

Abstract
The well established rat hepatocarcinogen N-nitrosopyrrolidine (NPYR, 1) requires metabolic activation to DNA adducts to express its carcinogenic activity. Among the NPYR-DNA adducts that have been identified, the cyclic 7,8-butanoguanine adduct 2-amino-6,7,8,9-tetrahydro-9-hydroxypyrido[2,1-f]purine-4(3H)-one (6), has been quantified using moderately sensitive methods, but its levels have never been compared to those of other DNA adducts of NPYR in rat hepatic DNA. Therefore, in this study, we developed a sensitive new LC-ESI-MS/MS-SRM method for quantitation of adduct 6, and compared its levels to those of several other NPYR-DNA adducts formed by different mechanisms. The new method was shown to be accurate and precise, with good recoveries and low fmol detection limits. Rats were treated with NPYR by gavage at doses of 46, 92, or 184 mg/kg body weight and sacrificed 16 h later. Hepatic DNA was isolated and analyzed for NPYR-DNA adducts. Adduct 6 was by far the most prevalent, with levels ranging from about 900 – 3000 μmol/mol Gua and responsive to dose. Levels of adducts formed from crotonaldehyde, a metabolite of NPYR, were about 0.2 – 0.9 Pmol/mol dGuo, while those of adducts resulting from reaction with DNA of tetrahydrofuranyl- like intermediates were in the range of 0.01 – 4 μmol/mol deoxyribonucleoside. The results of this study demonstrate that, among typical NPYR-DNA adducts, adduct 6 is easily the most abundant in hepatic DNA. Since previous studies have shown that it can be detected in the urine of NPYR-treated rats, the results suggest that it is a potential candidate as a biomarker for assessing human exposure to and metabolic activation of NPYR.
Keywords: N-nitrosopyrrolidine, DNA-adducts, mass spectrometry
Introduction
N-Nitrosopyrrolidine (NPYR, 1, Scheme 1) is a firmly established hepatocarcinogen in rats, with carcinogenic activity exceeding that of many well known carcinogens such as vinyl chloride, styrene oxide, various red dyes, 2-acetylaminofluorene, and 2-amino-3-methylimidazo[4,5-f]quinoline (IQ) (1–4). Its carcinogenicity has been confirmed in extensive dose response studies (5). NPYR also induces respiratory tract tumors in mice and Syrian golden hamsters (6).
Scheme 1.

Overview of adduct formation from NPYR (1) and α-acetoxyNPYR (3).
NPYR has been detected in human urine in several studies, with typical levels in one study ranging from 1–2 nmol/24 h, and influenced by nitrate consumption (7–9). Studies in rats demonstrate that only about 1% of NPYR is excreted unchanged, so the actual levels present in humans are likely to be far higher than those reported in urine, perhaps reaching 100–200 nmol/day (10). The presence of these amounts of NPYR in humans is plausible because the precursors pyrrolidine and nitrite are abundant in the human body (11), and endogenous formation of the related nitrosamine, N-nitrosoproline, has been confirmed in many studies (12). Exogenous exposure to preformed NPYR through the diet from foods such as bacon and cured meats, from drinking water, and from tobacco smoke also occurs, but the levels are likely to be lower than through in vivo formation (13–15). Since most NPYR metabolites are ultimately excreted as volatiles such as CO2 and N2, they cannot be readily measured as an indicator of NPYR levels in the body(10).. An alternate approach is to quantify DNA adducts as biomarkers.
NPYR is metabolically activated by α-hydroxylation, catalyzed by cytochrome P450 enzymes (16). All available data support the hypothesis that DNA adducts formed as a result of this process are critical to NPYR carcinogenicity. The structures of multiple DNA adducts from NPYR α-hydroxylation have been characterized by using α-acetoxyNPYR (3, Scheme 1) as a stable precursor to α-hydroxyNPYR (2) in vitro, and by analyses of DNA from rats treated with NPYR (17–28). An overview of these reactions is presented in Scheme 1. Three major types of adducts are formed, represented by 5 and 6, 9, and 13 – 17. Adducts such as 5 and 6 likely result from the reaction of diazonium ion 7 with the 7- and/or 8-positions of dGuo (18,20,24,26). The solvolysis product crotonaldehyde (8) reacts with DNA producing diastereomers of adduct 9 (17,20,26,29). Adducts 13 – 17 are formed by the reaction of the oxonium ion 12, or related intermediates, with DNA bases (22,23,26,27). Adducts 13, 14, and 17 are also produced when 2-hydroxytetrahydrofuran (11) or oxidized tetrahydrofuran reacts with dGuo, dAdo, and dCyd (22,27,30).
The 7,8-butanoguanine adduct 6 was first observed in hepatic DNA of NPYR-treated rats by Hunt and Shank, using HPLC with fluorescence detection, and was characterized structurally by our group (18,31). Subsequent studies used HPLC-fluorescence or radio-labeled NPYR for detection of adduct 6 in DNA from various tissues of NPYR-treated animals and in most cases reported substantial adduct levels (32). Adducts 9 and 13–16 have also been quantified in hepatic DNA of NPYR-treated rats, by 32P-postlabelling and LC-ESI-MS/MS-SRM, respectively (19,28). However, there is no information in the literature on the relative amounts of these three different types of adducts in DNA of NPYR-treated rats. Therefore, we addressed this problem in the current study by developing a new LC-ESI-MS/MS-SRM method for adduct 6, and comparing its levels to those of the other adduct types, which were also determined by LC-ESI-MS/MS-SRM. Data obtained will be important for the choice of biomarkers to be used in human studies to assess human exposure to NPYR and related cancer risk, and in further understanding of mechanisms of NPYR carcinogenesis in rats.
Materials and Methods
Chemicals and Enzymes
All chemicals were of the highest purity grade commercially available. α-AcetoxyNPYR (2) and 4-iodobutyraldehyde were prepared as described (17,20,33). [15N5]dGuo was obtained from Cambridge Isotope Laboratories (Andover, MA). Ethanol was procured from AAPER Alcohol and Chemical Co. (Shelbyville, KY). 2-Propanol was purchased from Acros Organics (Morris Plains, NJ). Puregene DNA purification solutions were obtained from QIAGEN (Valencia, CA). Alkaline phosphatase (from calf intestine) was purchased from Roche Diagnostics Corp. (Indianapolis, IN). 2′,3′,5′-Triacetylguanosine, herring testes DNA, DNase I, phosphodiesterase I, dGuo, dAdo, and dThd were obtained from Sigma-Aldrich Co. (St. Louis, MO). All other chemicals were obtained from Sigma-Aldrich or Fisher Scientific (Fairlawn, NJ). Water was purified in a Milli-Q system (Millipore, Bedford, MA).
