

Abstract
A fluorinated detergent, CF3(CF2)5C2H4-O-maltose, was reconstituted into a lipid bilayer model membrane system to demonstrate the feasibility of determining solvent accessibility and membrane immersion depth of each fluorinated group by 19F NMR. Apolar oxygen, which is known to partition with an increasing concentration gradient toward the hydrophobic membrane interior, exhibits a range of paramagnetic relaxation effects on 19F nuclei, depending on its depth in the membrane. This effect, which is predominately associated with spin-lattice relaxation rates (R1) and chemical shifts, can be amplified greatly with minimal line broadening by increasing the partial pressure of O2 at least 100-fold (i.e., PO2 greater than 20 bar). The differences of longitudinal relaxation rates at 20 bar of oxygen pressure to those under ambient pressure (R120bar − R10) are largest for those fluorine groups expected to be most deeply buried in the membrane bilayer. This result contrasts with the reverse trend, which is observed on addition of a membrane surface-associated paramagnetic species, 4-(N,N-dimethyl-N-hexadecyl) ammonium-2,2,6,6-tetramethylpiperidine-1-oxyl iodide (CAT-16) at ambient pressures. Thus, differential relaxation rates may be observed in 19F-labeled membrane-associated molecules resulting from the addition of apolar oxygen under high pressure. The results demonstrate that the degree of solvent accessibility and membrane immersion depth of specific fluorinated species in membrane-associated macromolecules can be probed by 19F NMR.
Keywords: paramagnetic O2, spin-lattice relaxation times, bicelles, model membranes
The degree to which site-specific labels penetrate a biological membrane represents critical information in understanding the interaction of small organic molecules, drugs, and proteins with membranes. One way of ascertaining relative immersion depth in a membrane by NMR would be to measure the paramagnetic contribution to the spin-lattice relaxation rates of specific nuclei arising from oxygen, whose diffusional accessibility is known to increase with immersion depth in the bilayer (1). This paper explores the potential of such an approach by using the 19F nucleus as a probe and a semiperfluorinated detergent that inserts into the lipid bilayer. The prospects of applying this technique to larger membrane-associated molecules and proteins are discussed in light of these results.
Membrane-associated proteins, now known to constitute nearly one-third of all proteins (2–3), represent an important class of macromolecules for which there is no clear technique for the determination of three-dimensional structure with atomic resolution. Although significant progress has been made by both high-resolution NMR by using detergent micelles (4–5) and electron and x-ray diffraction from two- and three-dimensional crystals (6–7), each method relies on stringent sample conditions. High-resolution NMR samples must consist of small micellar aggregates without intermediate timescale motions (8), whereas electron and x-ray diffraction techniques require diffractable membrane protein crystals. Solid-state NMR is less restrictive in the sense that it requires only that the membrane protein be uniaxially aligned in the magnetic field to achieve acceptable resolution.
The combination of site-directed spin labeling and ESR spectroscopy provides a means of studying membrane proteins without the need for alignment or crystallization. In this case, a spin-labeled derivative such as (1-oxyl-2,2,5,5,-tetramethylpyrroline-3-methyl) methanethiosulfonate may be reacted readily with the sulfhydryl group of a single cysteine residue in the protein or its mutant. By comparing ESR spectra and saturation decay rates, in the presence of both a water-soluble shift reagent [typically Ni(II)ethylenediaminediacetate or Ni(II)acetylacetonate] and apolar (membrane-soluble) oxygen, the immersion depth and solvent accessibility of the residue-specific nitroxide may be determined reliably (9–13). This approach relies on the property that oxygen preferentially partitions into membranes with increasingly higher concentrations at greater depths in the membrane (1). By studying versions of the protein in which each residue is separately replaced by a cysteine spin-label conjugate, it is possible to map out secondary structure and topography of (reconstituted and fully active) membrane proteins by ESR. In the spirit of this approach, we have used a fluorinated detergent incorporated into a model membrane to investigate the sensitivity of 19F NMR spectra and spin-lattice relaxation times to a water-soluble paramagnetic shift reagent anchored to the membrane and to membrane-soluble paramagnetic oxygen under high oxygen partial pressures.
