

Abstract
Selenoprotein P (Sepp1) is a secreted protein that is made up of 2 domains. The larger N-terminal domain contains 1 selenocysteine residue in a redox motif and the smaller C-terminal domain contains the other 9 selenocysteines. Sepp1 isoforms of varying length occur but quantitation of them has not been achieved. Hepatic synthesis of Sepp1 affects whole-body selenium content and the liver is the source of most plasma Sepp1. ApoER2, a member of the lipoprotein receptor family, binds Sepp1 and facilitates its uptake into testis and retention of its selenium by the brain. Megalin, another lipoprotein receptor, facilitates uptake of filtered Sepp1 into proximal tubule cells of the kidney. Thus, Sepp1 serves in homeostasis and distribution of selenium. Mice with deletion of Sepp1 suffer greater morbidity and mortality from infection with Trypanosoma congolense than do wild-type mice. Mice that express only the N-terminal domain of Sepp1 have the same severity of illness as wild-type mice, indicating that the protective function of Sepp1 against the infection resides in the N-terminal (redox) domain. Thus, Sepp1 has several functions. In addition, plasma Sepp1 concentration falls in selenium deficiency and, therefore, it can be used as an index of selenium nutritional status.
1.0 Introduction
Selenoprotein P (Sepp1) is a secreted glycoprotein that contains most of the selenium in plasma [1, 2]. With respect to its selenium content, Sepp1 can be divided into two domains. The N-terminal domain, approximately two-thirds of the amino acid sequence, contains 1 selenocysteine (U) in a U-x-x-C redox motif (Fig. 1A). The shorter C-terminal domain contains multiple selenocysteines—9 in rats, mice, and humans (Fig. 1B).
Figure 1.

Domains of rat Sepp1. Panel A depicts the N-terminal domain (residues 1–244) that contains one selenocysteine residue (U) in a thioredoxin-like motif (U-x-x-C). A typical heparin-binding site (HBS) and 2 histidine-rich stretches (H-rich) are also present in this domain as are 3 occupied N-glycosylation sites. Panel B depicts the C-terminal selenium-rich domain (residues 245–366) containing 9 selenocysteine residues. It has a single occupied O-glycosylation site. Sepp1 isoforms terminate before the selenocysteine residues at positions 245, 263, and 352 as indicated by ▼
Sepp1 binds to heparin [3, 4] in a pH-sensitive manner [5]. This property has been attributed to a heparin-binding site in the N-terminal domain that contains histidine as 2 of its 5 basic residues [6]. Histidine becomes protonated, and therefore able to bind heparin, only when pH drops below the normal physiologic range. This confers on Sepp1 the property of binding to heparin under acidic conditions but remaining unbound at physiologic pH.
Two histidine-rich stretches are present in the distal N-terminal domain of Sepp1 (Fig. 1A). These stretches contain up to 10 sequential basic amino acids and likely contribute to the heparin-binding properties of Sepp1 [6, 7]. Sepp1 also contains carbohydrate, with 3 occupied N-glycosylation sites in the N-terminal domain and 1 occupied O-glycosylation site in the C-terminal domain (Fig. 1). Several disulfide and selenenylsulfide bonds have been identified in rat Sepp1 as purified [8]. These bonds might have structural functions; they also might serve to protect the selenium atoms by reducing their reactivity.
Sepp1 purified from rat plasma is present as 4 isoforms. In addition to the full-length isoform that contains 10 selenocysteine residues, shorter isoforms are present that terminate at the second, third, and seventh selenocysteine positions (Fig. 1). These isoforms contain 1, 2, and 6 selenocysteine residues, respectively [9, 10]. There is evidence for the existence of Sepp1 isoforms in the mouse [11] and the human [12] but separation and characterization of them in these species has not been reported.
The liver produces most of the Sepp1 in plasma, where its turnover is rapid. Sepp1 is also expressed in other tissues and is presumably secreted by them [13, 14].
Several reviews on Sepp1 are available [15, 16]. We reviewed research on Sepp1 published through 2004 [17]. Since then additional work, including experiments using mice with genetic alterations of Sepp1, has provided new insights into the metabolism and function of this selenoprotein. The present review covers selected topics from this recent period.
2.0 Genetic Characteristics of Sepp1
2.1 Origin and evolution
Analysis of genomes with respect to Sepp1 has been revealing [18]. The proximal portion of the Sepp1 N-terminal domain, which contains the redox motif U-x-x-C, contains conserved sequences with similarity to bacterial thioredoxin-fold proteins. This observation indicates that Sepp1 is a member of the thioredoxin superfamily and thereby strongly suggests that its N-terminal domain has a thiol-redox function. This strengthens earlier suggestions of such a function [17, 19–21].
The selenium-rich C terminus, and therefore Sepp1 as we know it, probably first appeared in early metazoan species [18]. The suggestion has been made that a C-terminal selenocysteine residue was acquired by mutation of the protein’s original termination codon to UGA, which codes for selenocysteine in selenoproteins. With read-through of the new UGA as selenocysteine, the protein would have been extended to the next in-frame termination codon. Repetition of this process would add selenocysteine residues to the C terminus.
Marine animals evolved Sepp1 molecules with as many as 27 selenocysteine residues in the C-terminal domain, presumably to supply selenium for their large number of selenoproteins. Terrestrial animals have fewer selenoproteins and their Sepp1 molecules correspondingly contain fewer selenocysteine residues as a consequence in most cases of replacement of selenocysteines by cysteine residues. This bio-informatic analysis [18] is consistent with the two domains of Sepp1 having distinct functions. This concept is now strongly supported by experimental results as will be discussed below.
2.2 Expression
Insertion of selenocysteine into selenoproteins is less efficient than is incorporation of other amino acids. Ten selenocysteines must be inserted into full-length Sepp1, so there has been great interest in determining how this is accomplished. Evidence has been presented that the 2 selenocysteine insertion sequence (SECIS) elements in the 3′ untranslated region (utr) of Sepp1 mRNA have distinct functions [22]. SECIS 2 is less efficient than SECIS 1 [23]. SECIS 2 was shown to be responsible for insertion of the single selenocysteine in the N-terminal domain (Fig. 2). This process occurs slowly, so ribosomes pass this UGA relatively infrequently [22]. Once the ribosome reaches the second UGA it interacts with SECIS 1, which rapidly facilitates insertion of the remaining selenocysteines. The authors postulate that the first UGA serves as a checkpoint to ensure that all the ingredients for selenoprotein synthesis are present before allowing synthesis to proceed to the selenium-rich domain. Also because it utilizes the inefficient SECIS 2, the first UGA serves as a hindrance to ribosomal progress and prevents ribosomes from arriving at the second UGA while SECIS 1 is engaged with the preceding ribosome. This model is the first detailed mechanism postulated for translation of Sepp1. Extension of this work will need to consider how isoforms of Sepp1 are produced and whether synthesis of Sepp1 produces the same products in all tissues.