Adduct Standards
N2-(4-hydroxybutyl)dGuo (N2-(4-HOB)dGuo, 18), N6-(4-hydroxybutyl)dAdo (N6-(4-HOB)dAdo, 19), O2-(4-hydroxybutyl)dThd (O2-(4-HOB)dThd, 20), and the respective isotopic standards [15N5]N2-(4-HOB)dGuo ([15N5]18), [15N5]N6-(4-HOB)dAdo ([15N5]19), and [15N2]O2-(4-HOB)dThd ([15N2]20) were prepared as described (27,28,30). These were used for quantitation of adducts 13 – 15, after NaBH3CN reduction. [An interfering peak prevented quantitation of the NaBH3CN reduction product of adduct 16 in this study, and levels of adduct 17 in vivo were too low for quantitation previously (28)]. The 7,8-butanoguanine adduct 2-amino-6,7,8,9-tetrahydro-9-hydroxypyrido[2,1-f]purine-4(3H)-one (6) was prepared as described and its spectral properties agreed with those reported (18). 1H NMR, 600 MHz, (DMSO-d6): δ 1.85(m, H7,8 partially obscured by solvent), 4.03(m, 1H, H6a), 4.22 (m, 1H, H6b), 4.64 (m, 1H, H9), 6.10 (s, 2H, NH2); UV, λmax 285 nm; positive ESI-MS: m/z 222 [M + H]+, m/z 204 [M – H2O + H]+. 3-(2′-Deoxyribos-1′-yl)-5,6,7,8-tetrahydro-8-hydroxy-6-methylpyrimido[1,2-a]purine-10(3H)one (adduct 9) and [15N5]9 were prepared as described (34).
[15N5]7,8-butanoguanine 6([15N5]6)
α-AcetoxyNPYR (3) (48 mg, 0.3 mmol) was allowed to react with [15N5]Guo (12 mg, 0.04 mmol) in 6 mL of 0.1 M phosphate buffer (pH 7) at 37 °C for 17 h. Then, more α-acetoxyNPYR (30 mg) was added and the reaction mixture was incubated an additional 6 h, then heated at 100 °C for 1 h. The product, [15N5]6, was collected and purified by HPLC systems 2 and 1, respectively, and then neutralized, concentrated to dryness, and extracted 3 times with 10 mL of 1/1:CH3OH/C2H5OH. The extracts were concentrated to dryness and redissolved in H2O. The resulting [15N5]6 adduct was purified, identified and quantified by comparison to a standard in HPLC Systems 1 and 2.
HPLC Separations
HPLC analyses were carried out with Waters Associates (Waters Division, Millipore, Milford, MA) instruments with a model RF10 AXL fluorescence detector (Shimadzu Scientific Instruments, Columbia, MD) for HPLC systems 1 and 2, or a model 996 photodiode array UV detector (Waters) for HPLC system 3, or a model SPD-10 A UV-vis detector (Shimadzu) for HPLC system 4. Elution systems were as follows.
System 1 consisted of two 25 cm × 4.6 mm Partisil-10 SCX strong cation exchange columns (Whatman, Clifton, NJ) eluted isocratically with 5% CH3OH in 40 mM ammonium phosphate buffer, pH 2.0, at a flow rate of 1 mL/min, with detection by fluorescence (excitation 290 nm, emission 380 nm) for identification and quantitation of 6 and [15N5]6.
System 2 was a 9.4 mm × 50 cm Partisil 10 SCX column eluted isocratically with 40 mM ammonium phosphate buffer, pH 2.0, at a flow rate of 3 mL/min, and detection as in system 1, for collection and purification of [15N5]6.
System 3 was two 4.6 mm × 25 cm Supelcosil LC 28-BD columns (Supelco, Bellefonte, PA) with isocratic elution by 5% CH3OH in 40 mM ammonium acetate buffer, pH 6.6, for 5 min and then a gradient from 5 to 35% CH3OH in 40 min at a flow rate of 1 mL/min with UV detection (285 nm) for determination of the UV spectrum of adduct 6.
System 4 used a 250 mm × 4.6 mm i.d., 5 μm, Luna C18(2) column (Phenomenex, Torrance, CA) eluted with a gradient of H2O and CH3OH at a flow rate of 0.7 mL/min: from 0 to 15 min, 5 to 22% CH3OH; from 15 to 17 min, 22 to 80% CH3OH; from 17 to 20 min, 80% CH3OH; from 20 to 22 min, 80 to 5% CH3OH; from 22 to 35 min, 5% CH3OH. The UV detector was set at 254 nm for dGuo, dThd, dAdo, and Gua quantitation in DNA samples, essentially as described previously (35).
LC-MS Analysis of Synthetic Adduct 6
LC-MS analyses were carried out on a Thermo Finnigan LCQ Deca instrument (Thermoquest LC/MS Division, San Jose CA) interfaced with a Waters Alliance 2690 HPLC multi-solvent delivery system and equipped with an SPD 10 A UV-vis detector (Shimadzu). The ESI source was set as follows: voltage, 2.0 kV; current, 10 μA; and capillary temperature, 250 °C. HPLC system 3 was used, except that the flow rate was 0.7 mL/min.
Capillary HPLC-ESI-MS/MS-SRM Analyses of Adducts 6 and 18–20
Adduct quantitation in DNA samples was carried out with a Finnigan Quantum Discovery Max (ThermoElectron Division, San Jose, CA) triple quadrupole mass spectrometer interfaced with an Agilent 1100 Series capillary flow HPLC. A 250 mm × 0.5 mm i.d., 5 μm, Zorbax SB C18 (Agilent) column was used with different solvent elution programs for quantitation of 7,8-butanoguanine 6 or 18 – 20. The elution systems were as follows.