Fluorine NMR offers distinct advantages in the study of membrane protein structure (14–17). In particular, sensitivity is nearly that obtained by 1H, whereas the dispersion of chemical shifts is nearly 100-fold larger than that of 1H. 19F chemical shifts are also known to be very sensitive to local van der Waals and electrostatic environments. In particular, the addition of paramagnetic additives has a pronounced effect on 19F chemical shifts and relaxation times (17–19). In some cases, chemical shift changes can be observed from solvent-accessible 19F-labeled sites simply by exchanging D2O for H2O (16, 20). Fluorine labeling is relatively straightforward, and a variety of 19F-labeled residues are commercially available (17). Certain fluorinated residues may also be incorporated biosynthetically and nearly fully expressed (21–23). Alternatively, in a manner similar to that described above for site-directed ESR spin labeling, fluorinated substituents such as trifluoroethylthio (TET) may be attached readily to the sulfhydryl groups of cysteine residues under nondenaturing conditions (24). Fluorine labels such as TET are unquestionably smaller and therefore less perturbing than comparable ESR spin labels. One would expect this to result in more uniform activities for site-directed mutants of membrane proteins (25). The second potential advantage of site-directed labeling by 19F NMR is that the chemical shift range of 19F is sufficiently large to avoid significant spectral overlap from biosynthetically labeled proteins with more than one label per molecule. Here we report our first measurements, which demonstrate the feasibility of 19F NMR to determine relative immersion depth of specifically fluorinated species in membrane-associated molecules by using apolar paramagnetic oxygen under pressure.
Materials and Methods
3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecylfluoro β-d-octyl maltoside (TFOM) was a gift from C. Sanders (Case Western Reserve University, Cleveland, OH). Bicellar dispersions [15% (wt/vol)] consisting of phospholipid, 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and detergent, 1,2-dihexanoyl-sn-glycero-3-phosphocholine (DHPC) were prepared in 99.9% D2O (Isotec). DMPC/DHPC molar ratios (referred to hereafter as q) of 0.5 or 1.0 were used. Typically, TFOM (2–5 mg) was combined with DMPC/DHPC (approximately 250 mg) giving a DMPC/TFOM molar ratio of between 20 and 50. The samples were vortexed followed by a few cycles of freeze thawing and centrifuging, after which a clear solution was obtained. Small unilamellar vesicles (SUVs) consisting of a 15% (wt/wt) dispersion of DMPC and 1,2-dimyristoyl-sn-glycero-3-phosphoglycerol (DMPG) in a 15:1 molar ratio, and TFOM (2 mg) per 600 μl solution, were obtained by sonicating the lipid/TFOM dispersion over an ice bath for several minutes, followed by centrifugation to remove heavy metal particles resulting from sonication. All lipids (DMPC, DHPC, and DMPG) were obtained from Avanti Polar Lipids. To study TFOM in the absence of a membrane, approximately 0.6 mg of TFOM was added to 600 μl deuterated methanol (Isotec). For the studies of differential paramagnetic relaxation from a surface-bound ESR spin label, 4-(N,N-dimethyl-N-hexadecyl) ammonium-2,2,6,6-tetramethylpiperidine-1-oxyl iodide (CAT-16) (Molecular Probes) (approximately 4 mg) was added to the NMR sample, which consisted of DMPC (60 mg) and DHPC (40 mg). For studies on paramagnetic relaxation from oxygen, the sample was first equilibrated to 20 bar PO2 for 1 week. NMR experiments were performed by using a 5 mm OD (1.6 mm i.d.) high-pressure NMR tube, purchased from Wilmad (Buena, NJ). The threaded Teflon fitting (Wilmad) in the NMR tube was permanently epoxied to a standard 1/8-inch Swagelok copper fitting, which was connected via 1/8-inch copper tubing to a three-way valve (for purging) and standard oxygen cylinder. An identical setup was constructed for studying TFOM in bicelles under pressurized nitrogen. To further prevent leakage between the Teflon connection and Swagelok fitting, a small O-ring was seated over the Swagelok ferrel, and the Swagelok nut was in turn soldered to the copper tubing. On completion of the NMR measurements, the pressurized sample was degassed by repeated freeze thawing and studied under ambient oxygen partial pressures.