Figure 2.

Messenger RNA of rat Sepp1. The open reading frame (ORF) has 10 UGAs that code for selenocysteine residues. The 3′utr has 2 stem loops designated SECIS 1 and 2. SECIS 2 is postulated to decode the lone UGA in the N-terminal region while SECIS 1 decodes the other 9 UGAs [22].
Several studies of the Sepp1 promoter indicated that it interacts with cytokine and growth factor pathways, suggesting that inflammation alters its activation [24–26]. More recently a report detailed a number of hepatic factors that make the liver the primary organ expressing Sepp1 [27]. That study implicated insulin and possibly glucocorticoids as regulators of Sepp1 expression. These studies were carried out in cell lines; extension of them to the intact organism will be important to determine the physiologic significance of their results.
Sepp1 mRNA is present in most tissues, implying that the protein is expressed and secreted by cells throughout the organism [28]. Therefore in order to gain insight into Sepp1 function, additional tissue factors that affect its expression need to be determined.
Two relatively common single nucleotide polymorphisms (SNPs) of SEPP1 have been reported. One is located in the N-terminal domain and results in conversion of the alanine at position 234 to threonine (A234T) [29, 30]. This SNP introduces a potential O-glycosylation site between the histidine-rich sequences. The other SNP is at nucleotide position 25191, which is located in the 3′utr between the termination codon and SECIS 1 (see Fig. 2). Unlike the SECIS, which has a stem-loop structure, the sequence containing this SNP has not been described to have a secondary structure. However, this sequence is conserved between species [13], and can therefore be speculated to have a function. Thus, both these SNPs might be important in the expression and/or function of SEPP1.
Factors besides the Sepp1 gene affect its translation. The production of a selenoprotein requires several gene products [31, 32] in addition to the usual protein translation machinery. A mutation in one of those proteins, SECIS binding protein-2 (SBP2), a protein that binds to the SECIS element and facilitates translation of selenoproteins, has been shown to affect SEPP1 concentration in plasma [33, 34]. Finally, selenium deficiency decreases synthesis of Sepp1 and causes its plasma concentration to decline [35].
3.0 Sepp1 Roles in Selenium Homeostasis and Transport
3.1 Selenium homeostasis
The liver acquires selenium from several sources and apportions it between selenoprotein synthesis and excretion from the organism (Fig. 3). Specifically, liver synthesizes its intrinsic selenoproteins as well as the secreted selenium molecules Sepp1 and excretory metabolites [36].
Figure 3.
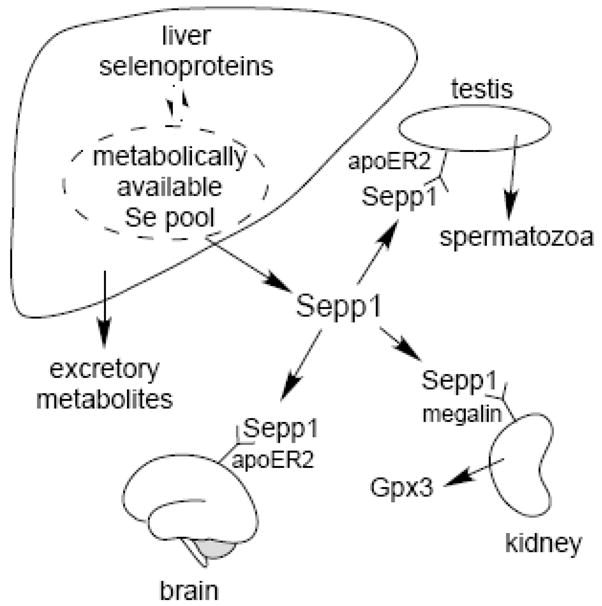
Sepp1 in selenium homeostasis and transport to testis, brain, and kidney. Whole-body selenium is controlled by selenium excretion in the urine. Sepp1 and selenium excretory metabolites compete for metabolically available selenium in the liver, determining urinary selenium excretion. The lipoprotein receptor apoER2 binds Sepp1 and facilitates its uptake into the testis where selenium is incorporated into spermatozoa. ApoER2 also maintains brain selenium. Sepp1 is filtered by the kidney into the glomerular filtrate and binds to megalin on the brush border of PCT cells. Those cells endocytose the Sepp1 bound to megalin and presumably use its selenium to synthesize Gpx3.
Deletion of Sepp1 causes increased excretion of selenium in the urine and, as a result, decreased whole-body selenium [37] (Fig. 4A). It is likely that competition for the hepatic metabolically available selenium pool between synthesis of Sepp1 and synthesis of excretory metabolites underlies this phenomenon (Fig. 3). Synthesis of intrinsic liver selenoproteins would not be expected to affect this selenium pool because their catabolism would return selenium to it. The loss of competition between selenium-containing molecules secreted by the liver, however, would explain the increased production of excretory metabolites and, hence, the increased urinary loss of selenium in Sepp1−/− mice.
Figure 4.

Comparison of the effects of Sepp1 deletion, selenium deficiency, and apoER2 deletion on tissue and whole-body selenium concentrations. The Sepp1−/− and Sepp1+/+ mice (panel A) and the apoER2−/− and apoER2+/+ mice (panel C) were fed Torula yeast-based diet supplemented with 0.25 mg selenium/kg [38]. They were studied 4 weeks after weaning. The selenium-deficient and control mice (panel B) were fed the Torula yeast-based diet in selenium-deficient form or supplemented with 0.25 mg selenium/kg (control), respectively, for 18 weeks beginning at weaning. The mice were exsanguinated and tissues were removed and weighed. Selenium was determined in tissues, blood, and carcass [38]. Selenium values were added to obtain the whole-body selenium. Values are means ± S.D., n=5. Values in pairs that were different from each other by the Student t-test have the percentage of difference indicated. Panel C is a modification of results presented in [41], presented here with permission.
Whole-body selenium, thus, appears to be regulated in the liver by the distribution of metabolically available selenium between the pathways of selenoprotein synthesis and selenium excretory metabolite synthesis. This process might be a passive one, with the selenium that is incorporated into Sepp1 being destined for retention by the organism and the selenium that cannot be accommodated by the selenoprotein synthesis pathway entering the excretory pathway. Active regulation of excretory metabolite synthesis is a possibility, however, and must be considered in future work.