System 5 was used for quantitation of 7,8-butanoguanine 6. The column was eluted with a gradient of 25 mM ammonium acetate (solvent A) and 25% CH3CN in CH3OH (solvent B) at a flow rate of 10 μL/min and a column temperature of 40 °C, as follows: from 0 to 10 min, 2 to 10% of solvent B; from 10 to 15 min, 10 to 30% of solvent B; from 15 to 22 min, 30% of solvent B; from 22 to 25 min, 30 to 70% of solvent B; from 25 to 30 min, 70% of solvent B; from 30 to 35 min, 70 to 2% of solvent B; from 35 to 45 min, 2% of solvent B. The first 12 min of eluant was directed to waste and the 12 – 45 min fraction was diverted to the ESI source.
System 6 was used for quantitation of adducts 18–20, from NaBH3CN reduction of adducts 13 – 15. The column was eluted with a gradient of 15 mM ammonium acetate, pH 6.6, and CH3CN at a flow rate of 10 μL/min and 25 °C, as follows: from 0 to 10 min, 0% of CH3CN; from 10 to 39 min, 0 to 20% CH3CN; from 39 to 44 min, 20 to 75% CH3CN; from 44 to 49 min, 75% CH3CN; from 49 to 54 min, 75 to 0% CH3CN; from 54 to 64 min, 0% of CH3CN. The first 31 min of eluant was directed to waste and the 31 – 49 min fraction was diverted to the ESI source.
The ESI-MS parameters for quantitation of 7,8-butanoguanine 6 were set in the positive ion mode as follows: spray voltage, 3 kV; sheath gas pressure, 20 psi; capillary temperature, 325 °C; collision energy, 25 V; scan width, 0.1 amu; scan time, 0.5 s; Q1 peak width, 0.7 amu; Q3 peak width, 0.7 amu; Q2 collision cell gas pressure, 1.0 mTorr; source CID, 8 V; and tube lens offset, 85 V.
The ESI-MS parameters for quantitation of 18 – 20 were set in the positive ion mode as follows: spray voltage, 4 kV; sheath gas pressure, 30 psi; capillary temperature, 250 °C; collision energy, 13V; scan width, 0.1 amu; scan time, 0.2 s; Q1 peak width, 0.7 amu; Q3 peak width, 0.7 amu; Q2 collision cell gas pressure, 1.0 mTorr; source CID, 10 V; and tube lens offset, 85V.
Adduct analyses were carried out with selected reaction monitoring (SRM) as follows: 6 m/z 222 → m/z 204; [15N5]6 m/z 227 → m/z 209; 18 m/z 340 → m/z 224; [15N5]18 m/z 345 → m/z 229; 19 m/z 324 → m/z 208; [15N5]19 m/z 329 → m/z 213; 20 m/z 315 m/z 199; [15N2]20 m/z 317 → m/z 201. Data were acquired and processed with Xcalibur software version 1.4 (ThermoElectron).
HPLC-ESI-MS/MS-SRM Analysis of Adduct 9 Diastereomers
This was carried out essentially as described previously (34).
Animal Experiments
Six week old male F344 rats were purchased from Charles River (Wilmington, MA) and housed in the Research Animal Resources facility of the University of Minnesota. They were maintained on NIH-07 diet (Harlan, Madison, WI) and tap water.
Three groups of five rats, weighing approximately 250 g each, were treated by gavage with a single dose of NPYR (46, 92, or 184 mg/kg body weight) in 0.5 mL of saline. Another group of five rats received only 0.5 mL of saline by gavage. They were sacrificed after 16 h and their organs were collected and frozen at −80 °C.
DNA Extraction
Hepatic DNA was isolated as described in the Puregene® protocol for 1 g animal tissue (QIAGEN) with several modifications. Briefly, rat liver tissue samples (0.5 g) were homogenized with 15 mL of Puregene cell lysis solution on ice. Proteinase K (75 μL of a 20 mg/mL solution) was added and the homogenate was allowed to stand at room temperature overnight. RNase A (75 μL of a 4 mg/mL solution) was added to the cell lysate and, after 2 h at room temperature, protein was precipitated by addition of 5 mL of Puregene protein precipitation solution and centrifugation at 2000 × g for 10 min. The supernatant was poured into a tube containing cold 2-propanol and the precipitated DNA was collected, dissolved in 4 mL of 10 mM Tris/1 mM EDTA pH 7.0 buffer, and the solution was extracted 3 times with 4 mL of CHCl3 containing 4% isoamyl alcohol. The DNA was precipitated from the aqueous phase by addition of 0.4 mL of 5 M NaCl and 8 mL of ice-cold ethanol, washed three times with 3 mL of 70% ethanol and three times with 3 mL of 100% ethanol, and dried with a stream of N2. DNA concentration was determined by measuring UV absorption at 260 nm, and DNA purity was assessed by the UV absorbance ratios at 260/230 nm and 260/280 nm.
Sample Preparation for MS/MS Analyses
DNA samples were subjected to neutral thermal hydrolysis for quantitation of 7,8-butanoguanine 6 and to enzymatic hydrolysis for analysis of the other adducts. For neutral thermal hydrolysis, DNA (0.1 – 1 mg weighed) was dissolved in 1 mL of 10 mM sodium cacodylate buffer, pH 7.0, containing 5000 fmol of [15N5]6. The solution was incubated at 100 °C for 1 h, an aliquot of 50 μL was separated for Gua quantitation (see below), and the remaining 950 μL hydrolysate was partially purified by centrifugation using a Centrifree MPS device (MW cutoff of 30,000 Da, Amicon, Beverly, MA) to remove high molecular weight substances, and by solid-phase extraction (Strata-X cartridges, 33 μm, 30 mg/1 mL, Phenomenex) to desalt and partially isolate the adduct from the normal base pool. The Strata-X cartridge was washed with 1 mL of 5% CH3OH in H2O and 1 mL of 40% CH3OH in H2O. The 40% CH3OH fraction was collected and evaporated to dryness. The residue was dissolved in 95 μL of H2O, and 8 μL aliquots were analyzed by HPLC-ESI-MS/MS-SRM as described above. Prior to each set of analyses, a calibration curve containing varying amounts of 7,8-butanoguanine 6 (10, 50, 100, 200, 400, 800, 1600, 3200, and 6400 fmol) and a fixed amount of [15N5]6 (400 fmol) was constructed. Quantitation of Gua was carried out by a method similar to that described previously (35). A 50 μL aliquot reserved from the hydrolysate was added to 150 μL of 0.1 N HCl and incubated at 80 °C for 1 h. After dilution with 300 μL of H2O, 10 μL was injected in HPLC system 4.