19F NMR experiments were performed on a Varian INOVA 500 MHz high-resolution spectrometer by tuning the proton channel of a 5-mm probe to the 19F resonance frequency (470.327 MHz). All experiments were performed at 40°C, where DMPC is expected to adopt bilayer domains within the isotropic bicelle sample (26). Spin-lattice relaxation times were measured by a simple inversion-recovery sequence {π−τ−π/2−acquire} by using a repetition time of 3.5 s. A total of 64 τ values were used to characterize the inversion-recovery decay curves. Each of the decay curves proved to fit very well to a single exponential, and the fitted uncertainties in T1 were typically less than 2%. However, after repeating these measurements with several samples, we estimate the uncertainty of the relaxation rates to be 5%. 19F π/2 pulse lengths were 5.2 μs. A spectral width of 32 kHz and acquisition time of 250 ms were used, with a delay of 100 μs after the last pulse preceding acquisition. Spectra were usually processed with 2–20 Hz line broadening and referenced to an internal standard of trifluoroacetic acid (−76.55 ppm). T1 relaxation times and line widths were measured by using Varian software. Usually, no more than 64 scans were necessary to obtain the signal-to-noise ratios demonstrated below. To assign the 19F NMR spectrum, nuclear Overhauser effect (NOE) difference (27) and double quantum-filtered (DQF)–correlated spectroscopy (COSY) (28–29) experiments were performed. The NOE difference experiment involved selectively saturating each of the six TFOM resonances by using mixing times between 100 ms and 500 ms. The (19F,19F) DQF-COSY experiment consisted of 1,024 complex points in the indirect dimension and 8,192 complex points in the direct dimension with a spectral width of 23,229 Hz in each dimension.
Results and Discussion
The structures of the fluorinated membrane-associating species, TFOM, the membrane-bound surface-associated spin label, CAT-16, and the phospholipid, DMPC, are shown in Fig. 1. The second constituent of the isotropic bicelle dispersion, DHPC, is identical to DMPC except that the fatty acid chains each consist of 6, rather than 14, carbon atoms. TFOM, which is readily solubilized in the isotropic bicelle, is expected to anchor into the membrane such that the fluorines at positions 3 through 8 would be progressively immersed in the membrane interior. In this study, all measurements were performed by using DMPC/DHPC molar ratios of either 0.5 or 1.0. In bicellar dispersions, the long chain lipid, DMPC, is known to organize as a bilayer, whereas DHPC coats the hydrophobic edges of the bilayer to form a disk-shaped aggregate, referred to as a bilayered micelle or bicelle (30–34). If the molar ratio of DMPC to DHPC is between roughly 2.5 and 5, these bicelles are observed to align in a magnetic field. However, if a molar ratio of between 0.5 and 1.0 is used, the bicelle dimensions are believed to be sufficiently reduced such that the system no longer aligns and the fast tumbling rates of the bicelles lead to high-resolution NMR spectra (26, 35). The 19F NMR spectrum in Fig. 2A was obtained from TFOM in a 15% isotropic bicellar dispersion consisting of DMPC and DHPC (DMPC/DHPC = 0.5). The spectrum in Fig. 2B, which represents an identical concentration of TFOM in a 15% DMPC/DMPG (15:1 molar ratio) SUV system, reveals a similar distribution of chemical shift resonances (Table 1), although with poorer resolution than that in Fig. 2A. The most striking difference between the spectrum of TFOM in an isotropic bicelle dispersion and an SUV is that the trifluoromethyl resonance, which appears furthest downfield, is a doublet in the SUV spectrum. These observations suggest that TFOM inserts into the isotropic bicelle system in a manner similar to that in the SUVs and that the trifluoromethyl doublet in the SUV spectrum is related to packing differences of TFOM between the inner and outer monolayers of the SUV. Assignments of TFOM in the isotropic bicelle (Fig. 2A) were made on the basis of chemical shifts, NOEs, and J-couplings (36). The furthest downfield peak, assigned to position 8, whose chemical shift is characteristic of a trifluoromethyl resonance, gave a positive NOE to the most upfield peak, assigned to position 7, in a difference NOE experiment. The difference NOE experiment, which involved selective saturation, mixing, and detection of signal (subtracted from off-resonance saturation, mixing, and detection), revealed only one NOE from the trifluoromethyl (8-position) to the 7-position and vice versa. The trifluoromethyl resonance is a triplet, with splittings characteristic of the four-bond J-coupling to the 6-difluoromethylene group (36). The 4-bond J-couplings between fluorinated substituents are typically much larger than three-bond couplings in perfluorinated aliphatics (36–38). Although the triplet in the trifluoromethyl resonance can be observed, no other J-couplings can be observed directly in the spectrum. However, the four-bond couplings can be seen easily from the (19F,19F) double quantum-filtered–correlated spectroscopy experiment, as shown in Fig. 3. In this case, the trifluoromethyl (8-position) clearly couples to position 6, which in turn couples to position 4, whereas position 7, identified by the difference NOE experiment, couples to position 5, which in turn couples to position 3.