3.2 Selenium transport
Because deletion of Sepp1 causes a moderate selenium deficiency state, clues to function of Sepp1 can be sought by comparing its deletion with dietary selenium deficiency. Dietary selenium deficiency caused a profound decrease in liver selenium concentration (Fig. 4B), presumably because the liver exports a fraction of its metabolically available selenium as Sepp1, even when selenium supply to the liver is drastically decreased [35]. It is not surprising, then, that deletion of Sepp1 had little effect on liver selenium in selenium-replete mice (Fig. 4A) and even rendered it resistant to depletion by dietary selenium deficiency [38, 39].
Brain and testis stand out as responding differently to Sepp1 deletion than to dietary selenium deficiency [38, 39]. Brain selenium concentration fell with Sepp1 deletion to 45% of the wild-type value, a decrease that was greater than the fall to 64% of wild-type by the whole-body selenium concentration (Fig. 4A). Brain selenium fell only to 56% of control in severe selenium deficiency in spite of whole-body selenium concentration declining to 13% of control (Fig. 4B). This demonstrates that the presence of Sepp1 helps preserve brain selenium when dietary selenium is limiting. Testis selenium concentration was also protected by the presence of Sepp1.
Both kidney and muscle selenium concentrations fell approximately to the same extent as did whole-body selenium with deletion of Sepp1 and with dietary selenium deficiency. Thus, Sepp1 appears to be responsible for maintaining selenium in brain and testis by a mechanism that is distinct from its effect on whole-body selenium. That mechanism has recently been identified as receptor-mediated uptake of Sepp1.
3.3 Receptor-mediated uptake
Sepp1-containing vesicles were identified in Sertoli cells by immunocytochemistry [40], consistent with receptor-mediated endocytosis of the protein in the testis. Analysis of a testis membrane preparation for Sepp1-binding proteins identified a lipoprotein receptor family member, apolipoprotein E receptor-2 (apoER2) [40]. Sertoli cells express apoER2 and those cells did not contain Sepp1-positive vesicles in apoER2−/− mice [40]. Moreover, testis selenium content was very low in apoER2−/− mice (Fig. 4C). Thus, most of the selenium in the testis is derived from apoER2-mediated uptake of Sepp1 by Sertoli cells (Fig. 3).
Further study of apoER2−/− mice demonstrated that brain selenium concentration in them was depressed to 44% of that in wild-type mice but that kidney and whole-body selenium concentrations were not affected (Fig. 4C) [41]. This indicates that brain selenium is apoER2 dependent but that kidney selenium is not. The finding that apoER2−/− mice have the same whole-body selenium content as do wild-type mice is consistent with the hypothesis discussed above that regulation of whole-body selenium is determined in the liver, a tissue that does not express apoER2.
Brain selenium content is much lower than testis selenium content [42, 43] (Fig. 4). Moreover, testis exports selenium as sperm selenoproteins, while the brain is not known to export the element. These considerations are consistent with the brain taking up much less selenium than the testis. Immunocytochemistry of the brain did not reveal Sepp1-containing vesicles like the ones found in the testis, compatible with a postulated low uptake of Sepp1. Thus, a role for apoER2 in taking up Sepp1 at the blood-brain barrier is inferred but has not been demonstrated directly.
Sepp1 is expressed inside the brain and has been proposed to serve as a storage form of selenium there, explaining the low brain selenium in Sepp1−/− mice [44]. However, insertion into Sepp1−/− mice of a transgene directing hepatic synthesis of human SEPP1 led to selenium delivery to the mouse brain [45]. This confirms that Sepp1 transports selenium to the brain and increases the likelihood that apoER2 facilitates Sepp1 uptake at the blood-brain barrier. Storage of selenium in the form of Sepp1 within the brain remains a possibility, however.
Immunocytochemical study of the kidney revealed Sepp1-containing vesicles in proximal convoluted tubule (PCT) cells, suggesting that these cells take up Sepp1 from the glomerular filtrate [46]. PCT cells have a special need for selenium because they synthesize and secrete glutathione peroxidase-3 (Gpx3), an extracellular selenoprotein [47]. Thus, PCT cells appear to prevent Sepp1 from being lost in the urine and, coincidentally, can utilize Sepp1 selenium for Gpx3 synthesis.
ApoER2 is not responsible for Sepp1 uptake by PCT cells. Deletion of apoER2 had no effect on the Sepp1-containing vesicles in PCT cells and no effect on kidney selenium level (Fig. 4C) [46]. Megalin, another member of the lipoprotein receptor family that is also referred to as Lrp2, is present on the brush border of PCT cells and facilitates endocytosis of a number of glomerular filtrate proteins and other ligands [48]. 18-Day megalin−/− fetuses did not have Sepp1-containing vesicles in PCT cells, while wild-type fetuses did have such vesicles [46]. Fetuses were studied because megalin−/− mice typically die at birth. Thus, megalin is responsible for PCT cell uptake of Sepp1 from the glomerular filtrate.
3.4 Sepp1 isoforms in receptor-mediated uptake
ApoER2 and megalin have different ligand-binding properties so it is reasonable to ask whether they distinguish between isoforms of Sepp1. Mice with deletion of the C-terminal domain of Sepp1 (see Fig. 1), Sepp1Δ240–361 mice, were produced and studied for the presence of Sepp1-containing vesicles in testis and kidney. Such vesicles were not detected in testis [Olson, G.E. V.P. Winfrey, K.E. Hill, and R.F. Burk, unpublished] but they were present in kidney PCT cells [46]. Because only one isoform of Sepp1 is present in Sepp1Δ240–361 mice [11], these results indicate that Sepp1Δ240–361 is not taken up by apoER2-mediated endocytosis but is taken up by the megalin-mediated process.
Sepp1Δ240–361 and other short isoforms are small enough to be filtered by the kidney glomerulus. The full-length form of Sepp1 has a combined peptide and carbohydrate weight of approximately 50 kDa but elutes from a gel filtration column at a position corresponding to 79 kDa [49]. Thus, shorter isoforms would be expected to enter the glomerular filtrate to a greater extent than the full-length isoform.
These results raise the possibility that apoER2 is selective for full-length Sepp1. Such selectivity would maximize selenium uptake by tissues that express apoER2. Megalin, on the other hand, prevents loss of Sepp1 in the urine, so it would need to bind the shorter isoforms.