For quantitation of 18 – 20, the procedure was essentially as described (28) with some modifications. Briefly, DNA samples (~ 1 mg weighed) were dissolved in 1 mL of 10 mM Tris/5 mM MgCl2 buffer, pH 7.0, containing DNase I (650 units). NaBH3CN (10 mg) was added to the mixture three times. After the first two additions, the sample was allowed to stand at room temperature for 30 min. After the final addition, it was incubated at 37 °C for 30 min. The pH was adjusted to 7 with 40 μL of HCl, and the internal standards [15N5]18 (250 fmol), [15N5]19 (1250 fmol), and [15N2]20 (2500 fmol) were added. The resulting solution was incubated with an additional amount of DNase I (650 units) at 37 °C for 10 min. Phosphodiesterase I (0.02 units) and alkaline phosphatase (150 units) were then added and the incubation was continued for another 60 min (final volume = 1435 μL). A 20 μL aliquot was separated for dGuo, dThd, and dAdo quantitation using HPLC System 4 after dilution with 80 μL of H2O. The remaining solution (1415 μL) was applied to a solid-phase extraction cartridge (Strata-X, 33 μm, 30 mg/1 mL, Phenomenex) that was washed with 1 mL of H2O, 2 mL of 20% CH3OH in H2O, and 2 mL of 100% CH3OH. The methanol containing eluants were combined, evaporated to dryness, redissolved in 200 μL of 50% CH3OH in H2O, transferred into an autosampler vial with an infused 300 μL insert (Chrom Tech), and dried again. The residue was dissolved in 49.3 μL of H2O, and 8 μL aliquots were analyzed by HPLC-ESI-MS/MS-SRM as described above.
Blank samples with and without purchased herring testes DNA, containing all the solutions, enzymes, and internal standards used in the procedure were processed concomitantly to all DNA samples following the procedures described above to check for any contamination.
Validation of Method for Quantitation of 7,8-Butanoguanine 6
Method accuracy was determined by adding varying amounts of 7,8-butanoguanine 6 and a fixed amount of [15N5]6 to 1 mg of herring testes DNA and carrying out the analysis. The intraday precision was determined by adding 6 and [15N5]6 to 1 mg herring testes DNA (N = 4) and carrying out the analysis. Recovery was calculated by adding the internal standard [15N5]6 (5000 fmol) to 1 mg herring testes DNA and processing the samples as described above (N = 8). The internal standard areas were compared to those of the same quantities (1 mg) of herring testes DNA processed without addition of the internal standard (N = 4), which was added just prior to the HPLC-ESI-MS/MS-SRM analysis. The limit of detection (LOD) was estimated from the lowest amount of 6 added to the herring testes DNA sample.
Results
Calibration curves in the 10–200 and 200–12800 fmol range of 6, with a fixed amount of the internal standard [15N5]6 are presented in Figure 1, showing that the MS response was linear in the range measured. The accuracy of the method for adduct quantitation in DNA was determined by adding varying amounts of 6 to samples of herring testes DNA and carrying out the analysis. Representative chromatograms showing the detection of 6 in a herring testes DNA sample to which 50 fmol of this adduct had been added prior to the hydrolysis procedure and a corresponding control sample are presented in Figure 2. Excellent agreement was observed between added and detected amounts of 6, as shown in Table 1.
Figure 1.

Calibration curves for LC-ESI-MS/MS-SRM analysis of 7,8-butanoguanine adduct 6: A, 10–200 fmol 6 and 400 fmol [15N5]6; B, 200–12, 800 fmol 6 and 400 fmol [15N5]6.
Figure 2.

Chromatograms obtained upon LC-ESI-MS/MS-SRM analysis of a neutral thermal hydrolysate of herring testes DNA to which (A) 50 fmol of adduct 6 had been added and (B) the same sample with no addition of adduct 6: upper panels, analyte; lower panels, internal standard.
Table 1.
Accuracy and precision of the method for quantitation of 7,8-butanoguanine 6a
| 7,8-butanoguanine 6 (fmol) | Intraday precision CV (%) | |
|---|---|---|
| Added | Detected | |
| 10 | 12 | |
| 50 | 51.3 ± 2.9b | 5.7 |
| 100 | 87 | |
| 200 | 187 | |
| 400 | 408 ± 6.6b | 1.6 |
| 800 | 741 | |
| 1600 | 1520 | |
| 3200 | 3180 ± 78.9b | 2.5 |
Adduct 6 was added to 1 mg of herring testes DNA and the analysis was performed. Untreated herring testes DNA did not contain 6.
Mean ± S.D. of four samples
Method precision was determined by quadruplicate analysis of some samples. The coefficient of variation averaged less than 6% (Table 1). In a further test of precision, hepatic DNA was isolated from two rats, each treated with 46 mg/kg NPYR by gavage. Two separate aliquots of each DNA sample were analyzed one week apart. Adduct levels were 712 and 879 μmol/mol Gua for the aliquots of the DNA isolated from the first rat and 981 and 1043 μmol/mol Gua for those from the second rat, further demonstrating the precision of the method. This is consistent with the results of previous studies which have shown that the release of adduct 6 from DNA by neutral thermal hydrolysis is quantitative and reproducible (18,24).
The average recovery of adduct 6 was 78%. The LOD for 6 injected on column was 3 fmol (S/N = 3). The LOD for the detection of adduct 6 in DNA was 10 fmol (S/N = 2).
The method was applied to hepatic DNA from rats treated with NPYR. Analysis for adduct 6 provided clear chromatographic peaks co-eluting with the internal standard in DNA samples from NPYR-treated rats, but no peak was observed in control rat DNA (Figure 3). Levels of adduct 6 in the rat liver DNA samples are summarized in Table 2. A clear dose/response relationship was observed.
Figure 3.

Chromatograms obtained upon LC-ESI-MS/MS-SRM analysis for adduct 6 in a neutral thermal hydrolysate of (A) hepatic DNA from a rat treated with NPYR by gavage and (B) a control rat treated only with saline; upper panels, analyte; lower panels, internal standard.
Table 2.