Figure 1.

Structural formulae of the fluorinated detergent TFOM, the phospholipid DMPC, and the nitroxide spin probe CAT-16. Note that positions 3–8 designate the specific fluorinated sites on TFOM.
Figure 2.

19F NMR spectra of TFOM in a 15% (wt/wt) isotropic bicelle (DMPC/DHPC = 0.5) (A), a 15% (wt/wt) small unilamellar vesicle dispersion consisting of DMPC/DMPG = 15:1 (B), and a 15% (wt/wt) isotropic bicelle (DMPC/DHPC = 0.5), under 20 bar of oxygen partial pressure (C).
Table 1.
Chemical shifts, referenced to trifluoroacetic acid (−76.55 ppm), and relaxation rates from inversion recovery experiments of TFOM in the isotropic bicelle sample and SUV
| Position | δ0, ppm | δSUV | δ20bar − δ0 | R10, s−1 | R120 bar − R10 | R1CAT-16 − R10 |
|---|---|---|---|---|---|---|
| 3 | −114.083 | −114.4 | 0.000 | 2.496 ± .008 | 2.45 ± .05 | 18.2 ± .66 |
| 4 | −124.091 | −124.4 | 0.019 | 2.056 ± .004 | 2.91 ± .05 | 12.93 ± .20 |
| 5 | −122.934 | −123.1 | 0.070 | 2.047 ± .006 | 3.15 ± .06 | 12.72 ± .27 |
| 6 | −123.939 | −124.2 | 0.098 | 1.972 ± .005 | 3.20 ± .05 | 12.93 ± .20 |
| 7 | −127.345 | −127.5 | 0.155 | 1.588 ± .003 | 3.71 ± .05 | 7.48 ± .16 |
| 8 | −82.39 | −82.2, −82.4 | 0.183 | 1.200 ± .001 | 4.18 ± .02 | 6.61 ± .04 |
δ0 and δSUV represent chemical shifts in the isotropic bicelle and SUV, and δ20bar − δ0 represents chemical shift changes in the TFOM at 20 bar (PO2), relative to the isotropic bicelle at ambient pressure. R10 represents the spin-lattice relaxation rates of TFOM at ambient pressure. R120 bar − R10 and R1CAT − 16 − R10 represent changes in the relaxation rates by introducing oxygen partial pressures of 20 bar or by addition of the surface nitroxide spin label, CAT-16.
Figure 3.

Contour plot from a (19F,19F) double quantum-filtered–correlated spectroscopy experiment of TFOM in an isotropic q = 0.5 bicelle at 40°C. Indices 3–8 refer to the specific chain position on TFOM.
Attempts to identify resonances associated with sites close to the surface of the membrane, either by differential shifts or line broadening resulting from the addition of paramagnetic shift reagents (Dy3+, Dy3+ EDTA, or Dy3+ DTPA or equivalent complexes with Gd3+) all failed. Although large downfield shifts could be observed, each resonance gave almost exactly the same shift with increasing amounts of shift reagent added. We attribute this result to either fast exchange of the detergent, TFOM, with coexisting micelles or penetration of the shift reagent into the membrane. By comparison, in an oriented bicelle medium consisting of a 25% (wt/wt) dispersion of TFOM and DMPC/DHPC = 3, differential downfield shifts resulting from addition of Dy3+ EDTA could be seen for position 3, confirming our assignment and the notion that position 3 lies closest to the membrane–water interface.