4.0 Sepp1 and Spermatogenesis
Male animals with severe selenium deficiency produce defective spermatozoa and have reduced fertility [50, 51]. Abnormalities of the mitochondrial capsule, which contains the selenoprotein Gpx4 as a structural component [52], underlie at least some of the sperm defects [53]. Sepp1−/− mice and apoER2−/− mice have reduced fertility and produce spermatozoa that have the same appearance as selenium-deficient ones [54]. Thus, Sepp1 supplies selenium for spermatogenesis through apoER2-mediated endocytosis and the absence of either protein blocks selenium supply and leads to the production of defective spermatozoa.
In addition to maintaining selenium, apoER2 has non-endocytotic signaling functions in the brain that are activated by its ligand Reelin [55]. Studies with 4 strains of apoER2 knock-in mice, each of which has a different alteration in the cytoplasmic tail of the receptor, have characterized the signaling pathway activated by Reelin [56]. A recent report indicated that none of the 4 knock-in strains had decreases in brain or testis selenium that were comparable to those in apoER2−/− mice [57]. However, morphologic abnormalities were present in spermatozoa from one of the knock-in strains and sperm motility was depressed in two others. Thus, some abnormalities in sperm function appear to be related to a signaling function of apoER2 in the testis. The affected signal might involve Sepp1 and/or selenium trafficking, or it might be unrelated to selenium. It is clear, however, that more details remain to be learned about Sepp1 and apoER2 effects on spermatogenesis.
5.0 Sepp1 and Brain Injury
That supply of selenium to the brain is very important derives from the fact that the organism maintains brain selenium at the expense of selenium in other tissues (Fig. 4B) [43]. As discussed above, apoER2 and Sepp1-facilitated maintenance of brain selenium appears to be the major mechanism by which the brain competes with other tissues for selenium. However, Sepp1 is also expressed inside the brain [58], primarily by glial cells [14, 59], and has therefore been postulated to store and/or transport brain selenium [44].
Discovery of the Sepp1-apoER2 pathway provided a strategy for the production of selenium deficiency in the brain. That strategy employed the deletion or alteration of Sepp1 or apoER2 and then the feeding of a selenium-deficient diet. Early studies using non-congenic Sepp1−/− mice demonstrated that feeding a diet containing 0.25 mg selenium (as selenite)/kg produced no neurological dysfunction [60]. When fed a 0.10 mg selenium/kg diet (which is considered to satisfy the selenium requirement of wild-type mice), Sepp1−/− mice developed spasticity and abnormal movements in addition to poor performance on the rotarod test and pole climb. Feeding selenium-deficient diet to Sepp1−/− mice caused severe neurological impairment that led to death within weeks. Thus, the absence of Sepp1 raised the selenium intake needed to prevent brain injury.
When selenium was re-fed to Sepp1−/− mice after neurological abnormalities had developed, the neurological status stabilized, but it did not improve [60]. Thus, lack of selenium causes irreversible brain injury. Similar neurological abnormalities were observed in apoER2−/− mice and Sepp1Δ240–361 mice fed selenium-deficient diet [11, 41]. These experiments indicate that decrease of selenium uptake into the brain, not lack of Sepp1 itself, was the principal cause of the brain injury.
Brain injury was assessed in the 3 strains of genetically altered mice by silver staining serial brain sections from them [61]. Feeding selenium-deficient diet caused severe axonal injury that was mitigated (but not completely eliminated) by feeding high-selenium diet. No injury at all was detected in the brains of wild-type mice fed selenium-deficient diet. The pattern of injury was similar in Sepp1−/−, apoER2−/−, and Sepp1Δ240–361 mice, affecting mainly areas involved in motor and auditory functions. Severity of the injury was greatest in Sepp1−/− mice, consistent with this deletion causing more severe selenium deficiency in the brain through directly decreasing brain selenium and indirectly decreasing whole-body selenium content. Thus, this study demonstrated that interruption of selenium supply to the brain by impairing the Sepp1-apoER2 pathway causes neurodegeneration.
Many questions remain about Sepp1 in the brain. Do receptors other than apoER2 bind Sepp1? Does Sepp1 have functions in the brain other than transporting selenium? Does the putative thiol-redox function of Sepp1 serve as an extracellular oxidant defense in the brain? Might Sepp1 be a signaling molecule in the brain [62]? It is obvious from what is already known that subtle defects in the Sepp1-receptor pathway might play a role in neurodegenerative diseases, as is already being suggested [63, 64].
6.0 Sepp1 in Parasitic Infection
Selenium nutritional status affects the severity of infections. Aside from the role of Gpx1 in coxsackie virus infection [65], little is known about how specific selenoproteins affect infection outcomes. A recent report, however, indicated that Sepp1 protects mice against trypanosomiasis [66].
Trypanosomiasis is a parasitic disease that is often fatal. After inoculation of mice with Trypanosoma congolense, an initial parasitemia occurs that is controlled by gamma-interferon activation of immune cells. An interleukin-10 dependent phase ensues that ends with death of the mice approximately 150 days after inoculation. Because Sepp1 expression was increased in myeloid cells by interleukin-10, deletion of Sepp1 was studied in the experimental infection [66]. In Sepp1−/− mice the initial parasitemia was worsened and caused some mortality. The Sepp1−/− mice that survived the initial parisitemia died sooner than did infected wild-type mice. Thus, Sepp1 protects mice against the infection.
The investigators studied Sepp1Δ240–361 mice to assess which domain of Sepp1 protected the mice. Results in Sepp1Δ240–361 mice were indistinguishable from those in wild-type mice [66]. This result implicates the N-terminal domain of Sepp1 in the protection against illness caused by trypanosomal infection. It suggests that an enzymatic function and not selenium transport was the protective mechanism. In addition, this result clearly establishes that the N-terminal domain of Sepp1 has a function in vivo.
7.0 Human SEPP1
7.1 Marker of selenium nutritional status
Because selenium functions through selenoproteins, it has been proposed that optimum health would be achieved if enough of the element were supplied to prevent selenium from becoming the limiting factor in selenoprotein synthesis. Determination of selenoprotein optimization has become the major technique used to assess the selenium nutritional requirement. Tissue selenoproteins cannot be measured without performing a biopsy, so plasma selenoproteins have been used as surrogates for all the selenoproteins.
Gpx3 and Sepp1 are the plasma selenoproteins. They serve as selenium nutritional markers and their plasma concentrations fall as the severity of selenium deficiency increases [35, 43]. Plasma glutathione peroxidase activity and Sepp1 concentration decline to less than 5% of selenium-replete values in animals with severe selenium deficiency. Thus, either of these selenoproteins can be used to assess the severity of selenium deficiency in animals.