Adduct levels in hepatic DNA of rats treated with NPYRa
| Dose (mg/kg bw) | 7,8-Butanoguanine 6 (μmol/mol Gua) | Adduct 9 |
N2-(4-HOB)dGuo (18)b (μmol/mol dGuo) | N6-(4-HOB)dAdo (19)b (μmol/mol dAdo) | O2-(4-HOB)dThd (20)b (μmol/mol dThd) | ||
|---|---|---|---|---|---|---|---|
| (6S,8S)-(μmol/mol dGuo) | (6R,8R)-(μmol/mol dGuo) | Total (μmol/mol dGuo) | |||||
| 46 | 952 ± 180 | 0.154 ± 0.057 | 0.077 ± 0.021 | 0.231 ± 0.078 | 0.369 ± 0.134c | 0.013 ± 0.006 | 1.065 ± 0.349 |
| 92 | 1742 ± 245 | 0.341 ± 0.066 | 0.158 ± 0.026 | 0.499 ± 0.091 | 0.478 ± 0.114c | 0.008 ± 0.004 | 2.049 ± 0.361 |
| 184 | 3032 ± 855 | 0.634 ± 0.188 | 0.271 ± 0.054 | 0.905 ± 0.240 | 0.951 ± 0.193c | 0.016 ± 0.008 | 3.767 ± 0.840 |
Rats were given a single dose of NPYR by gavage and sacrificed 16 h later. Limits of quantitation were: 6, 0.1 μmol/mol Gua; 9, 0.004 μmol/mol dGuo; 18, 0.02 μmol/mol dGuo; 19, 0.002 μmol/mol dAdo; 20, 0.01 μmol/mol dThd.
DNA was treated with NaBH3CN during enzyme hydrolysis to convert adducts 13–15 to 18–20.
Control amount was subtracted from each value.
The hepatic DNA samples were next analyzed for adduct 9, which is formed from crotonaldehyde (8, Scheme 1). The (6S,8S)- and (6R,8R)-diastereomers of adduct 9 are separable under the conditions of this analysis.
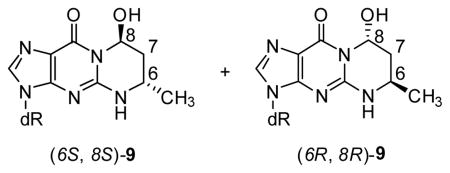
A typical chromatogram is illustrated in Figure 4. Clear peaks corresponding to (6S,8S)-9 and (6R,8R)-9 were observed. These peaks were not seen in hepatic DNA from control rats. Adduct levels increased with dose (Table 2). Levels of (6S,8S)-9 were 2–3 times as great as those of (6R,8R)-9.
Figure 4.

Chromatograms obtained upon LC-ESI-MS/MS-SRM analysis for adduct 9 in an enzymatic hydrolysate of hepatic DNA from a rat treated with NPYR by gavage (46 mg/kg body weight): upper panel, analyte; lower panel, internal standard.
The final analysis was for adducts 13–15, as their NaBH3CN-reduced forms 18 – 20. (Adduct 16 was not quantified due to an interfering peak).
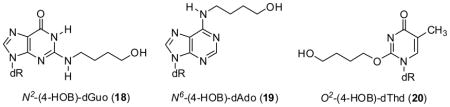
Clear chromatographic peaks co-eluting with the internal standards for adducts 18 – 20 were observed in samples from NPYR treated rats, as reported previously (28). No adduct peaks, or sometimes small peaks close to background noise, were detected in DNA samples from control rats. Blank samples containing all solutions, enzymes, and internal standards used in the analysis, but no DNA, were processed concomitantly to the DNA samples. The background noise levels corresponding to adduct retention times were subtracted to obtain the adduct levels presented in Table 2. Clear dose-response relationships were observed for adducts 18 and 20 in the rats treated with 46, 92, or 184 mg NPYR/kg bw by gavage. Levels of 19 were lower and did not clearly increase with dose.
Levels of adduct 6 were 3400–4100 times as great as those of adduct 9 and 800–220,000 times as great as those of adducts 13 – 15 (quantified as 18 – 20) in the same rat liver DNA samples.
Discussion
A major goal of this study was to develop a quantitative LC-ESI-MS/MS-SRM method for the 7,8-butanoguanine adduct 6. This was accomplished. Clear peaks with the correct mass transition and retention time were observed for adduct 6, and these were not present in control DNA samples. Using known addition and analysis of replicates, we demonstrated that the method was accurate and precise, with good recovery. The detection limit of 10 fmol of adduct 6 starting with 1 mg of DNA was easily sensitive enough for its analysis in hepatic DNA of rats treated with NPYR at the doses used here, and may be adequate for analysis of human DNA samples. In previous in vivo studies of NPYR-DNA adducts, 6 has been analyzed only by HPLC with fluorescence or radioflow detection (18,21,24,31,32). These methods would not be applicable for analysis of human DNA samples. HPLC with fluorescence detection is likely not sensitive or specific enough, while radioflow detection is clearly impractical.
A second goal of our study was to analyze three major types of NPYR DNA adducts - represented by adduct 6 from the oxocarbocation pathway, adduct 9 from the crotonaldehyde pathway, and adducts 13–15 from the oxonium ion/2-hydroxytetrahydrofuran pathway (Scheme 1) - in the same DNA samples from rats treated with NPYR, using LC-ESI-MS/MS-SRM methods. All previous studies of NPYR-DNA adduct formation in vivo are summarized in Table 3. None of these studies investigated the three major adduct types in the same samples, and a variety of different doses, treatment protocols, and methods of analysis were used, making comparisons difficult. These studies did suggest however that adduct 6 was formed in the greatest quantities. Our results clearly confirm this impression, as levels of adduct 6 in hepatic DNA of our NPYR-treated rats were hundreds to thousands of times higher than those of adducts 9 or 13–15. The levels of adduct 6 observed here are quite consistent with those reported in studies which used HPLC-fluorescence for detection, as indicated in Table 3.
Table 3.