Membrane-soluble and water-soluble ESR spin labels have been used widely in NMR studies of membrane proteins and membrane peptides, primarily in detergent micelles to determine surface exposure and immersion depth (39–44). In general, these experiments involve the addition of spin-labeled probes, in which the nitroxide or paramagnetic species is located (on a lipid or fatty acid chain) at a particular depth in the micelle. Alternatively, the spin label can be attached to the headgroup of a lipid or simply solubilized in the water region. Because the paramagnetic spin labels affect NMR relaxation times and hence peak heights of resonances associated with nuclear spins within an approximate 15-Å radius of the spin label, changes in peak heights of one- or two-dimensional NMR spectra relate directly to water accessibility or depth of the specific nuclear spins in the micelle. By using paramagnetic species that are confined to either the membrane interior or the water region, membrane immersion depth may be determined similarly by power-saturation ESR experiments of spin labels attached directly to cysteine residues, introduced at a site of interest in the membrane protein. However, the changes in the nitroxide ESR spin-lattice relaxation time, T1, are the result of collision-mediated (Heisenberg exchange) interactions between the paramagnetic species and nitroxide label. In contrast, NMR spin-lattice relaxation times are shortened by the addition of paramagnetic species through dipolar relaxation mechanisms and therefore depend on the motionally averaged distance, rIS, between the nuclear and paramagnetic species. Therefore, the power saturation ESR experiments provide information on local mobility and collisional access of paramagnetic species, whereas the NMR measurements more accurately reflect distance. In particular, the paramagnetic contribution to the nuclear spin-lattice relaxation rate, 1/T1P, is given in the short correlation time limit (ωlτs ≪ 1) as (45–46)
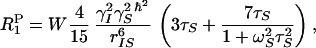 |
1 |
where ωI and ωS define the nuclear and electronic Larmor frequencies, whereas γI and γS represent the respective gyromagnetic ratios, τS characterizes the motion of the rIS vector, and W is proportional to the local concentration of the paramagnetic species. The concentration of molecular oxygen in the membrane, CO2M, is given by Henry's law (at least up to partial pressures of 100 bar) and may be expressed in terms of a partition coefficient, Kp, between the membrane and water (43), such that
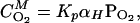 |
2 |
where αH is Henry's Law constant (5.9 × 1017 bar−1⋅cm−3). By using a membrane partition coefficient of 2.9 (46), the concentration of molecular oxygen in the membrane at a partial pressure of 20 bar is estimated to be 3.6 × 1019⋅cm−3, or 4.5% of the concentration of the lipids. Thus, we can express the observed spin-lattice relaxation rate at 20 bar, R120bar, in terms of a paramagnetic component, R1P, and a nonparamagnetic component, R10. By measuring the spin-lattice relaxation rates at 20 bar and at ambient oxygen partial pressures after degassing the sample, we can determine R1P from the difference,
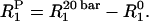 |
3 |
This procedure assumes there are no conformational or dynamic changes introduced by the pressure. Generally, conformational or dynamical effects of pressure are not observed in model membranes by NMR below 200 bar (47–48). Furthermore, Kimmich and Peters have made careful control measurements of 1H T1 relaxation times in lipid vesicles under equivalent N2 partial pressures to demonstrate that changes in relaxation times arise from paramagnetic relaxation effects with oxygen (46). In our hands, 19F T1 measurements of TFOM in bicelles at equivalent N2 pressures resulted in slightly enhanced relaxation rates from the equivalent measurement at ambient pressure, although there were no depth-dependent effects.
One important advantage of the use of paramagnetic oxygen to influence fluorine spin-lattice relaxation times effectively in the model membrane is that this effect is not observed to contribute to severe line broadening at the pressure studied with the exception of a marginal broadening observed in the trifluoromethyl group. Here, the spin-lattice relaxation times are considerably longer than spin-spin relaxation times, and the paramagnetic oxygen is predominantly T1 effective. Furthermore, as evidenced by the spectrum in Fig. 2C of TFOM in an isotropic bicelle dispersion under 20 bar PO2, the chemical shift resonances change only slightly in comparison to the line widths. However, a clear downfield shift is observed to increase with chain position on introduction of oxygen at 20 bar partial pressure (Table 1). The most significant effect of the higher oxygen concentrations for this sample involves spin-lattice relaxation rate enhancement. These changes in spin-lattice relaxation rates are reported in Table 1 for positions 3 through 8 because of the introduction of oxygen at 20 bar partial pressure or because of the addition of the surface-associated nitroxide spin label, CAT-16. The changes in relaxation rates (R120bar − R10) provide a measure of the paramagnetic relaxation term alone, R1P, which through Eq. 1 is directly proportional to the local oxygen concentration. Similarly, the difference, R1CAT-16 − R10, reflects the paramagnetic interactions between the various fluorine spins and the surface-bound nitroxide probe. In this way, it is possible to factor out relaxation processes possibly not related to immersion or surface accessibility. The difference, R120bar − R10, is exquisitely sensitive to chain position. R120bar − R10 is largest for the trifluoromethyl position (i.e., R120bar − R10 = 4.2 s−1), which is expected to be most buried in the membrane. Moreover, this difference decreases monotonically within the uncertainty toward the 3-position (i.e., R120bar − R10 = 2.45 s−1), consistent with the anticipated immersion depth profile of the fluorine labels in the membrane. These results are summarized in Fig. 4, where the paramagnetic relaxation rates arising from oxygen are observed clearly to increase with chain position or equivalently membrane immersion depth. In contrast, the paramagnetic relaxation rates resulting from the addition of CAT-16 generally decrease with increasing immersion depth. The third profile in Fig. 4 represents the dependence of paramagnetic relaxation rates of TFOM in methanol because of the addition of oxygen at 100 bar. Because the concentration of TFOM was such that it was below the critical micelle concentration, oxygen should be equally accessible to each position, leading to an almost flat line as shown in Fig. 4.