Our group studied the effect of selenium supplementation on plasma SEPP1 and GPX3 in a selenium-deficient human population in China that had a dietary selenium intake of 10 μg/d [67]. Plasma glutathione peroxidase activity reached its maximum value, implying that GPX3 had been optimized, when a supplement of 37 μg selenium per day in the form of selenomethionine was given. SEPP1 concentration was continuing to rise at that level of supplementation and had not reached a plateau at the highest supplement dose, 61 μg/d. Thus of the two plasma selenoproteins available for measurement, SEPP1 requires a greater selenium intake to reach its maximum concentration. This makes it a better marker than GPX3 for the maximization of all the selenoproteins in the human body.
Another supplementation study was performed in selenium-replete individuals in the U.S. [68]. Supplementing them with selenium did not cause an increase in plasma glutathione peroxidase activity or SEPP1 concentration. That study indicated that these plasma selenium biomarkers do not mirror selenium intake once the selenium requirement has been met.
The results of these studies and one carried out in Britain [30] indicate that SEPP1 plasma concentration is the best easily accessible marker of human selenium nutritional status. Once the nutritional requirement has been met, however, SEPP1 concentration does not reflect additional increases in selenium intake.
7.2 Decreased Sepp1 concentration in sepsis
Plasma selenium concentration falls acutely by as much as 40% in patients with sepsis or sepsis-like illness [69]. SEPP1 concentration also falls in sepsis and is presumably the cause of the decline in selenium concentration [70].
Neither the reason for SEPP1 decrease nor its mechanism is known with certainty. The decrease in plasma SEPP1 might be beneficial to the patient, perhaps by decreasing the availability of selenium to invading microorganisms or by decreasing reactive selenium in the circulation. The more common opinion, however, is that the decrease in SEPP1 is an indication that selenium is lacking and that supplementation with it is warranted. Selenium intervention studies have been carried out in these severely ill patients but have not shown that selenium supplementation is globally beneficial to patients with sepsis [71, 72]. Some of the data suggest, however, that septic patients with initial low selenium status might benefit from selenium administration. Because of the potential deleterious effects of administering selenium to seriously ill patients, clear evidence supporting its use is needed before recommendations for its administration in medical practice can be made.
Rat Sepp1 is a pH-dependent heparin-binding protein; it is possible that binding to heparan sulfate proteoglycans in acidotic tissues causes the sepsis-associated decline of plasma SEPP1. Such binding would be specific for host cells and might deliver selenium to cells in sites of inflammation and/or provide enzymatic protection of host cells against reactive metabolites.
Alternatively, the decline in plasma SEPP1 might be caused by decreased release of it by the liver. A recent study in mice suggests that decreased liver release is the mechanism of the decline [73]. Measurements of mRNA after endotoxin injection failed to show a decrease in mRNA for Sepp1 but did show decreases in mRNA for several proteins essential for selenoprotein synthesis. Synthesis of Sepp1 likely constitutes a large fraction of hepatic selenoprotein synthesis, so impairment of selenoprotein synthesis should affect Sepp1 release by the liver. This work with mRNA suggests a mechanism for the decrease in plasma Sepp1, but direct studies on the fate of plasma Sepp1 are needed to determine whether its utilization is increased by sepsis.
The results in trypanosomiasis indicate that Sepp1 plays a role in protection against that infectious illness [66]. Convincing evidence that it protects against other infections has not yet emerged.
8.0 Perspectives
The existence of Sepp1 was first reported over 30 years ago, but progress in understanding its functions has come mostly in the present decade. Much of that progress can be traced to the study of mice with Sepp1 deleted or modified.
Sepp1 is now known to consist of two domains. The N-terminal domain appears to be an enzyme with redox properties. The C-terminal domain contains selenium that is in transit. These statements, while helpful, do not capture the complexity of this protein, which has a number of characteristics that are as yet unexplained.
Full-length Sepp1is present in plasma but so are shortened forms that have reduced selenium content. The origin of these Sepp1 isoforms is not known. They might result from termination of Sepp1 translation at UGAs in the open reading frame of Sepp1 mRNA, or they might result from removal of C-terminal fragments of plasma Sepp1 in the extracellular space, perhaps as a prelude to uptake of the selenium-rich fragments by cells. No methods to quantitate isoforms have been reported and this is a great research need.
Sepp1 mRNA is present in most tissues, indicating that the protein is produced widely in the body. Only three tissues are known to export selenoproteins in large quantities—liver, testis, and kidney—and they all have higher selenium contents than other tissues (Fig. 4). Of these 3 tissues, liver exports Sepp1 and the other 2 take up Sepp1. Liver has several mechanisms for acquiring the selenium that it exports as Sepp1. Thus, it can be speculated that liver exports full-length Sepp1 to supply other tissues with selenium, and other tissues secrete mostly shorter isoforms of Sepp1, possibly for local use as a redox enzyme or as a signaling molecule.
The understanding of Sepp1 physiology will guide research on its health effects. Especially exciting is the possibility that Sepp1 protects host cells and extracellular matrix against oxidative damage at sites of inflammation. In addition to its role in transporting selenium, other functions of Sepp1 will surely be discovered.
Acknowledgments
The authors are grateful to Gary E. Olson for critical reading of the manuscript and to Amy K. Motley for assistance with the manuscript and figures. The authors’ research is supported by NIH grants ES02497 and DK58763.