Studies of NPYR-DNA adduct formation in laboratory animals
| NPYR-DNA adduct | Protocols used | Amounts Reported | Reference |
|---|---|---|---|
| 6 | rat: 175 or 350 mg/kg NPYR gavage, sac 16h later | Estimated levels: 500 μmol/mol dGuo (175 mg/kg dose) 650 μmol/mol dGuo (350 mg/kg dose) |
(18) |
| rat: 3.1 mg/kg [3,4-3H] NPYR gavage, sac 16h later | 1.2 μmol/mol dGuo | (18) | |
| rat: 14–900 mg/kg NPYR gavage, sac 12h later | 0.1–3 “7-methylGua equivalents”/mol Gua (no units given) | (31) | |
| rat, mouse, hamster: 900 mg/kg NPYR gavage, sac 12–24h later | liver: 3983 (rat), 9000 (hamster), 9987 (mouse) μmol/mol Gua; kidney and lung – lower | (32) | |
| rat: 56–900 mg/kg NPYR gavage, sac 1–96h later | adduct levels maximum 12–24h after gavage and dose-responsive in liver, lung, kidney | (32) | |
| hamster: 225 mg/kg NPYR gavage, sac 12–120h later | adduct levels maximum 24h after treatment in lung and liver; 12h in kidney; declined thereafter. Highest levels in liver, followed by lung/kidney | (32) | |
| 5, 6 | rat: 450 mg/kg NPYR i.p., sac 16h later | 643 μmol/mol Gua adduct 5 vs.1792 μmol/mol Gua adduct 6 | (21) |
| rat: 450 mg/kg NPYR gavage, sac 24h later | 603 μmol/mol Gua adduct 5 vs.1430 μmol/mol Gua adduct 6 | (24) | |
| 9 | rat: 6 mM in drinking water, 14 days, total dose 1 mmol/rat | 0.06 μmol/mol Gua | (19) |
| 13 (as 18) | rat: 6 mM NPYR in drinking water, 7 days | 3.4 μmol/mol dGuo | (28) |
| rat: 2 mM NPYR in drinking water, 28 days | 4.8 μmol/mol dGuo | (28) | |
| rat: 2 mM NPYR in drinking water, 91 days | 5.4 μmol/mol dGuo | (28) | |
| 14 (as 19) | rat: 6 mM NPYR in drinking water, 7 days | 0.02 μmol/mol dGuo | (28) |
| rat: 2 mM NPYR in drinking water, 28 days | 0.03 μmol/mol dGuo | (28) | |
| rat: 2 mM NPYR in drinking water, 91 days | 0.04 μmol/mol dGuo | (28) | |
| 15 (as 20) | rat: 6 mM NPYR in drinking water, 7 days | 2.6 μmol/mol dGuo | (28) |
| rat: 2 mM NPYR in drinking water, 28 days | 3.9 μmol/mol dGuo | (28) | |
| rat: 2 mM NPYR in drinking water, 91 days | 3.5 μmol/mol dGuo | (28) | |
This is the first study to quantify the crotonaldehyde derived adducts 9 by LC-ESI-MS/MS-SRM analysis of DNA samples from NPYR-treated rats. Only one previous study estimated these adduct levels, using 32P-postlabelling (Table 3). Although levels of adduct 9 were considerably lower than those of adduct 6, it was readily detectable (Figure 4) and clearly responsive to dose. Levels of adduct 9 were generally comparable to those of adducts 13 and 15 (quantified as their reduced forms 18 and 20) (Table 2). We also observed that levels of the 6S,8S- isomer of adduct 9 were about twice as great as those of the 6R,8R- isomer (Table 2). This is consistent with data from previous studies of the reaction of crotonaldehyde with DNA in vitro (36), supporting the hypothesis that these adducts result directly from reaction of crotonaldehyde, a metabolite of NPYR, with DNA in vivo. The LC-ESI-MS/MS-SRM method for adduct 9 has also been applied to human DNA samples. Adduct 9 diastereomers were observed in about 17% of the liver and 35% of the lung DNA samples analyzed, at levels approximately 10,000 times lower than seen here (34). There are potential sources of crotonaldehyde adducts in human DNA other than NPYR exposure, including lipid peroxidation and direct exposure to crotonaldehyde.
Levels of adducts 13–15 (quantified as 18–20) were consistent with our previous study, with the amounts of the dGuo adduct 13 and dThd adduct 15 being considerably higher than those of the dAdo adduct 14 (28). Levels of adducts 13 and 15 were also clearly responsive to dose. Although these adduct levels were far lower than those of adduct 6, there is evidence that dAdo or dThd adducts such as 14 and 15 may be biologically important. Thus, Kanki et al found that A:T to G:C transition mutations in hepatic DNA were common in NPYR-treated rats, and that this was the only mutation more frequently observed than in controls (2).
The clear prevalence of the 7,8-butanoguanine adduct 6 as the major NPYR adduct in this study is consistent with the known DNA alkylation chemistry of alkyl diazonium ions, carbocations and SN2 alkylating agents, in which products of alkylation at the 7-position of Gua often predominate (37). This has been observed in many studies of simple alkylating agents, including the 1-propanediazonium ion, which is the closest analogue to intermediates 4 and 7 of Scheme 1, but lacking the aldehyde group, that has been studied in detail (38). Clearly, the aldehyde group is important in the chemistry of NPYR-DNA adduct formation, as it leads to the structurally unique adduct 6, as well as to the secondary intermediates such as crotonaldehyde and oxonium ion 12 which produce adducts 9 and 13–15. While the aldehyde group is responsible for these unique properties, the main driving force in terms of yield seems to be the alkyl carbocation properties of intermediates 4 and 7.
In summary, the results of this study clearly show that, among adducts formed in hepatic DNA of NPYR-treated rats, the 7,8-butanoguanine adduct 6 is far more prevalent than the crotonaldehyde-derived adducts 9 or the oxonium ion-derived adducts 13–15. A previous study has shown that adduct 6 is excreted in the urine of rats treated with NPYR (24). With this in mind, the LC-ESI-MS/MS-SRM method developed here should be useful for analysis of human samples for adduct 6, which would be a specific biomarker for endogenous formation or exogenous exposure to the carcinogen NPYR.
Acknowledgments
This study was supported by grant CA-85702 from the National Cancer Institute, and by FAPESP and CNPq (Brazil). Ana Paula M. Loureiro held a fellowship from Pró-Reitoria de Pós-Graduação da Universidade de São Paulo, Brazil for development of this work at the University of Minnesota. We thank Bob Carlson for outstanding editorial assistance.