Figure 4.

Paramagnetic relaxation rates (R1P) of TFOM in an isotropic q = 0.5 bicelle under 20 bar of oxygen partial pressure (solid squares). Paramagnetic relaxation rates at ambient pressures on addition of a surface-anchored nitroxide spin label, CAT-16, are shown as open squares. The corresponding rates in methanol under 100 bar of oxygen partial pressure are shown as crosses. Note that the left-hand scale applies to the solid squares, and the right-hand scale applies to the open squares and crosses.
Conclusions
In summary, the changes in 19F NMR spin-lattice relaxation rates induced by the presence of oxygen at 20 bar or greater partial pressure are a sensitive indicator of membrane immersion depth of fluorinated membrane-associated molecules in model membranes, such as detergent micelles and isotropic bicelles. The trend in which the paramagnetic relaxation rate, R120bar − R10, increases with position along the detergent chain is attributed to the fact that positions 3 through 8 are increasingly immersed in the membrane, in keeping with the depth-dependent solubility properties of oxygen. The opposite trend of paramagnetic relaxation rates with chain position was observed on addition of CAT-16, which is consistent with the fact that the paramagnetic spin label is located on the surface of the membrane. It is important to emphasize that molecular oxygen does not lead to deleterious line broadening, as is the case in NMR applications of spin labels to probe membrane depth. This is partly because of the favorably short electronic relaxation time of oxygen and the differences in spin-lattice and spin-spin relaxation rates in these systems. Finally, in contrast with membrane immersion studies that combine spin labels and NMR, molecular oxygen can be added easily and removed, thereby requiring a single sample. The effects of pressure on local conformation or dynamics can also be neglected safely at 20 bar.
The results suggest that it should be possible to make use of changes in oxygen-induced 19F NMR spin-lattice relaxation times to probe immersion depth of site-directed 19F-labeled membrane proteins in detergent micelle and model membrane systems. These conclusions are supported by similar relaxation studies of specifically fluorinated cholesterols in model membranes, which will be reported in a subsequent paper. Recently, a similar study showed that proton spin-lattice relaxation rates can be used to monitor accessibility of oxygen to the interior of a water-soluble protein (49). However, it should be emphasized that our methodology is intended to probe the surface or topography of a membrane-associated macromolecule or membrane protein whose labels are exposed to the lipid bilayer milieu rather than the more heterogeneous interior of a water-soluble protein.
Acknowledgments
R.S.P. gratefully acknowledges Research Corporation (RI0322) for making this research possible. We also thank Ad Bax, Chris Roe, J. Brechtelsbauer, M. Tubergen, and L. Holland for their assistance with technical aspects of high pressure experiments.
