific DNA replication of bovine papillomavirus in vivo and in vitro. The EMBO Journal. 1995;14(24):6218–6228. doi: 10.1002/j.1460-2075.1995.tb00312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedman J, Stenlund A. The initiator protein E1 binds to the bovine papillomavirus origin of replication as a trimeric ring-like structure. The EMBO Journal. 1996;15(18):5085–5092. [PMC free article] [PubMed] [Google Scholar]
- Sedman J, Stenlund A. The Papillomavirus E1 Protein Forms a DNA-Dependent Hexameric Complex with ATPase and DNA Helicase Activities. Journal of Virology. 1998;72(8):6893–6897. doi: 10.1128/jvi.72.8.6893-6897.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalholz BA, Yang YC, Howley PM. Transactivation of a bovine papilloma virus transcriptional regulatory element by the E2 gene product. Cell. 1985;42(1):183–191. doi: 10.1016/s0092-8674(85)80114-8. [DOI] [PubMed] [Google Scholar]
- Stanway CA, Snowden MP, Wilson LE, Kingsman AJ, Kingsman SM. Efficient activation of transcription in yeast by the BPV1 E2 protein. Nucleic Acids Research. 1989;17(6):2187–2196. doi: 10.1093/nar/17.6.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Thorner L, Lentz M, MacPherson P, Botchan M. Identification of a 68-Kilodalton Nuclear ATP-Binding Phosphoprotein Encoded by Bovine Papillomavirus Type 1. Journal of Virology. 1990;64(10):5093–5105. doi: 10.1128/jvi.64.10.5093-5105.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ustav E, Ustav M, Szymanski P, Stenlund A. The Bovine Papillomavirus origin of replication requires a binding site for the E2 transcriptional activator. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:898–902. doi: 10.1073/pnas.90.3.898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ustav M, Stenlund A. Transient replication of BPV-1 requires two viral polypeptides encoded by the E1 and E2 open reading frames. The EMBO Journal. 1991;10(2):449–457. doi: 10.1002/j.1460-2075.1991.tb07967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SY, Lee AY, Hou SL, Kemper JK, Erdjumant-Bromage H, Tempst P, Chiang CM. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes and Development. 2006;20:2383–2396. doi: 10.1101/gad.1448206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xeros N. Deoxyriboside control and synchronization of mitosis. Nature. 1962;194:682–683. doi: 10.1038/194682a0. [DOI] [PubMed] [Google Scholar]
- Yang L, Mohr IJ, Fouts E, Lim DA, Nohaile M, Botchan MR. The E1 protein of bovine papilloma virus 1 is an ATP-dependent DNA helicase. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:5086–5090. doi: 10.1073/pnas.90.11.5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You J, Croyle JL, Nishimura A, Ozato K, Howley PM. Interaction of the Bovine Papillomavirus E2 Protein with Brd4 Tethers the Viral DNA to Host Mitotic Chromosomes. Cell. 2004;117:349–360. doi: 10.1016/s0092-8674(04)00402-7. [DOI] [PubMed] [Google Scholar]
- Zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nature Reviews in Cancer. 2002;2(5):342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]