References
- 1.Gold LS, Slone TH, Ames BN. Summary of the carcinogenic potency database by chemical. In: Gold LS, Zeiger E, editors. Handbook of Carcinogenic Potency and Genotoxicity Databases. CRC Press; Boca Raton, FL: 1997. pp. 621–660. [Google Scholar]
- 2.Kanki K, Nishikawa A, Masumura K, Umemura T, Imazawa T, Kitamura Y, Nohmi T, Hirose M. In vivo mutational analysis of liver DNA in gpt delta transgenic rats treated with the hepatocarcinogens N-nitrosopyrrolidine, 2-amino-3-methylimidazo[4,5-f]quinoline, and di(2-ethylhexyl)phthalate. Mol Carcinog. 2005;42:9–17. doi: 10.1002/mc.20061. [DOI] [PubMed] [Google Scholar]
- 3.Druckrey H, Preussmann R, Ivankovic S, Schmähl D. Organotrope Carcinogen Wirkungen bei 65 verschiedenen N-Nitrosoverbindungen an BD-ratten. Z Krebsforsch Klin Onkol. 1967;69:103–201. [PubMed] [Google Scholar]
- 4.Lijinsky W. Chemistry and Biology of N-Nitroso Compounds. Cambridge University Press; Cambridge, England: 1992. [Google Scholar]
- 5.Gray R, Peto R, Branton P, Grasso P. Chronic nitrosamine ingestion in 1040 rodents: the effect of the choice of nitrosamine, the species studied, and the age of starting exposure. Cancer Res. 1991;51:6470–6491. [PubMed] [Google Scholar]
- 6.Preussmann R, Stewart BW. N-Nitroso Carcinogens. In: Searle CE, editor. Chemical Carcinogens, Second Edition, ACS Monograph 182, vol. 2. American Chemical Society; Washington, DC: 1984. pp. 643–828. [Google Scholar]
- 7.van Maanen JMS, Welle IJ, Hageman G, Dallinga JW, Mertens PLJM, Kleinjans JCS. Nitrate contamination of drinking water: relationship with HPRT variant frequency in lymphocyte DNA and urinary excretion of N-nitrosamines. Environ Health Perspect. 1998;104:522–528. doi: 10.1289/ehp.96104522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mostafa MH, Helmi S, Badawi AF, Tricker AR, Spiegelhalder B, Preussmann R. Nitrate, nitrite and volatile N-nitroso compounds in the urine of Schistosoma haematobium and Schistosoma mansoni infected patients. Carcinogenesis. 1994;15:619–625. doi: 10.1093/carcin/15.4.619. [DOI] [PubMed] [Google Scholar]
- 9.Tricker AR, Stickler DJ, Chawla JC, Preussmann R. Increased urinary nitrosamine excretion in paraplegic patients. Carcinogenesis. 1991;12:943–946. doi: 10.1093/carcin/12.5.943. [DOI] [PubMed] [Google Scholar]
- 10.Hecht SS, McCoy GD, Chen CB, Hoffmann D. The metabolism of cyclic nitrosamines. In: Scanlan RA, Tannenbaum SR, editors. N-Nitroso Compounds. American Chemical Society; Washington, DC: 1981. pp. 49–75. [Google Scholar]
- 11.Tricker AR, Pfundstein B, Kalble T, Preussmann R. Secondary amine precursors to nitrosamines in human saliva, gastric juice, blood, urine, and faeces. Carcinogenesis. 1992;13:563–568. doi: 10.1093/carcin/13.4.563. [DOI] [PubMed] [Google Scholar]
- 12.Bartsch H, Ohshima H, Pignatelli B, Calmels S. Human exposure to endogenous N-nitroso compounds: quantitative estimates in subjects at high risk for cancer of the oral cavity, oesophagus, stomach and urinary bladder. Cancer Surv. 1989;8:335–362. [PubMed] [Google Scholar]
- 13.Tricker AR, Pfundstein B, Theobald E, Preussmann R, Spiegelhalder B. Mean daily intake of volatile N-nitrosamines from foods and beverages in West Germany in 1989–1990. Food Chem Toxicol. 1991;29:729–732. doi: 10.1016/0278-6915(91)90180-f. [DOI] [PubMed] [Google Scholar]
- 14.Zhao YY, Boyd J, Hrudey SE, Li XF. Characterization of new nitrosamines in drinking water using liquid chromatography tandem mass spectrometry. Environ Sci Technol. 2006;40:7636–7641. doi: 10.1021/es061332s. [DOI] [PubMed] [Google Scholar]
- 15.Roemer E, Stabbert R, Rustemeier K, Veltel DJ, Meisgen TJ, Reininghaus W, Carchman RA, Gaworski CL, Podraza KF. Chemical composition, cytotoxicity and mutagenicity of smoke from US commercial and reference cigarettes smoked under two sets of machine smoking conditions. Toxicology. 2004;195:31–52. doi: 10.1016/j.tox.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 16.Hecht SS. N-Nitrosamines. In: Rom WB, Markowitz SB, editors. Environmental and Occupational Medicine. 4. Wolters Kluwer/Lippincott Williams & Wilkins; Philadelphia: 2007. pp. 1226–1239. [Google Scholar]
- 17.Chung FL, Hecht SS. Formation of cyclic 1,N2-adducts by reaction of deoxyguanosine with α-acetoxy-N-nitrosopyrrolidine, 4-(carbethoxynitrosamino)butanal, or crotonaldehyde. Cancer Res. 1983;43:1230–1235. [PubMed] [Google Scholar]
- 18.Chung FL, Wang M, Hecht SS. Detection of exocyclic guanine adducts in hydrolysates of hepatic DNA of rats treated with N-nitrosopyrrolidine and in calf thymus DNA reacted with α-acetoxy-N-nitrosopyrrolidine. Cancer Res. 1989;49:2034–2041. [PubMed] [Google Scholar]
- 19.Chung FL, Young R, Hecht SS. Detection of cyclic 1, N2-propanodeoxyguanosine adducts in DNA of rats treated with N-nitrosopyrrolidine and mice treated with crotonaldehyde. Carcinogenesis. 1989;10:1291–1297. doi: 10.1093/carcin/10.7.1291. [DOI] [PubMed] [Google Scholar]