The authors’ work is supported by NIH grants ES02497 and DK58763.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hill KE, Xia Y, Åkesson B, Boeglin ME, Burk RF. Selenoprotein P concentration in plasma is an index of selenium status in selenium-deficient and selenium-supplemented Chinese subjects. J Nutr. 1996;126:138–145. doi: 10.1093/jn/126.1.138. [DOI] [PubMed] [Google Scholar]
- 2.Read R, Bellew T, Yang JG, et al. Selenium and amino acid composition of selenoprotein P, the major selenoprotein in rat serum. J Biol Chem. 1990;265:17899–17905. [PubMed] [Google Scholar]
- 3.Åkesson B, Mårtensson B. Heparin interacts with a selenoprotein in human plasma. J Inorg Biochem. 1988;33:257–261. doi: 10.1016/0162-0134(88)80004-7. [DOI] [PubMed] [Google Scholar]
- 4.Herrman JL. The properties of a rat serum protein labelled by the injection of sodium selenite. Biochim Biophys Acta. 1977;500:61–70. doi: 10.1016/0304-4165(77)90046-0. [DOI] [PubMed] [Google Scholar]
- 5.Chittum HS, Himeno S, Hill KE, Burk RF. Multiple forms of selenoprotein P in rat plasma. Arch Biochem Biophys. 1996;325:124–128. doi: 10.1006/abbi.1996.0015. [DOI] [PubMed] [Google Scholar]
- 6.Hondal RJ, Ma S, Caprioli RM, Hill KE, Burk RF. Heparin-binding histidine and lysine residues of rat selenoprotein P. J Biol Chem. 2001;276:15823–15831. doi: 10.1074/jbc.M010405200. [DOI] [PubMed] [Google Scholar]
- 7.Arteel GE, Franken S, Kappler J, Sies H. Binding of selenoprotein P to heparin: characterization with surface plasmon resonance. Biol Chem. 2000;381:265–268. doi: 10.1515/BC.2000.034. [DOI] [PubMed] [Google Scholar]
- 8.Ma S, Hill KE, Burk RF, Caprioli RM. Mass spectrometric identification of N- and O-glycosylation sites of full-length rat selenoprotein P and determination of selenide-sulfide and disulfide linkages in the shortest isoform. Biochemistry. 2003;42:9703–9711. doi: 10.1021/bi0346300. [DOI] [PubMed] [Google Scholar]
- 9.Himeno S, Chittum HS, Burk RF. Isoforms of selenoprotein P in rat plasma. Evidence for a full-length form and another form that terminates at the second UGA in the open reading frame. J Biol Chem. 1996;271:15769–15775. doi: 10.1074/jbc.271.26.15769. [DOI] [PubMed] [Google Scholar]
- 10.Ma S, Hill KE, Caprioli RM, Burk RF. Mass spectrometric characterization of full-length rat selenoprotein P and three isoforms shortened at the C terminus. Evidence that three UGA codons in the mRNA open reading frame have alternative functions of specifying selenocysteine insertion or translation termination. J Biol Chem. 2002;277:12749–12754. doi: 10.1074/jbc.M111462200. [DOI] [PubMed] [Google Scholar]
- 11.Hill KE, Zhou J, Austin LM, et al. The selenium-rich C-terminal domain of mouse selenoprotein P is necessary for supply of selenium to brain and testis but not for maintenance of whole-body selenium. J Biol Chem. 2007;282:10972–10980. doi: 10.1074/jbc.M700436200. [DOI] [PubMed] [Google Scholar]
- 12.Åkesson B, Bellew T, Burk RF. Purification of selenoprotein P from human plasma. Biochim Biophys Acta. 1994;1204:243–249. doi: 10.1016/0167-4838(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 13.Hill KE, Lloyd RS, Burk RF. Conserved nucleotide sequences in the open reading frame and 3′-untranslated region of selenoprotein P. Proc Natl Acad Sci USA. 1993;90:537–541. doi: 10.1073/pnas.90.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang X, Hill KE, Maguire MJ, Burk RF. Synthesis and secretion of selenoprotein P by cultured rat astrocytes. Biochim Biophys Acta. 2000;1474:390–396. doi: 10.1016/s0304-4165(00)00035-0. [DOI] [PubMed] [Google Scholar]
- 15.Mostert V. Selenoprotein P: properties, functions, and regulation. Arch Biochem Biophys. 2000;376:433–438. doi: 10.1006/abbi.2000.1735. [DOI] [PubMed] [Google Scholar]
- 16.Persson-Moschos M. Selenoprotein P. Cell Mol Life Sci. 2000;57:1836–1845. doi: 10.1007/PL00000665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burk RF, Hill KE. Selenoprotein P: An extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annu Rev Nutr. 2005;25:215–235. doi: 10.1146/annurev.nutr.24.012003.132120. [DOI] [PubMed] [Google Scholar]
- 18.Lobanov AV, Hatfield DL, Gladyshev VN. Reduced reliance on the trace element selenium during evolution of mammals. Genome Biol. 2008;9:R62. doi: 10.1186/gb-2008-9-3-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kryukov GV, Gladyshev VN. Selenium metabolism in zebrafish: multiplicity of selenoprotein genes and expression of a protein containing seventeen selenocyteine residues. Genes Cells. 2000:1049–1060. doi: 10.1046/j.1365-2443.2000.00392.x. [DOI] [PubMed] [Google Scholar]
- 20.Saito Y, Hayashi T, Tanaka A, et al. Selenoprotein P in human plasma as an extracellular phospholipid hydroperoxide glutathione peroxidase. J Biol Chem. 1999;274:2866–2871. doi: 10.1074/jbc.274.5.2866. [DOI] [PubMed] [Google Scholar]
- 21.Takebe G, Yarimizu J, Saito Y, et al. A comparative study on the hydroperoxide and thiol specificity of the glutathione peroxidase family and selenoprotein P. J Biol Chem. 2002;277:41254–41258. doi: 10.1074/jbc.M202773200. [DOI] [PubMed] [Google Scholar]
- 22.Stoytcheva Z, Tujebajeva RM, Harney JW, Berry MJ. Efficient incorporation of multiple selenocysteines involves an inefficient decoding step serving as a potential translational checkpoint and ribosome bottleneck. Mol Cell Biol. 2006;26:9177–9184. doi: 10.1128/MCB.00856-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berry MJ, Banu L, Harney JW, Larsen PR. Functional characterization of the eukaryotic SECIS elements which direct selenocysteine insertion at UGA codons. EMBO J. 1993;12:3315–3322. doi: 10.1002/j.1460-2075.1993.tb06001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Al-Taie OH, Seufert J, Mork H, et al. A complex DNA-repeat structure within the Selenoprotein P promoter contains a functionally relevant polymorphism and is genetically unstable under conditions of mismatch repair deficiency. Eur J Hum Genet. 2002;10:499–504. doi: 10.1038/sj.ejhg.5200811. [DOI] [PubMed] [Google Scholar]