Abbreviations
- TFOM
3,3,4,4,5,5,6,6,7,7,8,8,8-tridecylfluoro β-d-octyl maltoside
- DMPC
1,2-dimyristoyl-sn-glycero-3-phosphocholine
- DHPC
1,2-dihexanoyl-sn-glycero-3-phosphocholine
- SUV
small unilamellar vesicle
- DMPG
1,2-dimyristoyl-sn-glycero-3-phosphoglycerol
- CAT-16
4-(N,N-dimethyl-N-hexadecyl) ammonium-2,2,6,6-tetramethylpiperidine-1-oxyl iodide
- NOE
nuclear Overhauser effect
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.170295297.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.170295297
References
- 1.Windrem D A, Plachy W Z. Biochim Biophys Acta. 1980;600:655–665. doi: 10.1016/0005-2736(80)90469-1. [DOI] [PubMed] [Google Scholar]
- 2.Boyd D, Schierle C, Beckwith J. Protein Sci. 1998;7:201–205. doi: 10.1002/pro.5560070121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wallin E, von Heijne G. Protein Sci. 1998;7:1029–1038. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Riek R, Wider G, Pervushin K, Wuthrich K. Proc Natl Acad Sci USA. 1999;96:4918–4923. doi: 10.1073/pnas.96.9.4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pellecchia M, Sebbel P, Hermanns U, Wuthrich K, Glockshuber R. Nat Struct Biol. 1999;6:336–339. doi: 10.1038/7573. [DOI] [PubMed] [Google Scholar]
- 6.Walz T, Grigorieff N. J Struct Biol. 1998;121:142–161. doi: 10.1006/jsbi.1998.3945. [DOI] [PubMed] [Google Scholar]
- 7.Garavito R M, White S H. Curr Opin Struct Biol. 1997;7:533–536. doi: 10.1016/s0959-440x(97)80118-2. [DOI] [PubMed] [Google Scholar]
- 8.Czerski L, Vinogradova O, Sanders C R. J Magn Reson. 2000;142:111–119. doi: 10.1006/jmre.1999.1920. [DOI] [PubMed] [Google Scholar]
- 9.Altenbach C, Marti T, Khorana H G, Hubbell W L. Science. 1990;248:1088–1092. doi: 10.1126/science.2160734. [DOI] [PubMed] [Google Scholar]
- 10.Altenbach C, Greenhalgh D A, Khorana H G, Hubbell W L. Proc Natl Acad Sci USA. 1994;91:1667–1671. doi: 10.1073/pnas.91.5.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hubbell W L, Altenbach C. Curr Opin Struct Biol. 1994;4:566–573. [Google Scholar]
- 12.Steinhoff H J, Mollaaghababa R, Altenbach C, Khorana H G, Hubbell W L. Biophys Chem. 1995;56:89–94. doi: 10.1016/0301-4622(95)00019-t. [DOI] [PubMed] [Google Scholar]
- 13.Hubbell W L, Mchaourab H S, Altenbach C, Lietzow M A. Structure (London) 1996;4:779–783. doi: 10.1016/s0969-2126(96)00085-8. [DOI] [PubMed] [Google Scholar]
- 14.Dettman H, Weiner J H, Sykes B D. Biophys J. 1982;37:243–251. doi: 10.1016/S0006-3495(82)84673-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson M L, Dahlquist F W. Biochemistry. 1985;24:1920–1928. doi: 10.1021/bi00329a018. [DOI] [PubMed] [Google Scholar]
- 16.Gerig J T. Prog NMR Spectrosc. 1994;26:293–370. [Google Scholar]
- 17.Danielson M A, Falke J J. Annu Rev Biophys Biomol Struct. 1996;25:163–195. doi: 10.1146/annurev.bb.25.060196.001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang F, Yu L, He D-Y, Yu C-A. J Biol Chem. 1991;266:20863–20869. [PubMed] [Google Scholar]
- 19.Birnbaum E R, Hodge J A, Grinstaff M W, Schaefer W P, Henling L, Labinger J A, Bercaw J E, Gray H B. Inorg Chem. 1995;34:3625–3632. [Google Scholar]
- 20.Hull W E, Sykes B D. Biochemistry. 1976;15:1535–1546. doi: 10.1021/bi00652a027. [DOI] [PubMed] [Google Scholar]
- 21.Sykes B D, Hull W E. Methods Enzymol. 1978;49:270–295. doi: 10.1016/s0076-6879(78)49015-9. [DOI] [PubMed] [Google Scholar]
- 22.Kim H-W, Perez J A, Ferguson S J, Campbell I D. FEBS Lett. 1990;272:34–36. doi: 10.1016/0014-5793(90)80442-l. [DOI] [PubMed] [Google Scholar]