- 20.Wang M, Chung FL, Hecht SS. Formation of acyclic and cyclic guanine adducts in DNA reacted with α-acetoxy-N-nitrosopyrrolidine. Chem Res Toxicol. 1989;2:423–428. doi: 10.1021/tx00012a011. [DOI] [PubMed] [Google Scholar]
- 21.Wang M, Chung FL, Hecht SS. Formation of 7-(4-oxobutyl)guanine in hepatic DNA of rats treated with N-nitrosopyrrolidine. Carcinogenesis. 1992;13:1909–1911. doi: 10.1093/carcin/13.10.1909. [DOI] [PubMed] [Google Scholar]
- 22.Young-Sciame R, Wang M, Chung FL, Hecht SS. Reactions of α-acetoxy-N-nitrosopyrrolidine and α-acetoxy-N-nitrosopiperidine with deoxyguanosine: formation of N2-tetrahydrofuranyl or N2-tetrahydropyranyl adducts. Chem Res Toxicol. 1995;8:607–616. doi: 10.1021/tx00046a016. [DOI] [PubMed] [Google Scholar]
- 23.Wang M, Young-Sciame R, Chung FL, Hecht SS. Formation of N2-tetrahydrofuranyl and N2-tetrahydropyranyl adducts in the reactions of α-acetoxy-N-nitrosopyrrolidine and α-acetoxy-N-nitrosopiperidine with DNA. Chem Res Toxicol. 1995;8:617–624. doi: 10.1021/tx00046a017. [DOI] [PubMed] [Google Scholar]
- 24.Wang M, Hecht SS. A cyclic N7, C-8 guanine adduct of N-nitrosopyrrolidine (NPYR): formation in nucleic acids and excretion in the urine of NPYR-treated rats. Chem Res Toxicol. 1997;10:772–778. doi: 10.1021/tx960193x. [DOI] [PubMed] [Google Scholar]
- 25.Wang M, Upadhyaya P, Dinh TT, Bonilla LE, Hecht SS. Lactols in hydrolysates of DNA reacted with α-acetoxy-N-nitrosopyrrolidine and crotonaldehyde. Chem Res Toxicol. 1998;11:1567–1573. doi: 10.1021/tx980165+. [DOI] [PubMed] [Google Scholar]
- 26.Wang M, McIntee EJ, Shi Y, Cheng G, Upadhyaya P, Villalta PW, Hecht SS. Reactions of α-acetoxy-N-nitrosopyrrolidine with deoxyguanosine and DNA. Chem Res Toxicol. 2001;14:1435–1445. doi: 10.1021/tx010097i. [DOI] [PubMed] [Google Scholar]
- 27.Wang M, Lao Y, Cheng G, Shi Y, Villalta PW, Nishikawa A, Hecht SS. Identification of adducts formed in the reaction of α-acetoxy-N-nitrosopyrrolidine with deoxyribonucleosides and DNA. Chem Res Toxicol. 2007;20:625–633. doi: 10.1021/tx600332p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang M, Lao Y, Cheng G, Shi Y, Villalta PW, Nishikawa A, Hecht SS. Analysis of adducts in hepatic DNA of rats treated with N-nitrosopyrrolidine. Chem Res Toxicol. 2007;20:634–640. doi: 10.1021/tx600333e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang M, McIntee EJ, Cheng G, Shi Y, Villalta PW, Hecht SS. A Schiff base is a major DNA adduct of crotonaldehyde. Chem Res Toxicol. 2001;14:423–430. doi: 10.1021/tx000234w. [DOI] [PubMed] [Google Scholar]
- 30.Hermida SA, Possari EP, Souza DB, de ACI, Gomes OF, Di Mascio P, Medeiros MH, Loureiro AP. 2′-Deoxyguanosine, 2′-deoxycytidine, and 2′-deoxyadenosine adducts resulting from the reaction of tetrahydrofuran with DNA bases. Chem Res Toxicol. 2006;19:927–936. doi: 10.1021/tx060033d. [DOI] [PubMed] [Google Scholar]
- 31.Hunt EJ, Shank RC. Evidence for DNA adducts in rat liver after administration of N-nitrosopyrrolidine. Biochem Biophys Res Commun. 1982;104:1343–1348. doi: 10.1016/0006-291x(82)91397-3. [DOI] [PubMed] [Google Scholar]
- 32.Hunt EJ, Shank RC. Formation and persistence of a DNA adduct in rodents treated with N-nitrosopyrrolidine. Carcinogenesis. 1991;12:571–575. doi: 10.1093/carcin/12.4.571. [DOI] [PubMed] [Google Scholar]
- 33.Nagy A, Armatis P, Schally AV. High yield conversion of doxorubicin to 2-pyrrolinodoxorubicin, an analog 500–1000 times more potent: structure-activity relationship of daunosamine-modified derivatives of doxorubicin. Proc Natl Acad Sci U S A. 1996;93:2464–2469. doi: 10.1073/pnas.93.6.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang S, Villalta PW, Wang M, Hecht SS. Analysis of crotonaldehyde- and acetaldehyde-derived 1, N2-propanodeoxyguanosine adducts in DNA from human tissues using liquid chromatography-electrsopray ionization-tandem mass spectrometry. Chem Res Toxicol. 2006;19:1386–1392. doi: 10.1021/tx060154d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuo KC, McCune RA, Gehrke CW. Quantitive reversed-phase high performance liquid chromatographic determination of major and modified deoxyribnucleosides in DNA. Nucleic Acid Research. 1980;8:4763–4777. doi: 10.1093/nar/8.20.4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang M, McIntee EJ, Cheng G, Shi Y, Villalta PW, Hecht SS. Identification of paraldol-deoxyguanosine adducts in DNA reacted with crotonaldehyde. Chem Res Toxicol. 2000;13:1065–1074. doi: 10.1021/tx000095i. [DOI] [PubMed] [Google Scholar]
- 37.Singer B, Grunberger D. Molecular Biology of Mutagens and Carcinogens. Plenum Press; New York: 1983. pp. 45–96. [Google Scholar]
- 38.Lu X, Heilman JM, Blans P, Fishbein JC. The structure of DNA dictates purine atom site selectivity in alkylation by primary diazonium ions. Chem Res Toxicol. 2005;18:1462–1470. doi: 10.1021/tx0501334. [DOI] [PubMed] [Google Scholar]