- 25.Dreher I, Jakobs TC, Köhrle J. Cloning and characterization of the human selenoprotein P promoter. J Biol Chem. 1997;272:29364–29371. doi: 10.1074/jbc.272.46.29364. [DOI] [PubMed] [Google Scholar]
- 26.Mostert V, Wolff S, Dreher I, Kohrle J, Abel J. Identification of an element within the promoter of human selenoprotein P responsive to transforming growth factor-beta. Eur J Biochem. 2001;268:6176–6181. doi: 10.1046/j.0014-2956.2001.02565.x. [DOI] [PubMed] [Google Scholar]
- 27.Speckmann B, Walter PL, Alili L, et al. Selenoprotein P expression is controlled through interaction of the coactivator PGC-1α with FoxO1a and hepatocyte nuclear factor 4α transcription factors. Hepatology. 2008;48:1998–2006. doi: 10.1002/hep.22526. [DOI] [PubMed] [Google Scholar]
- 28.Burk RF, Hill KE. Selenoprotein P. A selenium-rich extracellular glycoprotein. J Nutr. 1994;124:1891–1897. doi: 10.1093/jn/124.10.1891. [DOI] [PubMed] [Google Scholar]
- 29.Al-Taie OH, Uceyler N, Eubner U, et al. Expression profiling and genetic alterations of the selenoproteins GI-GPx and SePP in colorectal carcinogenesis. Nutr Cancer. 2004;48:6–14. doi: 10.1207/s15327914nc4801_2. [DOI] [PubMed] [Google Scholar]
- 30.Meplan C, Crosley LK, Nicol F, et al. Genetic polymorphisms in the human selenoprotein P gene determine the response of selenoprotein markers to selenium supplementation in a gender-specific manner (the SELGEN study) FASEB J. 2007;21:3063–3074. doi: 10.1096/fj.07-8166com. [DOI] [PubMed] [Google Scholar]
- 31.Driscoll DM, Copeland PR. Mechanism and regulation of selenoprotein synthesis. Annu Rev Nutr. 2003;23:17–40. doi: 10.1146/annurev.nutr.23.011702.073318. [DOI] [PubMed] [Google Scholar]
- 32.Hatfield DL, Carlson BA, Xu XM, Mix H, Gladyshev VN. Selenocysteine incorporation machinery and the role of selenoproteins in development and health. Prog Nucleic Acid Res Mol Biol. 2006;81:97–142. doi: 10.1016/S0079-6603(06)81003-2. [DOI] [PubMed] [Google Scholar]
- 33.Dumitrescu AM, Liao XH, Abdullah MS, et al. Mutations in SECISBP2 result in abnormal thyroid hormone metabolism. Nat Genet. 2005;37:1247–1252. doi: 10.1038/ng1654. [DOI] [PubMed] [Google Scholar]
- 34.Schomburg L, Dumitrescu AM, Liao XH, et al. Selenium supplementation fails to correct the selenoprotein synthesis defect in subjects with SBP2 gene mutations. Thyroid. 2009;19 doi: 10.1089/thy.2008.0397. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang JG, Hill KE, Burk RF. Dietary selenium intake controls rat plasma selenoprotein P concentration. J Nutr. 1989;119:1010–1012. doi: 10.1093/jn/119.7.1010. [DOI] [PubMed] [Google Scholar]
- 36.Kobayashi Y, Ogra Y, Ishiwata K, et al. Selenosugars are key and urinary metabolites for selenium excretion within the required to low-toxic range. Proc Natl Acad Sci U S A. 2002;99:15932–15936. doi: 10.1073/pnas.252610699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burk RF, Hill KE, Motley AK, Austin LM, Norsworthy BK. Deletion of selenoprotein P upregulates urinary selenium excretion and depresses whole-body selenium content. Biochim Biophys Acta. 2006;1760:1789–1793. doi: 10.1016/j.bbagen.2006.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hill KE, Zhou J, McMahan WJ, et al. Deletion of selenoprotein P alters distribution of selenium in the mouse. J Biol Chem. 2003;278:13640–13646. doi: 10.1074/jbc.M300755200. [DOI] [PubMed] [Google Scholar]
- 39.Schomburg L, Schweizer U, Holtmann B, et al. Gene disruption discloses role of selenoprotein P in selenium delivery to target tissues. Biochem J. 2003;370:397–402. doi: 10.1042/BJ20021853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olson GE, Winfrey VP, Nagdas SK, Hill KE, Burk RF. Apolipoprotein E receptor-2 (ApoER2) mediates selenium uptake from selenoprotein P by the mouse testis. J Biol Chem. 2007;282:12290–12297. doi: 10.1074/jbc.M611403200. [DOI] [PubMed] [Google Scholar]
- 41.Burk RF, Hill KE, Olson GE, et al. Deletion of apolipoprotein E receptor-2 in mice lowers brain selenium and causes severe neurological dysfunction and death when a low-selenium diet is fed. J Neurosci. 2007;27:6207–6211. doi: 10.1523/JNEUROSCI.1153-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Behne D, Hofer-Bosse T. Effects of a low selenium status on the distribution and retention of selenium in the rat. J Nutr. 1984;114:1289–1296. doi: 10.1093/jn/114.7.1289. [DOI] [PubMed] [Google Scholar]
- 43.Nakayama A, Hill KE, Austin LM, Motley AK, Burk RF. All regions of mouse brain are dependent on selenoprotein P for maintenance of selenium. J Nutr. 2007;137:690–693. doi: 10.1093/jn/137.3.690. [DOI] [PubMed] [Google Scholar]
- 44.Schweizer U, Streckfuss F, Pelt P, et al. Hepatically derived selenoprotein P is a key factor for kidney but not for brain selenium supply. Biochem J. 2005;386:221–226. doi: 10.1042/BJ20041973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Renko K, Werner M, Renner-Muller I, et al. Hepatic selenoprotein P (SePP) expression restores selenium transport and prevents infertility and motor-incoordination in Sepp-knockout mice. Biochem J. 2008;409:741–749. doi: 10.1042/BJ20071172. [DOI] [PubMed] [Google Scholar]
- 46.Olson GE, Winfrey VP, Hill KE, Burk RF. Megalin mediates selenoprotein P uptake by kidney proximal tubule epithelial cells. J Biol Chem. 2008;283:6854–6860. doi: 10.1074/jbc.M709945200. [DOI] [PubMed] [Google Scholar]
- 47.Avissar N, Ornt DB, Yagil Y, et al. Human kidney proximal tubules are the main source of plasma glutathione peroxidase. Am J Physiol. 1994;266:C367–C375. doi: 10.1152/ajpcell.1994.266.2.C367. [DOI] [PubMed] [Google Scholar]
- 48.Christensen EI, Willnow TE. Essential role of megalin in renal proximal tubule for vitamin homeostasis. J Am Soc Nephrol. 1999;10:2224–2236. doi: 10.1681/ASN.V10102224. [DOI] [PubMed] [Google Scholar]