- 23.Drake S K, Bourret R B, Luck L A, Simon M I, Falke J J. J Biol Chem. 1993;268:13081–13088. [PMC free article] [PubMed] [Google Scholar]
- 24.Klein-Seetharaman J, Getmanova E V, Loewen M C, Reeves P J, Khorana H G. Proc Natl Acad Sci USA. 1999;96:13744–13749. doi: 10.1073/pnas.96.24.13744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perozo E, Cortes D M, Cuello L G. Nat Struct Biol. 1998;5:459–469. doi: 10.1038/nsb0698-459. [DOI] [PubMed] [Google Scholar]
- 26.Vold R R, Prosser R S, Deese A J. J Biomol NMR. 1997;9:329–335. doi: 10.1023/a:1018643312309. [DOI] [PubMed] [Google Scholar]
- 27.Jeener J, Meier B H, Bachmann P, Ernst R R. J Chem Phys. 1979;71:4546–4553. [Google Scholar]
- 28.Piatini U, Sørensen O W, Ernst R R. J Am Chem Soc. 1982;104:6800–6801. [Google Scholar]
- 29.Shaka A J, Freeman R. J Magn Reson. 1983;51:169–173. [Google Scholar]
- 30.Ram P, Prestegard J H. Biochim Biophys Acta. 1998;940:289–294. doi: 10.1016/0005-2736(88)90203-9. [DOI] [PubMed] [Google Scholar]
- 31.Sanders C R, Prestegard J H. Biophys J. 1990;58:447–460. doi: 10.1016/S0006-3495(90)82390-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanders C R, Schwonek J P. Biochemistry. 1992;31:8898–8905. doi: 10.1021/bi00152a029. [DOI] [PubMed] [Google Scholar]
- 33.Sanders C R, Hare B J, Howard K P, Prestegard J H. Prog Nucleic Magn Reson Spectrosc. 1994;26:421–444. [Google Scholar]
- 34.Sanders C R, Prosser R S. Structure (London) 1998;6:1227–1234. doi: 10.1016/s0969-2126(98)00123-3. [DOI] [PubMed] [Google Scholar]
- 35.Vold R R, Prosser R S. J Magn Reson. 1996;113:267–271. [Google Scholar]
- 36.Mooney E F. An Introduction to 19F NMR Spectroscopy. New York: Heyden; 1970. [Google Scholar]
- 37.Mason J, editor. Multinuclear NMR. New York: Plenum; 1987. [Google Scholar]
- 38.Berger S, Braun S, Kalinowski H O. 19F NMR-Spektroskopiein, NMR-Spektroskopie von Nichtmetallen. Vol. 4. Stuttgart, Germany: Verlag Stuttgart; 1992. [Google Scholar]
- 39.Wertz S L, Savino Y, Cafiso D S. Biochemistry. 1996;35:11104–11112. doi: 10.1021/bi961248x. [DOI] [PubMed] [Google Scholar]
- 40.van den Hooven H W, Spronk C A E M, van de Kamp M, Konings R N H, Hilbers C W, van de Ven F J M. Eur J Biochem. 1996;235:394–403. doi: 10.1111/j.1432-1033.1996.00394.x. [DOI] [PubMed] [Google Scholar]
- 41.Papavoine C H M, Aelen J M A, Konings R N H, Hilbers C W, Vandeven F J M. Eur J Biochem. 1995;232:490–500. doi: 10.1111/j.1432-1033.1995.490zz.x. [DOI] [PubMed] [Google Scholar]
- 42.Ruan K-H, Li D, Ji J, Lin Y-Z, Gao X. Biochemistry. 1998;37:822–830. doi: 10.1021/bi971881y. [DOI] [PubMed] [Google Scholar]
- 43.Curtain C, Separovic F, Nielsen K, Craik D, Zhong Y, Kirkpatrick A. Eur Biophys J Biophys Lett. 1999;28:427–436. doi: 10.1007/s002490050225. [DOI] [PubMed] [Google Scholar]
- 44.Schibli D J, Hwang P M, Vogel H J. Biochemistry. 1999;38:16749–16755. doi: 10.1021/bi990701c. [DOI] [PubMed] [Google Scholar]
- 45.Abragam A. The Principles of Nuclear Magnetism. Oxford, U.K.: Clarendon; 1961. [Google Scholar]
- 46.Kimmich R, Peters A. Chem Phys Lipids. 1975;14:350–362. doi: 10.1016/0009-3084(75)90072-9. [DOI] [PubMed] [Google Scholar]
- 47.Jonas J, Xie C-L, Jonas A, Grandinetti P J, Campbell D, Driscoll D. Proc Natl Acad Sci USA. 1988;85:4115–4117. doi: 10.1073/pnas.85.12.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bonev B B, Morrow M R. Biophys J. 1995;69:518–523. doi: 10.1016/S0006-3495(95)79925-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Teng C L, Bryant R G. J Am Chem Soc. 2000;122:2667–2668. [Google Scholar]