- 49.Burk RF, Gregory PE. Some characteristics of 75Se-P, a selenoprotein found in rat liver and plasma, and comparison of it with seleno-glutathione peroxidase. Arch Biochem Biophys. 1982;213:73–80. doi: 10.1016/0003-9861(82)90441-6. [DOI] [PubMed] [Google Scholar]
- 50.Wallace E, Calvin H, Cooper G. Progressive defects observed in mouse sperm during the course of three generations of selenium deficiency. Gamete Res. 1983;4:377–387. [Google Scholar]
- 51.Wu AS, Oldfield JE, Shull LR, Cheeke PR. Specific effect of selenium deficiency on rat sperm. Biol Reprod. 1979;20:793–798. doi: 10.1095/biolreprod20.4.793. [DOI] [PubMed] [Google Scholar]
- 52.Ursini F, Heim S, Kiess M, et al. Dual function of the selenoprotein PHGPx during sperm maturation. Science. 1999;285:1393–1396. doi: 10.1126/science.285.5432.1393. [DOI] [PubMed] [Google Scholar]
- 53.Olson GE, Winfrey VP, Nagdas SK, Hill KE, Burk RF. Selenoprotein P is required for mouse sperm development. Biol Reprod. 2005;73:201–211. doi: 10.1095/biolreprod.105.040360. [DOI] [PubMed] [Google Scholar]
- 54.Andersen OM, Yeung CH, Vorum H, et al. Essential role of the apolipoprotein E receptor-2 in sperm development. J Biol Chem. 2003;278:23989–23995. doi: 10.1074/jbc.M302157200. [DOI] [PubMed] [Google Scholar]
- 55.May P, Herz J, Bock HH. Molecular mechanisms of lipoprotein receptor signalling. Cell Mol Life Sci. 2005;62:2325–2338. doi: 10.1007/s00018-005-5231-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beffert U, Weeber EJ, Durudas A, et al. Modulation of synaptic plasticity and memory by Reelin involves differential splicing of the lipoprotein receptor Apoer2. Neuron. 2005;47:567–579. doi: 10.1016/j.neuron.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 57.Masiulis I, Quill TA, Burk RF, Herz J. Differential functions of the Apoer2 intracellular domain in selenium uptake and cell signaling. Biol Chem. 2009;390:67–73. doi: 10.1515/BC.2009.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saijoh K, Saito N, Lee MJ, et al. Molecular cloning of cDNA encoding a bovine selenoprotein P-like protein containing 12 selenocysteines and a (His-Pro) rich domain insertion, and its regional expression. Mol Brain Res. 1995;30:301–311. doi: 10.1016/0169-328x(94)00007-2. [DOI] [PubMed] [Google Scholar]
- 59.Zhang Y, Zhou Y, Schweizer U, et al. Comparative analysis of selenocysteine machinery and selenoproteome gene expression in mouse brain identifies neurons as key functional sites of selenium in mammals. J Biol Chem. 2008;283:2427–2438. doi: 10.1074/jbc.M707951200. [DOI] [PubMed] [Google Scholar]
- 60.Hill KE, Zhou J, McMahan WJ, Motley AK, Burk RF. Neurological dysfunction occurs in mice with targeted deletion of selenoprotein P gene. J Nutr. 2004;134:157–161. doi: 10.1093/jn/134.1.157. [DOI] [PubMed] [Google Scholar]
- 61.Valentine WM, Abel TW, Hill KE, Austin LM, Burk RF. Neurodegeneration in mice resulting from loss of functional selenoprotein P or its receptor apolipoprotein E receptor 2. J Neuropathol Exp Neurol. 2008;67:68–77. doi: 10.1097/NEN.0b013e318160f347. [DOI] [PubMed] [Google Scholar]
- 62.Peters MM, Hill KE, Burk RF, Weeber EJ. Altered hippocampus synaptic function in selenoprotein P deficient mice. Mol Neurodegener. 2006;1:12. doi: 10.1186/1750-1326-1-12. (website: http://www.molecularneurodegeneration.com/content/1/1/12) [DOI] [PMC free article] [PubMed]
- 63.Bellinger FP, He QP, Bellinger MT, et al. Association of selenoprotein p with Alzheimer’s pathology in human cortex. J Alzheimers Dis. 2008;15:465–472. doi: 10.3233/jad-2008-15313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Scharpf M, Schweizer U, Arzberger T, et al. Neuronal and ependymal expression of selenoprotein P in the human brain. J Neural Transm. 2007;114:877–884. doi: 10.1007/s00702-006-0617-0. [DOI] [PubMed] [Google Scholar]
- 65.Beck MA. Selenium and vitamin E status: impact on viral pathogenicity. J Nutr. 2007;137:1338–1340. doi: 10.1093/jn/137.5.1338. [DOI] [PubMed] [Google Scholar]
- 66.Bosschaerts T, Guilliams M, Noel W, et al. Alternatively activated myeloid cells limit pathogenicity associated with African trypanosomiasis through the IL-10 inducible gene selenoprotein P. J Immunol. 2008;180:6168–6175. doi: 10.4049/jimmunol.180.9.6168. [DOI] [PubMed] [Google Scholar]
- 67.Xia Y, Hill KE, Byrne DW, Xu J, Burk RF. Effectiveness of selenium supplements in a low-selenium area of China. Am J Clin Nutr. 2005;81:829–834. doi: 10.1093/ajcn/81.4.829. [DOI] [PubMed] [Google Scholar]
- 68.Burk RF, Norsworthy BK, Hill KE, Motley AK, Byrne DW. Effects of chemical form of selenium on plasma biomarkers in a high-dose human supplementation trial. Cancer Epidemiol Biomarkers Prev. 2006;15:804–810. doi: 10.1158/1055-9965.EPI-05-0950. [DOI] [PubMed] [Google Scholar]
- 69.Forceville X, Vitoux D, Gauzit R, et al. Selenium, systemic immune response syndrome, sepsis, and outcome in critically ill patients. Crit Care Med. 1998;26:1536–1544. doi: 10.1097/00003246-199809000-00021. [DOI] [PubMed] [Google Scholar]
- 70.Hollenbach B, Morgenthaler NG, Struck J, et al. New assay for the measurement of selenoprotein P as a sepsis biomarker from serum. J Trace Elem Med Biol. 2008;22:24–32. doi: 10.1016/j.jtemb.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 71.Angstwurm MW, Engelmann L, Zimmermann T, et al. Selenium in Intensive Care (SIC): results of a prospective randomized, placebo-controlled, multiple-center study in patients with severe systemic inflammatory response syndrome, sepsis, and septic shock. Crit Care Med. 2007;35:118–126. doi: 10.1097/01.CCM.0000251124.83436.0E. [DOI] [PubMed] [Google Scholar]
- 72.Schomburg L. Selenium in intensive care (SIC) study: the XX files are still unresolved. Crit Care Med. 2007;35:995–996. doi: 10.1097/01.ccm.0000257479.88792.a5. [DOI] [PubMed] [Google Scholar]
- 73.Renko K, Hofmann PJ, Stoedter M, et al. Down-regulation of the hepatic selenoprotein biosynthesis machinery impairs selenium metabolism during the acute phase response in mice. FASEB J. 2009;23 doi: 10.1096/fj.08-119370. in press. [DOI] [PubMed] [Google Scholar]